

Abstract
Purpose
To optimize and streamline molecular genetics techniques in diagnosing choroideremia (CHM).
Methods
PCR primers were designed for exons 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, and 15 of the CHM gene. Each PCR protocol was optimized so that all exons could be amplified with the same component ratio and PCR conditions. Sense and antisense primers were tested for their ability to be used as sequencing primers. Fibroblast cells were cultured, and an immunoblot analysis was performed to detect the presence or absence of Rab escort protein 1 (REP-1) in a suspected CHM patient sample when no mutation was detected with sequencing. Multiplex ligation-dependent probe amplification (MLPA) of the CHM gene was performed and used to detect deletions and duplications in affected males and female carriers. RNA analysis using cDNA was used to detect the presence or absence of the CHM transcript and to search for splice defects.
Results
The newly designed PCR primers allow for more efficient PCR preparation and sequencing to detect point mutations in affected males and female carriers. Immunoblot successfully detects the absence of REP-1 in a CHM patient. MLPA identifies deletions and duplications spanning multiple exons in the CHM gene. RNA analysis aids in detecting splice variants.
Conclusions
The development of new molecular biology techniques and ongoing optimization of existing methods allows for an improved integrated approach to confirm CHM diagnosis and carrier status in consideration of patient family history and available patient sample materials. CHM can be confirmed with an immunoblot assay. To detect the molecular cause of CHM, an examination of the genomic DNA or the mRNA must be performed. Presymptomatic carriers with no identifiable fundus signs can be identified only through molecular analysis of genomic DNA or through quantitative assays.
Introduction
Choroideremia (CHM) is an X-linked eye disorder affecting 1 in 50,000 men [1]. The condition is caused by a mutation in the CHM gene that encodes Rab escort protein 1 (REP-1) [2]. Males with CHM suffer from progressive vision loss beginning with night blindness at a young age, leading to complete blindness later in life. Female carriers are generally asymptomatic; however, occasionally a heterozygous female experiences mild symptoms [2].
The CHM gene encodes the protein REP-1, an essential component of an enzyme complex formed with Rab geranylgeranyltransferase (GGT) [2]. A deficiency of GGT function caused by a mutation in CHM leads to insufficient transfer of geranylgeranylpyrophosphate groups onto Rab proteins. Rabs cannot participate in pathways of intracellular vesicular transport in the absence of REP. REP-1 is normally expressed ubiquitously, and the loss of functioning REP-1 appears to be compensated by REP-2 in all tissues, except in the eye [3]. REP-1 function is particularly crucial for the function of the retinal pigment epithelium and photoreceptors [4]. Ultimately, lack of REP-1 results in the degeneration of these cells, as well as associated choroidal tissue.
The CHM gene spans 186,382 bp on the X chromosome. The mRNA is made up of 15 exons and is 5,442 bp long. All exons are fewer than 400 bp long, with the exception of exon 15, which is 3,642 bp [5].
The open reading frame is 1,962 bp and produces a 653 amino acid long protein (95 kDa). A wide variety of CHM-causing mutations have been identified: small deletions, nonsense mutations, missense mutations, frameshifts, splice site defects, retrotransposon insertion and deletion of the entire CHM gene have been reported [6]. Thus, sequencing of the CHM gene supplemented with immunoblot analysis has emerged as a diagnostic tool used to identify mutations causing CHM [7]. Due to the large size of the gene’s introns, amplifying them with PCR and sequencing the entire gene region for every patient is time-consuming and technically impractical. Thus, only exons and intron–exon boundaries are amplified and sequenced from the genomic DNA. Analysis of RNA can also identify mutations in exons and may provide insights into splice defects and exon deletions and duplications. However, although CHM is expressed in all tissues, blood is most commonly used as the sample for analysis due to ease of collection and transport. The technical limitation in RNA extraction from blood samples that may not be fresh and the lack of availability of other tissues for analysis means that patient RNA is not always available for diagnostic testing.
Immunoblot analysis using patient fibroblast cells reveals the presence or absence of the REP-1 protein [7]. If a fibroblast cell line cannot be obtained or when identification of the genetic mutation is required, a genomic DNA sample can be used to amplify the exons of the CHM gene followed by sequencing to detect a mutation. Additionally, if no mutation is found in the genomic DNA, RNA can be extracted from patient cells, and cDNA of the CHM transcript can be created. The cDNA can be sequenced to verify any splice variations that cannot be predicted based on the sequencing results from the exon analysis. To detect copy number variation of exons in a patient, MLPA can be performed on genomic DNA, or an RNA analysis could be performed based on the availability of patient cells.
Primers suitable for PCR amplification and subsequent sequencing have been designed by Bokhoven et al. [8]. Multiple protocols were required to amplify all 15 exons, so we sought primer designs for a more efficient procedure. The labor-intensive process of screening all CHM exons can be streamlined to have one PCR reaction formulation and one uniform PCR condition that can be applied for all primers and can be processed at once on a single thermal cycler block. In addition, not all of these previously designed PCR primers work efficiently as sequencing primers; some exons can be sequenced in only one direction, which is suboptimal if the entire amplicon sequence is to be analyzed. In addition, the original method used two primer pairs to amplify the largest coding exon, exon 5. Through redesign, the reaction can be reduced to one primer pair resulting in a larger amplicon that can be sequenced using one primer pair [8].
Methods
Immunoblot
Protein was extracted from a confluent fibroblast culture from one 10 cm cell culture dish (1–2 × 106 cells) in 500 µl of lysis buffer containing 0.05 M Tris pH8, 0.15 M NaCl, 1% Triton X, and protein inhibitor cocktail (Roche, Basel, Switzerland). The cell debris was precipitated at 16,000 ×g for 15 min at 4 °C after 20 min incubation on ice. Aliquots were denatured in a 5× denaturing buffer containing 60 mM Tris pH 6.8, 25% glycerol, 2% sodium dodecyl sulfate, 5% β-mercaptoethanol, and 0.1% bromophenol blue, and boiled at 95 °C for 5 min. Proteins were separated via electrophoresis: 40 µl of denatured lysate was separated on a 10% sodium dodecyl sulfate-polyacrylamide gel with a PageRule Plus Prestained Protein Ladder (Thermo Fisher Scientific, Waltham, MA). Proteins were transferred onto a nitrocellose membrane (BioRad, Hercules, CA) by electroblotting (2.5–3 h at 80 V). The membrane was blocked with 2% skim milk, 0.05% Tween-20 in 150 mM Tris pH 7.6, and 730 mM NaCl (TBS) and then incubated overnight at 4 °C with primary mouse monoclonal anti-REP-1 antibody (2F1, sc-23905; Santa Cruz Biotechnology, Dallas, TX) diluted 1:500 in blocking buffer or with 1:2,500 monoclonal anti-beta-actin-horseradish peroxidase conjugated antibody (Clone AC-15; Sigma-Aldrich, St. Louis, MO). Blots were washed twice for 10 min with TBS containing 0.05% Tween-20 followed by two 10 min washes with TBS. The 2F1 incubated blots are then probed with secondary goat anti-mouse immunoglobulin 1-horseradish peroxidase conjugated antibody (sc-2060; Santa Cruz Biotechnology, Dallas, TX) diluted 1:1,000 in blocking buffer for 2.5 h and then washed as described above. The membranes were then incubated with Amersham ECL Plus Western Blotting Detection System (General Electric, Fairfield, CT) according to the manufacturer’s instructions, and protein bands were detected using KODAK BioMax MR Film in a Kodak X-OMAT 2000 X-ray film processor (Eastman Kodak Company, Rochester, NY).
Genomic DNA analysis
Primer design
The NetPrimer applet (PREMIER Biosoft International, Palo Alto, CA) was used to evaluate the predicted efficiency of a primer sequence for exons 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, and 15 (Table 1) and to determine features of the primer pair including hairpins, self-dimers, and cross-dimers. Primers for exon 1, 13, and 14 were selected from Bokhoven et al. [8]. Primers for exon 2 were designed by Song et al. [9].
Table 1. Details of the primer pairs used to amplify the exons of the CHM gene.
| Exon | Direction | Primer | Amplicon (bp) | Tm (°C) | |
|---|---|---|---|---|---|
| 1 |
* |
FWD |
GACCTTCCACCCAAGAACTAC |
216 |
55.0 |
| REV |
ACAGTCTTCCTAAACTTTGTCC |
52.4 |
|||
| 2 |
+ |
FWD |
CAGCAATGGCATGTATTGAACATT |
384 |
54.6 |
| REV |
TGATGCATTTGGTTTATTCTCAAAGA |
53.6 |
|||
| 3 |
Δ |
FWD |
AATTGTTACAGAAGAAGCTACTATGG |
160 |
52.9 |
| REV |
GGTTTTCTTCAGTGCAGGGTTA |
55.3 |
|||
| 4 |
Δ |
FWD |
TTTTTCTCCCCTTCCTTTGA |
411 |
51.5 |
| REV |
TGTAGCAACCAATGCCACAT |
55.1 |
|||
| 5 |
Δ |
FWD |
TTCCAATACAGTTCCCCTGT |
648 |
53.4 |
| REV |
TGTTTTCAATGCAAAGATGG |
49.4 |
|||
| 6 |
Δ |
FWD |
ATGGATCAGGTTTTGCTGCT |
481 |
54.9 |
| REV |
ACCACGGAGGACTGGAATTT |
56.3 |
|||
| 7 |
Δ |
FWD |
GGGAAAAAAGTGTAATTTGG |
330 |
47.4 |
| REV |
TGGCTAACCTATTGATAGTGC |
52.0 |
|||
| 8 |
Δ |
FWD |
TGTCCAAAATGTAATACCACC |
402 |
50.5 |
| REV |
TCAAGTATCACTTTTAGAAGGG |
49.7 |
|||
| 9 |
Δ |
FWD |
TCTGGTTTGCTCACAGTTCT |
346 |
54.0 |
| REV |
TGAAGGTTACTTATATCATCCTTAC |
49.9 |
|||
| 10 |
Δ |
FWD |
GCCCTCAAAATAGCAACAAGA |
452 |
53.4 |
| REV |
GGCTTCCCTAAAACCAGACC |
55.6 |
|||
| 11 |
Δ |
FWD |
GGGAGGTGACACTTTTATCC |
500 |
52.8 |
| REV |
AGAGAGTAAAACGGTGCTTG |
52.6 |
|||
| 12 |
Δ |
FWD |
AAATATGTTTCAAATTCTGTTCCAAA |
436 |
50.6 |
| REV |
AAGGGGATGGTGTGAAATGA |
54.0 |
|||
| 13 |
* |
FWD |
CGCTCAGCTCTCTATTATCCAT |
257 |
53.8 |
| REV |
CCGGAAGATTATGATGGTTACAT |
52.5 |
|||
| 14 |
* |
FWD |
TAGGCTACACAGTGTAGTAA |
322 |
49.6 |
| REV |
GACTTCTCTCCTCCCAGAGG |
56.1 |
|||
| 15 |
Δ |
FWD |
TGAGGTACTGCCATCCTTGA |
478 |
53.9 |
| REV | TGAGACCAGTCAGAATTTCCAA | 55.5 |
PCR amplification and sequencing using genomic DNA
For a 50 µl reaction, the following reagents were included: 10 µl of 10X Pfx Amplification Buffer (Invitrogen, Burlington, ON), 1.5 µl of 10 mM dNTP mix, 0.75 µl of 50 mM MgSO4, 1.5 µl each of 10 µM sense and antisense primer (Integrated DNA Technologies, Coralville, IA; Table 1), 0.4 µl of Platinum Pfx DNA polymerase (Invitrogen), 100 ng of genomic DNA template, and sterile H2O.
A touchdown PCR protocol was used in a thermal cycler (BioRad). The initial denaturation step was at 94 °C for 5 min followed by a cycle of denaturation at 94 °C for 15 s, first annealing at 60 °C for 30 s and extension at 68 °C for 45 s. The subsequent nine cycles were run with a decreasing annealing temperature of 1 °C each cycle, followed by 26 cycles at an annealing temperature of 50 °C. The final extension was at 68 °C for 5 min, and the temperature was held at 4 °C. Samples were loaded onto a 1.5% agarose gel and run for 25 min at 120 V. Subsequent treatment with ethidium bromide allowed visualization of the PCR product and gel purification.
The PCR product was gel extracted with the QIAEX II Gel Extraction Kit according to the manufacturer’s directions (Qiagen, Germantown, MD). The concentration of gel purified DNA was determined with a NanoDrop 1000 Spectrophotometer (Thermo Fisher Scientific). Samples were sequenced (Eurofins MWG Operon, Ebersberg, Germany) using sense and antisense PCR primers. Sequence chromatogram files were edited and aligned with NCBI reference sequence NC_000023.10 by ChromasPro (Technelysium, South Brisbane, Australia).
Multiplex ligation-dependent probe amplification
The MLPA analysis was performed as described by Chi et al. [10]. The data were analyzed with Coffalyser.Net software (MRC-Holland, Amsterdam, Netherlands).
Cell culture
Fibroblasts were cultured from skin biopsies obtained with consent from patient and control subjects. Written informed consent was obtained from all research participants. The protocols were approved by the Health Research Ethics Board of the University of Alberta and adhered to the tenets of the Declaration of Helsinki and the ARVO statement regarding research on human subjects. The skin biopsies were finely minced with a sterile scalpel blade and placed in Cascade Biologics attachment factor (Invitrogen) coated wells of a six-well culture dish and incubated in a high-glucose DMEM (Sigma-Aldrich Co., catalog number D5671, St. Louis, MO; supplemented with 20% fetal bovine serum, penicillin-streptomycin, non-essential amino acids, L-glutamine, and sodium pyruvate; Invitrogen) at 37 °C, 5% CO2. The cultures received fresh medium every 3 days, were observed for outgrowth of fibroblasts, and were transferred to a 6 cm culture dish at confluence. Fibroblasts were subsequently cultured in 10 cm culture dishes.
RNA analysis
cDNA synthesis
RNA was extracted from two confluent 10 cm culture dishes of fibroblast cells (approximately 1–2 × 106 cells) in 500 µl TRIzol reagent (Invitrogen) according to the manufacturer’s directions. The cDNA was synthesized using 2 µg of extracted RNA and 1 µl Maxima Reverse Transcriptase per 20 µl reaction with reverse transcription (RT) PCR, Maxima Universal First Strand cDNA Synthesis Kit (Thermo Fisher Scientific), according to the manufacturer’s directions.
Primer design
The NetPrimer applet (PREMIER Biosoft International) was used to evaluate the predicted efficiency of primer sequences flanking the open reading frame of the CHM cDNA (Table 2) and to determine features of the primer pair, including hairpins, self-dimers, and cross-dimers.
Table 2. CHM cDNA primers.
| Direction | CHM RT–PCR cDNA amplicon | Start bp | Tm (°C) |
|---|---|---|---|
| FWD |
TAATAGTCACATGACACGTTTCCC |
1 |
54.8 |
| REV |
TTTAAAATGAGCAAGTCAATGTGC |
2200 |
52.8 |
|
Direction |
CHM cDNA sequencing |
Start bp |
Tm (°C) |
| FWD |
CGGAGTTTGATGTGATCGTAATAG |
100 |
53.4 |
| FWD |
CAGCGATCCAGAGAATGC |
560 |
53.4 |
| FWD |
TACCAGGATTCTTGCATTTCG |
860 |
52.9 |
| FWD |
TTTGGTGGAATTTATTGTCTTCG |
1260 |
51.7 |
| FWD |
ATTTGACTTGCACATCTTCTAAAACA |
1600 |
53.7 |
| REV |
AGGCAGGAAGGCAGAATCT |
400 |
56.2 |
| REV |
CCTGTTCCACTCGTCCTTCT |
800 |
56.4 |
| REV |
AGGCACTGTACTGAATGGCG |
1200 |
57.6 |
| REV |
TTGTTCATTTTCTATCTCCATTTCAG |
1600 |
51.9 |
| REV | TAGACTCTGAGACCAGTCAGAATTTC | 2000 | 55.6 |
RT–PCR and cDNA sequencing primers for choroideremia open reading frame
PCR amplification and sequencing
For a 20 µl reaction, the following reagents were included: 4 µl of 5× Phusion HF Buffer (Thermo Fisher Scientific), 0.4 µl of 10 mM dNTP, 1 µl of 40 µM sense primer (Integrated DNA Technologies), 1 µl of 40 µM antisense primer (Integrated DNA Technologies), 0.2 µl of Phusion High-Fidelity DNA Polymerase (Thermo Fisher Scientific), 1 µl of 1/3 diluted cDNA template, and sterile H2O.
PCR was performed in a thermal cycler. The initial denaturation was 98 °C for 40 s followed by a cycle of denaturation at 98 °C for 20 s, annealing at 62 °C for 25 s and extension at 72 °C for 4 min. The cycle was repeated 30 times. A final extension was at 72 °C for 10 min. The PCR products were separated on a 1% agarose gel for approximately 1 h at 110 V and visualized with ethidium bromide.
The PCR products were gel extracted, and the concentration of DNA was measured according to the same procedure followed for products derived from genomic DNA (see above). Samples were sequenced using specific primers covering the entire range of PCR products (Table 2). Sequence chromatogram files where edited and aligned with NCBI reference sequence NM_000390.2 by ChromasPro (Technelysium).
Analysis of variants
Currently, 133 unique CHM mutations have been reported and catalogued (LOVD, Retinal and Hearing Impairment Genetic Mutation database). Missense mutations have rarely been identified in affected patients, with four noted in the database. Three-dimensional structures of the complexes REP1/RGGTase and REP1/Rab7 from Rattus norvegicus can provide a base for structural-functional modeling studies, due to the high sequence homology between rat Rep-1 and human REP-1 [11,12].
Results and Discussion
Immunoblot
Immunoblot analysis of the control and patient samples demonstrates the detection of REP-1 in the control patient and the absence of REP-1 in this patient with CHM (Figure 1).
Figure 1.
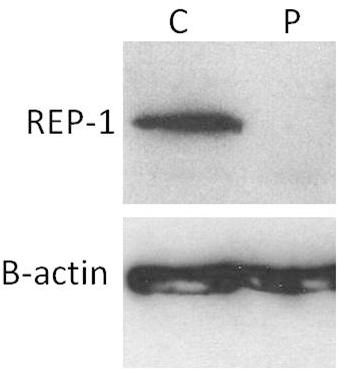
Lysates from a male healthy control subject (C) and from a male choroideremia patient (P) were analyzed with immunoblot as described in the Materials and Methods section, and probed with 2F1 antibody (upper panel) and anti-beta-actin (bottom panel). The figure demonstrates the absence of REP-1 in a CHM patient.
The absence of REP-1 protein detected by an immunoblot assay is the most direct method for molecular diagnosis of CHM (Figure 1). If REP-1 is present, it may be necessary to analyze the size of the protein product detected. Truncated REP-1 proteins have been reported [13]. If a protein of expected size is present, this suggests the patient may not have choroideremia, but it cannot be ruled out yet; further testing such as a genomic DNA analysis will provide more insight. If the protein is larger or smaller than expected, this suggests CHM. A larger protein suggests a large insertion within an exon or large duplication of an exon or a splice defect. The difficulty in obtaining cells (other than from peripheral blood) from which proteins are extracted for the immunoblot assay limits the use of this method. In practical terms, protein obtained from peripheral blood cells has proved to be a problem in allowing clear differentiation of bands when probed with anti-REP1 antibody. Although most clinical laboratories have the capability of drawing blood or extracting genomic DNA, a fibroblast culture from skin fibroblasts (or a lymphoblastoid cell line) is not always practical to set up. Since we receive patient samples from all over the world, practical issues related to shipping the samples and concerns in timely sample processing must be considered. This may limit the type of specimen available for diagnostic purposes, and immunoblotting is not always initially performed. In addition, this assay does not determine the nature of the CHM mutation, only whether the REP-1 protein is present, absent, or abnormal in size.
A normal protein product would be expected when analyzing a potential female carrier with immunoblot analyses. In the case of a large deletion or insertion, a secondary protein band of improper size may be detected, indicating a female carrier. If no secondary band is detected, the immunoblot results would be inconclusive, as it cannot distinguish whether there are one or two functional copies of CHM.
An additional complication arises because each female cell has only one active X chromosome attributable to lyonization [14], a process that occurs early in embryonic development and is random, resulting in a mixed cell population of cultured skin fibroblasts or lymphocytes. CHM expression analysis in cell cultures derived from heterozygous females could therefore be skewed. A portion of the cells in culture would be expected to express intact REP-1 and could produce immunoblot results representative of a healthy individual lacking CHM mutations [7]. Moreover, if a mutated and consequently misfolded REP-1 protein was produced, it would likely undergo degradation by the proteasome and go undetected. Therefore, in most cases no conclusion can be drawn about the results of the immunoblot analysis of protein from a presumed female carrier, and the assay is generally not used to diagnose these individuals.
Genomic DNA sequencing
PCR-based single-strand conformation polymorphism (PCR-SSCP) analysis was a method traditionally used in CHM molecular genetic diagnosis to identify exons with mutations [15]. The mutated exon was sequenced afterwards. The accessibility and cost of sequencing have decreased dramatically, and thus, it has become more efficient to sequence all exons without performing PCR-SSCP.
When combined with the newly designed primers for exons 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, and 15, all primer pairs produced an amplicon of expected size with no nonspecific products. A gel picture was created of all 15 exons after capillary electrophoresis with the QIAxcel DNA Screening Kit (Qiagen; Figure 2). Forward and reverse primers successfully sequenced the respective exons.
Figure 2.

QiaXcel capillary electrophoresis (QIAxcel DNA Screening Kit, Qiagen) demonstrates the PCR products obtained using all the CHM genomic primers under the same conditions, increasing the efficiency of this analytic tool. The sample analysed is from a choroideremia patient with no detectable mutation in the exon sequences.
Amplification and purification of CHM exons from genomic DNA primarily identify small mutations such as transitions, transversions, and small deletions that account for approximately 70% of mutations detected through sequencing [13] (Table 3). It is therefore possible to draw conclusions regarding the absence of a PCR product. The absence of multiple sequential exons suggests a large deletion, whereas the absence of a single exon may indicate a deletion or large insertion (which prevents amplification of the targeted template under the chosen PCR conditions). A mutation detected at the intron–exon boundary, especially changes to the invariant GT and AG sequences at the 5′ and 3′ exon and intron junctions, respectively, can be presumed to result in aberrant splicing. The effect of mutations situated elsewhere in the extensive consensus sequences spanning 5′ and 3′ splice sites, however, can be confirmed only with RNA/cDNA analysis [16]. Sequencing cannot rule out CHM, even if no mutations are found in the exon sequences, because in this analytic approach the introns and control regions (such as the promoter and 3′ untranslated region [UTR] likely involved in translational control) are not considered. This method is non-quantitative and may not detect increased copy numbers of exons or inversions of exons.
Table 3. Summary of the capabilities of molecular genetic diagnostic tools on choroideremia patients and carriers.
| Male |
Female |
||||||
|---|---|---|---|---|---|---|---|
| Immunoblot | Genomic sequencing | RNA (cDNA) sequencing | MLPA | Genomic sequencing | MLPA | ||
| Base Pair changes |
Transition / transversion |
• |
• |
• |
|||
| Small deletion |
• |
• |
|||||
| Small insertion |
• |
• |
• |
||||
| Large changes |
Large deletion |
• |
• |
• |
• |
||
| Large insertion |
• |
• |
|||||
| Exon copy number |
• |
• |
• |
||||
| Splice variants |
• |
||||||
| Protein presence | • | ||||||
The • indicates the general ability of the assay to detect a particular type of mutation. Exceptional cases discussed in the text. When referring to Genomic sequencing, mutations can only be detected within the exons and at the exon-intron boundary. cDNA sequencing will only detect mutations in the open reading frame.
The mutation spectrum of CHM is dominated by changes that result in the absence of REP-1 due to a premature stop codon and subsequent degradation of the inappropriately folded protein or truncated mRNA. Few disease-causing amino acid substitutions have been reported; the pathogenicity of these missense mutations may be confirmed with immunoblot analysis [17].
When a potential heterozygous female carrier is being analyzed, genomic DNA analysis can detect the same mutations as it does in a male with the exception of large deletions, which may go undetected. In the case of a large deletion, the sequence from the non-mutated copy of the gene would mask the deletion. Small deletions within an exon would be detected in sequencing, resulting in two overlapping chromatograms starting at the location of the deleted nucleotide.
Multiplex ligation-dependent probe amplification
MLPA is a quantitative assessment of genomic DNA. The presence or absence of amplicons and their relative amount indicates the copy number of the region being tested [18] (Table 3, Figure 3). If, after data normalization, the dosage quotient of amplicons from a patent sample is less than 0.7 times that of the control, the assay suggests a mutation is present at the binding site of the probe, including a deletion or possibly an insertion. If the dosage quotient for an amplicon from a patient sample compared to a control sample is at least 1.3 times greater, this indicates a duplication or multiplication of the probe binding site, which leads to a non-functional REP-1 protein resulting in CHM [10]. If the MLPA amplicons produced from the patient sample are detected at levels within the acceptable range (0.7–1.3 times) of a control sample, duplication or deletion of the probe binding site and an insertion within the binding site can be ruled out. This result alone cannot rule out or diagnose a patient with CHM; the case requires further testing as no mutations would be detected outside the probe-binding region. Therefore, this test is generally performed only when no other mutations have been identified in previous analyses. MLPA is also invaluable as the assay can detect copy number variations or deletions in heterozygous female carriers due to its quantitative nature. However, MLPA can detect mutations in male and female patients only if the mutations are within or contain the probe-binding region. In addition, although this method can detect duplications, deletions, and insertions in the region, the specific location of the breakpoints remains unknown. MLPA is not appropriate for detecting the insertion direction of a duplicated copy or its exact location in the genome.
Figure 3.
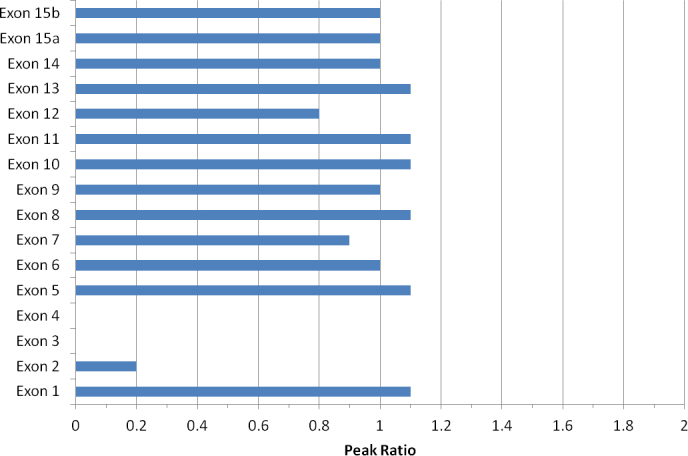
The multiplex ligation-dependent probe amplification peak ratios of choroideremia exons 2, 3 and 4 are significantly lower than those of the other exons for this patient, indicating a deletion on exons 2 through 4 in the CHM gene.
RNA analysis
Primers designed to amplify the open reading frame (ORF) of CHM RNA produce a PCR product of the expected size on a control sample (Figure 4). The forward and reverse sequencing primers produce sequences spanning the full length of the amplicon.
Figure 4.
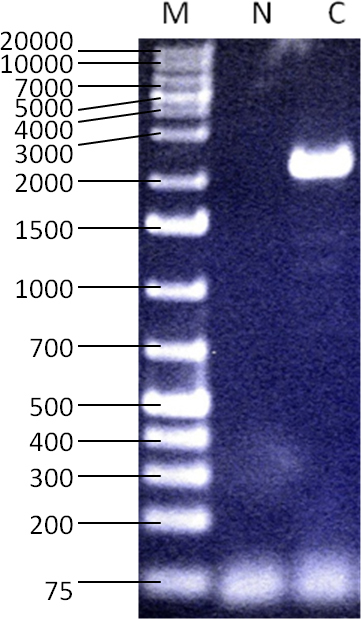
The RT-PCR result with primers specific to the coding region of the CHM mRNA showing a 2.2 kb amplicon. Size marker (“M”): GeneRuler 1kb+ DNA Ladder (Thermo Fisher Scientific, Waltham, MA), water control (“N”), RNA used from healthy male control subject (“C”). This demonstrates the ability to detect the presence of CHM RNA when it is expected to be present by creating and amplifying cDNA.
The process of isolating RNA and reverse transcribing it into cDNA may identify a patient with CHM in whom no mutation has been detected with genomic analysis. The absence of CHM cDNA suggests the patient is affected by CHM. This absence may be due to a complete or partial deletion of the gene (which could also be detected in genomic analysis), a mutation in a regulatory region, or a mutation resulting in an unstable and readily degradable RNA molecule [19]. If a transcript is detected, its size and sequence can lead to further conclusions. By analyzing the cDNA sequence, it is possible to confirm base substitutions, deletions, and insertions and detect splice variants. Similar to performing an immunoblot analysis, this method is selected on a limited basis, as samples appropriate for isolating high-quality RNA are not always available. In addition, this technique relies on PCR amplification of cDNA, and only the region within the primer pairs can be fully analyzed.
Although the entire open reading frame is contained, the current primer pair excludes the majority of the large 3′ UTR. Since mutations in the 3′ UTR could affect translational control [20], we may want to include analysis of the 3′ UTR of the genomic or mRNA sequence in future sample analyses where no other mutations have been identified.
Normal results would likely be obtained when the RNA of potential female carriers is analyzed. A second PCR product of improper size may be detected on an agarose gel indicating a female carrier. If no second band is detected, then no conclusion can be made because the woman could either have no RNA from one copy of the CHM gene or have two normal copies of the CHM gene producing normal RNA. Additionally, as with immunoblotting, a mixed population of cells arising from lyonization skews CHM expression. RNA analysis of cell cultures may not show any mutations, especially if mutated mRNA species go undetected due to nonsense-mediated RNA decay. It would be possible to sequence the cDNA product and find a point mutation; however, the same mutations can also be detected with a genomic DNA analysis. Consequently, RNA analysis is of limited utility in diagnosing female carriers; genomic DNA analysis and MLPA are the most reliable methods for identifying a female carrier.
Cell culture
The availability of fibroblast samples from patients and the challenges related to shipping and maintaining the samples limit the potential of diagnostic techniques for proteins and mRNA. Fibroblast cells have a finite life span; a cell culture can be maintained for only a limited number of passages [21]. As an alternative to fibroblast cells, lymphoblastoid cells can be immortalized through transformation with Epstein-Barr virus [22]. Lymphocytes for transformation are present in the blood, which is more easily obtained than fibroblast cells from a skin biopsy. Lymphocytes can be isolated and transformed even after sample shipping or long-term cryopreservation of blood samples.
Patient cells can also be obtained in the form of buffy coat leukocytes from peripheral blood samples for direct use in immunoblots [7]. This method is more time efficient than creating a cell line. However, to avoid protein degradation, the blood sample must be fresh (same day preferable). This cannot always be guaranteed due to transit times required to move the blood sample to the analyzing laboratory. In addition, serum components from the buffy coat may interfere with REP-1 detection on the immunoblot. Interpreting an immunoblot derived from buffy coat lysates relies on ascertaining the absence of a band in a background of non-specific protein bands. This presents a challenge that can be overcome by using lysates from a more homogenous source such as cell culture, where REP-1 blots show one clean distinctive protein band around 90 kDa.
Conclusions
Various techniques provide many tools for confirming a diagnosis in patients and carriers of choroideremia and providing molecular evidence to families. Streamlining the genomic analysis protocol improves the efficiency in diagnosing and detecting sequencing mutations. Each analysis method discussed (immunoblot, genomic analysis, MLPA, and RNA analysis) has benefits and limitations, but when combined, the methods are quite effective at detecting mutations considering the availability of sample materials for analysis. We hope that this manuscript will help guide molecular genetic diagnostic testing for families affected by this condition.
Acknowledgments
Grant Support from Canadian Institutes of Health Research (TR2 – 119190), Alberta Innovates-Health Solutions, Choroideremia Research Foundation Canada Inc. and The Foundation Fighting Blindness, Canada is gratefully acknowledged. The editing assistance of Dr. Paul Freund and Ms. Stephanie Chan is also gratefully acknowledged.
Bibliography
- 1.MacDonald IM, Sereda C, McTaggart K, Mah D. Choroideremia gene testing. Expert Rev Mol Diagn. 2004;4:478–84. doi: 10.1586/14737159.4.4.478. [DOI] [PubMed] [Google Scholar]
- 2.Seabra MC, Brown MS, Goldstein JL. Retinal degeneration in choroideremia: Deficiency of rab geranylgeranyl transferase. Science. 1993;259:377–81. doi: 10.1126/science.8380507. [DOI] [PubMed] [Google Scholar]
- 3.Seabra MC, Ho YK, Anant JS. Deficient geranylgeranylation of Ram/Rab27 in choroideremia. J Biol Chem. 1995;270:24420–7. doi: 10.1074/jbc.270.41.24420. [DOI] [PubMed] [Google Scholar]
- 4.Tolmachova T, Anders R, Abrink M, Bugeon L, Dallman MJ, Futter CE, Ramalho JS, Tonagel F, Tanimoto N, Seeliger MW, Huxley C, Seabra MC. Independent degeneration of photoreceptors and retinal pigment epithelium in conditional knockout mouse models of choroideremia. J Clin Invest. 2006;116:386–94. doi: 10.1172/JCI26617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.van Bokhoven H, van den Hurk JA, Bogerd L, Philippe C, Gilgenkrantz S, de Jong P, Ropers HH, Cremers FP. Cloning and characterization of the human choroideremia gene. Hum Mol Genet. 1994;3:1041–6. doi: 10.1093/hmg/3.7.1041. [DOI] [PubMed] [Google Scholar]
- 6.van den Hurk JA, van de Pol DJ, Wissinger B, van Driel MA, Hoefsloot LH, de Wijs IJ, van den Born LI, Heckenlively JR, Brunner HG, Zrenner E, Ropers HH, Cremers FP. Novel types of mutation in the choroideremia (CHM) gene: A full-length L1 insertion and an intronic mutation activating a cryptic exon. Hum Genet. 2003;113:268–75. doi: 10.1007/s00439-003-0970-0. [DOI] [PubMed] [Google Scholar]
- 7.MacDonald IM, Mah DY, Ho YK, Lewis RA, Seabra MC. A practical diagnostic test for choroideremia. Ophthalmology. 1998;105:1637–40. doi: 10.1016/S0161-6420(98)99031-5. [DOI] [PubMed] [Google Scholar]
- 8.van Bokhoven H, Schwartz M, Andreasson S, van den Hurk JA, Bogerd L, Jay M, Ruther K, Jay B, Pawlowitzki IH, Sankila EM. Mutation spectrum in the CHM gene of danish and swedish choroideremia patients. Hum Mol Genet. 1994;3:1047–51. doi: 10.1093/hmg/3.7.1047. [DOI] [PubMed] [Google Scholar]
- 9.Song J, Smaoui N, Ayyagari R, Stiles D, Benhamed S, MacDonald IM, Daiger SP, Tumminia SJ, Hejtmancik F, Wang X. High-throughput retina-array for screening 93 genes involved in inherited retinal dystrophy. Invest Ophthalmol Vis Sci. 2011;52:9053–60. doi: 10.1167/iovs.11-7978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chi JY, MacDonald IM, Hume S. Copy number variant analysis in CHM to detect duplications underlying choroideremia. Ophthalmic Genet. 2013;34:229–33. doi: 10.3109/13816810.2012.752016. [DOI] [PubMed] [Google Scholar]
- 11.Pylypenko O, Rak A, Reents R, Niculae A, Sidorovitch V, Cioaca MD, Bessolitsyna E, Thomä NH, Waldmann H, Schlichting I, Goody RS, Alexandrov K. Structure of Rab escort protein-1 in complex with Rab geranylgeranyltransferase. Mol Cell. 2003;11:483–94. doi: 10.1016/s1097-2765(03)00044-3. [DOI] [PubMed] [Google Scholar]
- 12.Rak A, Pylypenko O, Niculae A, Pyatkov K, Goody RS, Alexandrov K. Structure of the Rab7:REP-1 complex: insights into the mechanism of Rab prenylation and choroideremia disease. Cell. 2004;117:749–60. doi: 10.1016/j.cell.2004.05.017. [DOI] [PubMed] [Google Scholar]
- 13.McTaggart KE, Tran M, Mah DY, Lai SW, Nesslinger NJ, MacDonald IM. Mutational analysis of patients with the diagnosis of choroideremia. Hum Mutat. 2002;20:189–96. doi: 10.1002/humu.10114. [DOI] [PubMed] [Google Scholar]
- 14.Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature. 1961;190:372–3. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- 15.Nesslinger N, Mitchell G, Strasberg P, MacDonald IM. Mutation analysis in canadian families with choroideremia. Ophthalmic Genet. 1996;17:47–52. doi: 10.3109/13816819609057870. [DOI] [PubMed] [Google Scholar]
- 16.Krawczak M, Reiss J, Cooper DN. The mutational spectrum of single base-pair substitutions in mRNA splice junctions of human genes: causes and consequence. Hum Genet. 1992;90:41–54. doi: 10.1007/BF00210743. [DOI] [PubMed] [Google Scholar]
- 17.Sergeev YV, Smaoui N, Sui R, Stiles D, Gordiyenko N, Strunnikova N, Macdonald IM. The functional effect of pathogenic mutations in Rab escort protein 1. Mutat Res. 2009;665:44–50. doi: 10.1016/j.mrfmmm.2009.02.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G. Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res. 2002;30:e57. doi: 10.1093/nar/gnf056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mort M, Ivanov D, Cooper DN, Chuzhanova NA. A meta-analysis of nonsense mutations causing human genetic disease. Hum Mutat. 2008;29:1037–47. doi: 10.1002/humu.20763. [DOI] [PubMed] [Google Scholar]
- 20.Mazumder B, Seshadri V, Fox PL. Translational control by the 3′-UTR: The ends specify the means. Trends Biochem Sci. 2003;28:91–8. doi: 10.1016/S0968-0004(03)00002-1. [DOI] [PubMed] [Google Scholar]
- 21.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–36. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 22.Anderson MA, Gusella JF. Use of cyclosporin A in establishing epstein-barr virus-transformed human lymphoblastoid cell lines. In Vitro. 1984;20:856–8. doi: 10.1007/BF02619631. [DOI] [PubMed] [Google Scholar]