

Abstract
Activation of the GPCR sphingosine-1-phosphate receptor 1 (S1P1) by sphingosine-1-phosphate (S1P) regulates key physiological processes. S1P1 activation also has been implicated in pathologic processes, including autoimmunity and inflammation; however, the in vivo sites of S1P1 activation under normal and disease conditions are unclear. Here, we describe the development of a mouse model that allows in vivo evaluation of S1P1 activation. These mice, known as S1P1 GFP signaling mice, produce a S1P1 fusion protein containing a transcription factor linked by a protease cleavage site at the C terminus as well as a β-arrestin/protease fusion protein. Activated S1P1 recruits the β-arrestin/protease, resulting in the release of the transcription factor, which stimulates the expression of a GFP reporter gene. Under normal conditions, S1P1 was activated in endothelial cells of lymphoid tissues and in cells in the marginal zone of the spleen, while administration of an S1P1 agonist promoted S1P1 activation in endothelial cells and hepatocytes. In S1P1 GFP signaling mice, LPS-mediated systemic inflammation activated S1P1 in endothelial cells and hepatocytes via hematopoietically derived S1P. These data demonstrate that S1P1 GFP signaling mice can be used to evaluate S1P1 activation and S1P1-active compounds in vivo. Furthermore, this strategy could be potentially applied to any GPCR to identify sites of receptor activation during normal physiology and disease.
Introduction
Sphingosine-1-phosphate receptor 1 (S1P1), originally named EDG-1, is the founding member of a family of 5 GPCRs with high affinity for the lipid ligand sphingosine-1-phosphate (S1P) (1–3). Activation of S1P1 stimulates Gi-dependent and other intracellular signaling cascades, leading to cell type–specific responses that include migration, cytoskeletal changes, proliferation, and survival. S1P1 is widely expressed in tissues, is particularly enriched in endothelial cells, and is one of the most abundant receptors in the entire GPCR superfamily (4–6).
S1P1 is an important regulator of vascular development and function in endothelial cells. During embryogenesis, S1P1 expression in endothelial cells is essential for the formation of a stable, functional vasculature (7–9). In its absence, midgestation embryos exhibit massive hemorrhage and edema. Similar consequences result from a complete absence of the endogenous ligand S1P in sphingosine kinase-deficient embryos, indicating that S1P-induced activation of the S1P1-directed signaling pathways in endothelial cells is essential for proper vascular development (10). S1P1 also has an important role in the promotion of endothelial barrier function and integrity under both basal and inflammatory conditions (11–13). Furthermore, during infections, S1P1 functions on the endothelium to suppress inflammatory responses (14).
In addition to its essential functions in the vasculature, S1P1 has a pivotal role in regulating the recirculation of lymphocytes throughout the body (15). In a particularly critical function, S1P1 expression on lymphocytes enables their egress from lymphoid tissues into blood and lymph (16, 17). Intrinsic S1P1 expression also dictates the positioning and behavior of immune cells in the marginal zone (MZ) of the spleen (18, 19).
FTY720 (fingolimod) is a well-studied compound that is highly active on S1P1 (20, 21). It mimics sphingosine and targets S1P receptors after it is phosphorylated by sphingosine kinases in vivo (22, 23). FTY720 causes downmodulation of lymphocyte receptors (16), a process that is believed to block lymphocyte migratory responses toward elevated S1P concentrations in circulating fluids during egress from lymphoid tissues (15, 24). Because of its immunosuppressive properties, FTY720 is used in the treatment of multiple sclerosis (25). Its efficacy is attributed to its ability to block lymphocyte recirculation (15, 16) as well as its direct suppression of astrocyte responses (26).
S1P, the physiologic ligand for S1P1, is synthesized during sphingolipid metabolism, which is a process that occurs in all cells (27, 28). Levels of S1P are relatively high in circulating blood and lymph, ranging from low micromolar to high nanomolar concentrations (29, 30). However, it is believed that the S1P concentrations in interstitial spaces are substantially lower due to highly active degradation pathways (15, 31). The S1P concentration in circulation is necessary to enable the trafficking of lymphocytes (30) and for proper vascular functioning (12), suggesting that it directs S1P1 activation on immune and endothelial cells (32–34).
The major source of S1P in the blood is red blood cells (30). Endothelial cells are the major source of S1P in lymph and may play a minor role in maintaining blood S1P levels (30, 35). Pericytes also provide S1P to direct the egress of thymocytes from the thymus (36). How S1P is generated acutely for S1P1 activation is not understood.
Because S1P1 is widely expressed and the capacity to produce S1P ligand is a feature of essentially all cells, it has been difficult to identify precisely where and when S1P1 activation occurs in vivo. Furthermore, the cellular sites at which S1P1-specific compounds, such as FTY720, stimulate activation in vivo have not been clearly mapped. In order to gain insights into where and when S1P1 activation occurs in vivo, we have developed a mouse model that records S1P1 activation at a cellular level through a modified S1P1 signaling cascade that is distinct from the endogenous S1P1 signaling pathways. Using this mouse model, we have mapped the cellular sites of S1P1 activation that are present during homeostasis and in response to FTY720 and LPS-induced inflammation. We also provide evidence that hematopoietically derived S1P stimulates S1P1 activation both in endothelial cells and hepatocytes during systemically induced inflammation.
Results
S1P1 GFP signaling reporter mice.
We adapted elements of the “Tango” system (ref. 37 and Figure 1A), a cell-based assay that detects interactions between an activated GPCR and β-arrestin, to devise a recordable S1P1 signaling pathway in mice. In general outline of the Tango system, both the GPCR and β-arrestin are genetically modified as individual fusion proteins. The GPCR has its C terminus linked to a transcription factor via an amino acid sequence containing the tobacco etch virus (TEV) protease 7-residue cleavage site. The β-arrestin is fused to the TEV protease. Ligand activation of the modified GPCR induces phosphorylation on the receptor C terminus by GPCR-specific kinases (38–41). Next, the β-arrestin-TEV protease fusion protein is recruited to the modified receptor and, upon binding, proteolytically releases the transcription factor, which travels into the nucleus to transcriptionally activate a reporter gene (Figure 1A). The output of the reporter provides a measure of receptor activation.
Figure 1. Generation of S1P1 GFP signaling reporter mice.

(A) Schematic of the “Tango” design to monitor S1P1-β-arrestin interactions. Ligand activation of GPCRs leads to their phosphorylation and subsequent recognition by arrestins. The target GPCR, in this case S1P1, is modified by linking the tetracycline-controlled transactivator (tTA) to its C terminus through a TEV protease recognition sequence (tevs). Ligand binding to the receptor stimulates the recruitment of a β-arrestin-TEV protease fusion protein, triggering the release of tTA from the C terminus of modified S1P1. Free tTA enters the nucleus and stimulates histone H2B-GFP reporter gene activity. (B) Design of the S1pr1 knockin vector. Coding sequences for the 2 fusion proteins, S1P1-tTA and mouse β-arrestin-2-TEV (mArrb2-TEV) protease, connected by an IRES, were included along with the neomycin resistance gene (NeoR) flanked by loxP sites. This knockin segment was flanked by 2.4 kb of 5′ and 3.8 kb of homologous 3′ genomic sequences adjacent to the second exon of S1pr1. The herpes simplex virus thymidine kinase (TK) gene was added outside of the homologous sequence to minimize random integration. Schematics of the targeting vector, WT S1pr1 allele, and S1pr1 knockin allele are shown. PA, polyadenylation sequence. (C) Mouse mating scheme to obtain S1P1 GFP signaling (S1P1GS) mice. S1pr1 knockin mice were crossed with histone-EGFP reporter (H2B-GFP) mice, in which human histone 1 protein H2bj and EGFP fusion protein are expressed under the control of a tetracycline-responsive promoter element and cytomegalovirus minimal promoter.
We combined these basic modules in order to genetically encode a novel pathway in mice to stably record activated S1P1 (Figure 1B). We linked the well-characterized tetracycline transcriptional activator (tTA) (42–44) to the C terminus of S1P1 via a TEV protease cleavage motif (tevs). We created an S1P1 (S1pr1) gene-targeting vector that carried a bicistronic unit consisting of the S1P1-tTA fusion followed by a mouse β-arrestin-2-TEV protease fusion gene separated by an internal ribosome entry sequence (IRES). The bicistronic configuration ensured that both the receptor and β-arrestin fusion proteins would be expressed within the same cell, which is a requirement for the system to function. The bicistronic gene cassette was knocked in to the mouse S1pr1 locus in mouse ES cells by homologous recombination in order to place the gene cassette under control of the endogenous S1pr1 promoter elements, so that the expression of the modified genes would mirror the cellular expression characteristic of endogenous S1P1 (Figure 1B). Chimeric and, subsequently, heterozygous mice were derived, carrying modified S1P1 and β-arrestin knockin genes.
A well-characterized histone H2B-GFP reporter transgene, under control of a tetracycline-responsive promoter element (45, 46), was used for the readout of modified S1P1 signaling pathway. The H2B-GFP would be expressed when the tTA is proteolytically released from the activated S1P1 by the β-arrestin-TEV protease fusion protein. H2B-GFP incorporates into nucleosomes, giving rise to fluorescent nuclei as an indicator of activation of modified S1P1 signaling pathway. The H2B-GFP protein is relatively stable, and its fluorescent signal would be expected to persist for days in nondividing cells (45, 46).
In order to identify the expression profile of the activated H2B-GFP reporter transgene, mice carrying only this gene were crossed with R26-M2rtTA mice (47), which globally expressed the reverse tetracycline-controlled transactivator (rtTA-M2). The resulting bigenic mice were administered doxycycline in their drinking water to activate the rtTA-M2, and their tissues were examined for H2B-GFP expression. Thymi, spleens, lymph nodes, lungs, hearts, and livers all showed a widespread nuclear induction of GFP compared with the tissues of mice that were not administered doxycycline (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI71194DS1). This experiment demonstrates that the H2B-GFP reporter has the potential to be expressed in these tissues and, thus, should not limit the cellular detection of the modified S1P1 signaling pathway.
H2B-GFP reporter mice were crossed with S1P1-β-arrestin knockin mice to derive mice carrying one copy of each allele (Figure 1C). These mice are referred to as S1P1 GFP signaling mice. These mice were born in the expected numbers, indicating the absence of developmental lethality. Numbers of B220+ B cells, CD4+ T cells, CD8+ T cells, and CD11b+ monocytes were not significantly different between the S1P1 GFP signaling mice and the control H2B-GFP reporter mice (Supplemental Figure 3).
Validation of the modified S1P1 signaling pathway.
To determine whether the modified S1P1 signaling pathway genetically encoded into the S1P1 GFP signaling mice was functional in cells, primary mouse embryonic fibroblasts (MEFs) were derived from S1P1 GFP signaling mouse embryos and cultured. The addition of 10–7 M S1P to MEF cultures resulted in the appearance of fluorescently labeled nuclei by 6 hours (Figure 2A). Little background nuclear fluorescence was detected in the absence of exogenous S1P. Using a quantifiable flow cytometry assay (Figure 2C), the EC50 for the stimulation of nuclear fluorescence by S1P in the S1P1 GFP signaling MEFs was determined to be 43 nM, similar to the EC50 determined for the S1P-induced internalization of the S1P1 from the plasma membrane (48). The results indicate that the S1P1 GFP signaling pathway within MEFs was activated by S1P in a time course in line with the activation of transcription for the reporter gene (42) and with S1P concentrations known to induce receptor activation with subsequent internalization.
Figure 2. S1P1 activation in MEFs.

(A) Validation of modified S1P1 signaling pathway in MEFs. S1P1 GFP signaling MEFs were cultured for 16 hours in medium containing 10% charcoal-stripped FBS and received either S1P (10–7 M) or vehicle (4 mg/ml BSA in PBS). Nuclei were stained with Hoechst, and MEF cultures were imaged under an inverted laser-scanning confocal microscope. The experiment was repeated twice in duplicate, and a representative result is shown. (B) Treatment of MEFs with RP-001. S1P1 GFP signaling MEFs were cultured for 16 hours in medium containing 10% charcoal-stripped FBS and received either S1P or RP-001. After 24 hours, nuclei were stained with Hoechst, and MEF cultures were imaged under an inverted laser-scanning confocal microscope. The experiment was performed in duplicate, and a representative result is shown. (C) Flow cytometry analysis of S1P1 GFP signaling MEFs. S1P1 GFP signaling MEFs were cultured for 16 hours in medium containing 10% charcoal-stripped FBS and various concentrations of S1P were added. After 24 hours, the number of GFP+ cells was determined by flow cytometry. The experiment was repeated twice. Data represent mean ± SEM (n = 3). (D) Endogenous S1P1 signaling pathway in MEFs. S1P1 GFP signaling MEFs were cultured for 16 hours in medium containing 0.1% FBS and received 1 μM of S1P1 receptor ligands (RP-001, S1P, or SEW2871) or vehicle (4 mg/ml BSA in PBS). After 10 minutes, the cell lysate was harvested and then Akt and phospho-Akt were identified by Western blotting (see complete unedited blot in the supplemental material). The experiment was performed in triplicate, and a representative result is shown. Scale bars: 100 μm.
In order to verify the specificity of modified S1P1 signaling pathway, the S1P1 GFP signaling MEFs were exposed to various lipids and S1P1 active compounds (Figure 2B and Supplemental Figure 2). Selective S1P1 agonist RP-001, which was reported to activate S1P1 in the picomolar range (6), induced nuclear fluorescence at concentrations much lower than S1P (Figure 2B). Both selective S1P1 agonist, SEW2871 (49), and natural ligand, dihydro-S1P (dhS1P; ref. 50), induced nuclear fluorescence with an effectiveness comparable to that of S1P (Supplemental Figure 2). The S1P1 GFP signaling MEFs were barely responsive or not responsive to 1 μM sphingosine or LPA, neither of which are ligands for S1P1, although sphingosine can be converted to S1P. The results demonstrate that the S1P1 GFP signaling pathway within MEFs reports receptor activation, with the ligand specificity expected for native S1P1.
The S1P1 GFP signaling MEFs were treated with RP-001 and SEW2871, and cell extracts were assayed for phosphorylated Akt, a downstream target of S1P1-Gi signaling (51). Both RP-001 and SEW2871 induced phosphorylation of Akt comparably to S1P, showing the existence of functional native S1P1-directed signaling pathway and supporting the relevance of the genetically modified signaling pathway for the detection of activated S1P1 as a surrogate for endogenous signaling (Figure 2D).
The importance of S1P1 and S1P in the development of the embryonic vascular system has been established through analysis of null mice (7–9). In the absence of either the receptor, S1P1, or the ligand, S1P, the vasculature development is defective, implying that S1P1 activation by S1P is needed for this process. E9.5 and E10.5 control mouse embryos carrying only the H2B-GFP transgene displayed diffuse background fluorescence. In E9.5 S1P1 GFP signaling mouse embryos, intense GFP expression was noted in the dorsal aorta. In E10.5 S1P1 GFP signaling mouse embryos, strong GFP expression was displayed in the heart and telencephalon, in addition to that in the dorsal aorta (Figure 3). This result indicates that the S1P1 GFP signaling pathway system reports receptor activation in vivo in regions of presumptive endogenous S1P1 activation.
Figure 3. S1P1 activation in embryos.
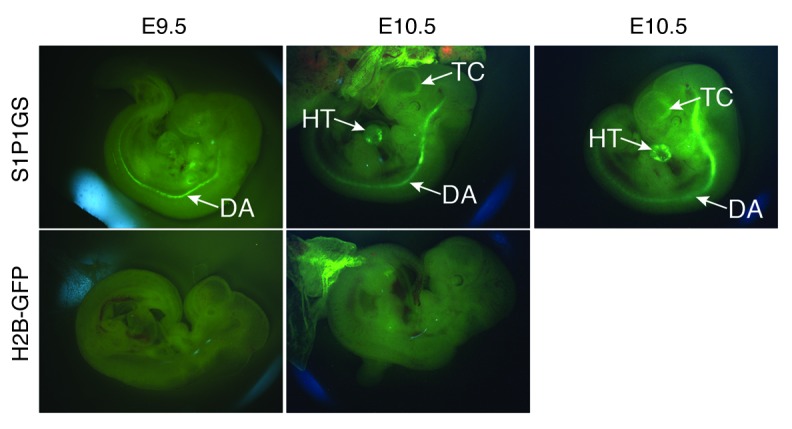
S1P1 GFP signaling and H2B-GFP E9.5 and E10.5 mouse embryos were imaged using a fluorescence stereomicroscope. TC, telencephalon; HT, heart; DA, dorsal aorta. Four litters each of E9.5 and E10.5 embryos were examined.
S1P1 activation in adult tissues.
Frozen sections of tissues from S1P1 GFP signaling mice and controls carrying only the H2B-GFP gene were examined for GFP fluorescence by confocal microscopy. Thymi, spleens, and lymph nodes from S1P1 GFP signaling mice contained the highest density of GFP+ cells (Figure 4). Sparse GFP+ cells were found in lungs and hearts, while livers were largely negative. In comparable sections from control H2B-GFP mice, infrequent, scattered GFP+ cells were noted in the immune tissues; otherwise, the tissues were GFP negative.
Figure 4. S1P1 activation in tissues.
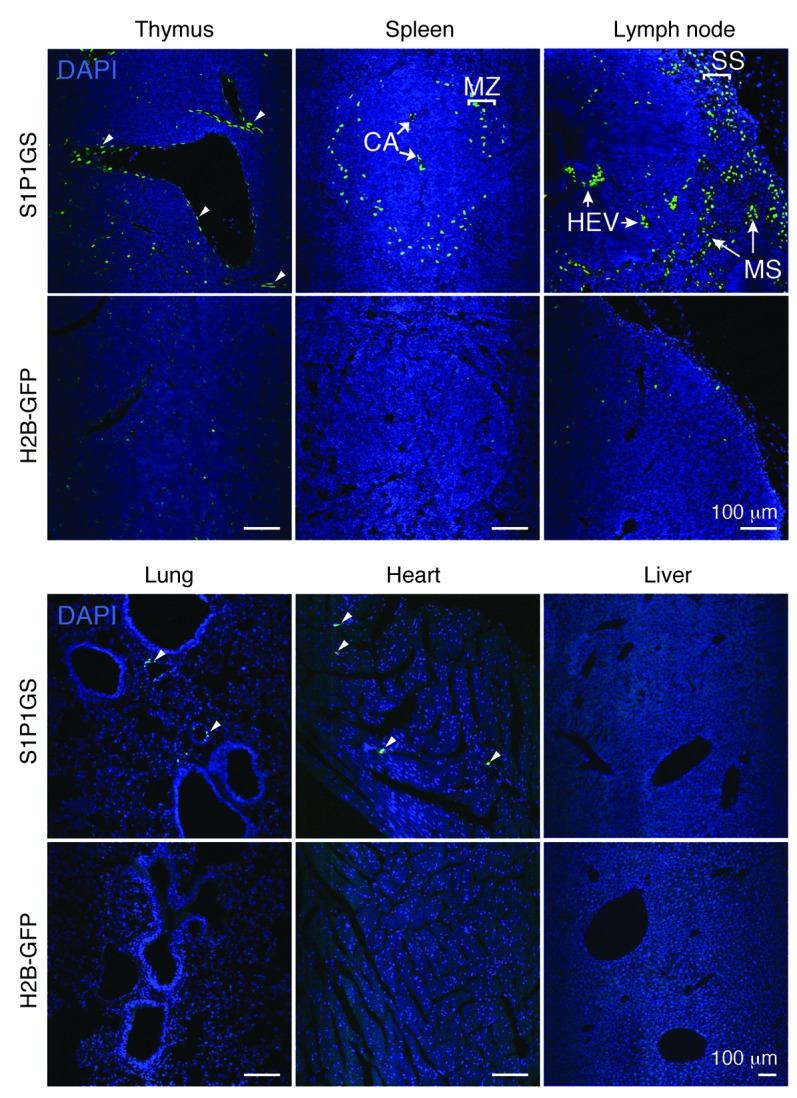
Histological sections from S1P1 GFP signaling and H2B-GFP mice were stained with DAPI, and the images were captured with an inverted laser-scanning confocal microscope. Small arrowheads point to GFP+ vascular structures. The tissues of 5 mice for each genotype were examined. CA, central arteries; HEV, high endothelial venules; MS, medullary sinuses; SS, subcapsular sinuses. Scale bars: 100 μm.
In thymi of the S1P1 GFP signaling mice, the majority of the GFP+ cells appeared to surround vascular structures, with some weakly positive cells within the parenchyma (Figure 4). Immunostaining with antibody to CD31 confirmed the identity of many of the thymic GFP+ cells as endothelial cells (Figure 5). The bulk of the thymic lymphocytes, as determined by immunostaining with antibody to CD45, were negative for GFP expression (data not shown). This finding was confirmed by flow cytometry, which showed a low but significant level of GFP+ thymocytes (CD3+ CD4+ CD8– and CD3+ CD4– CD8+) in S1P1 GFP signaling mice compared with that in control H2B-GFP mice (Supplemental Figure 4).
Figure 5. Identification of cell type–specific S1P1 activation.

Histological sections from S1P1 GFP signaling and H2B-GFP mice were immunostained with antibodies to CD31, PNAd, LYVE-1, B220, and MARCO, and the images were captured with an inverted laser-scanning confocal microscope. Scale bars: 50 μm.
In spleens of the S1P1 GFP signaling mice, GFP+ cells were concentrated in the vicinity of the MZ (Figure 4). Some of the GFP+ cells in the MZ were identified as B cells by immunostaining with antibodies to B220 (Figure 5). In addition, strongly GFP+ cells were identified as resident MZ macrophages that expressed macrophage receptor with collagenous (MARCO) structure (Figure 5 and refs. 52, 53). The central artery endothelial cells were also GFP+ in the spleens of the S1P1 GFP signaling mice (Figure 5). Flow cytometry of splenocytes revealed that significant percentages of B220+ B cells and CD11b+ macrophages were GFP+ in S1P1 GFP signaling mice compared with those in control H2B-GFP mice. CD3+ CD4+ and CD3+ CD8+ T cells that were GFP+ were increased marginally in the S1P1 GFP signaling mice compared with those in control H2B-GFP mice (Supplemental Figure 4).
In lymph nodes of S1P1 GFP signaling mice, a high density of GFP+ cells was localized in the vicinity of the subcapsular and medullary sinuses (Figure 4). These cells were identified as predominantly lymphatic endothelial cells by immunostaining with CD31 and LYVE-1 antibodies (Figure 5 and refs. 54, 55). High endothelial venules (PNAd+), which are specialized endothelial cells at the sites of lymphocyte entry into the lymph nodes (56), also displayed strong GFP expression (Figure 5). Flow cytometry indicated that a low percentage of B220+ B cells, CD11b+ macrophages, and CD3+ CD4+ T cells were GFP+ in the lymph nodes of S1P1 GFP signaling mice; however, these levels were significantly higher than those observed in the lymph nodes of control H2B-GFP mice (Supplemental Figure 4).
In lungs, scattered GFP+ cells in S1P1 GFP signaling mice also stained positively for CD31 and LYVE-1 (Figures 4 and 5). Flow cytometry experiments confirmed a significantly increased number of CD31+ GFP+ cells in the lungs of S1P1 GFP signaling mice compared with that in control H2B-GFP mice (Supplemental Figure 5A).
In livers, GFP+ cells were rare in the vasculature and in the parenchyma (Figure 4), but they were found at statistically higher levels in S1P1 GFP signaling mice compared with control H2B-GFP mice (Supplemental Figure 5, B and C). Nuclei in the hearts of S1P1 GFP signaling mice were largely negative for GFP expression, with rare GFP+ endothelial cells (Figures 4 and 5).
As an alternative method for identifying cellular S1P1 activation, we used immunohistochemical detection of GFP expression on paraffin-embedded tissue sections (Supplemental Figure 6). The results showed specific GFP expression (blue staining) in the tissues of the S1P1 GFP signaling mice and little or no GFP expression in tissues of the control H2B-GFP mice. The frequency and distribution of GFP+ cells detected by immunohistochemistry in the S1P1 GFP signaling mice were consistent with the results obtained by detection of direct GFP fluorescence on the frozen sections described above. This method also allowed for direct detection of S1P1 with a well-characterized antibody (57) to determine its expression in comparison to GFP expression on serial tissues sections (Supplemental Figure 6). We found that S1P1 was broadly expressed in all of the tissues examined. In particular, S1P1 appeared to be most highly expressed in vascular regions of the tissues, including in the lung and MZ of the spleen, as has been described previously (4); S1P1 was also well expressed in the parenchyma of the tissues.
Induction of S1P1 activation by FTY720.
To determine whether the S1P1 GFP signaling pathway can be induced in vivo, either FTY720, a well-characterized S1P receptor agonist compound, or vehicle was administered to S1P1 GFP signaling mice. After 24 hours, their tissues were evaluated for nuclear GFP expression. Overall, the number of endothelial cells with GFP+ nuclei in all tissues examined from FTY720-treated S1P1 GFP signaling mice was increased compared with that in S1P1 GFP signaling mice treated with vehicle (Figure 6 and Supplemental Figure 7). In lungs, FTY720 treatment of S1P1 GFP signaling mice induced an increase in GFP+ endothelial cells (CD31+ and LYVE-1+) (Figure 6). The increased number of GFP+ endothelial cells from the lungs of FTY720-treated S1P1 GFP signaling mice was verified by flow cytometry (Supplemental Figure 8, A and B).
Figure 6. FTY720 induced activation of S1P1 in endothelial cells and hepatocytes.

FTY720 (1 mg/kg) or vehicle (ethanol/PBS, 1:1) was intraperitoneally injected into S1P1 GFP signaling mice, and the tissues were harvested 1 day after injection. Histological sections were immunostained with antibodies to CD31, LYVE-1, or albumin, and the images were captured using an inverted laser-scanning confocal microscope. The tissues of 3 mice for each treatment were examined. Scale bars: 50 μm.
In livers, nuclear GFP expression was remarkably elevated in FTY720-treated S1P1 GFP signaling mice compared with that in vehicle-injected S1P1 GFP signaling mice (Supplemental Figure 7). Many hepatocytes (albumin+ cells) expressed GFP+ nuclei, indicating that they had been stimulated by the FTY720 treatment (Figure 6 and Supplemental Figure 8, A and D). The number of GFP+ endothelial cells was also increased in liver after FTY720 treatment (Supplemental Figure 8, A and C).
Immunochemical detection of GFP in tissues after FTY720 treatment afforded results compatible with those obtained by fluorescent detection of GFP. Staining with anti-S1P1 in serial sections revealed that treatment with FTY720 did grossly alter S1P1 expression (Supplemental Figure 9).
FTY720 treatment of S1P1 GFP signaling mice did not significantly alter the percentages of GFP+ lymphocytes in the thymi (Supplemental Figure 10). Similarly, no statistically significant differences in the percentage of GFP+ CD4+ T cells, CD8+ T cells, B220+ B cells, and CD11b+ macrophages in spleens and lymph nodes were observed after FTY720 treatment (Supplemental Figure 10).
S1P1 activation in response to inflammation.
To identify cellular sites of S1P1 activation during systemic inflammation, S1P1 GFP signaling mice were injected with a sublethal dose of LPS, and tissues were examined after 3 days. LPS-treated mice showed an increase in the density of GFP+ cells surrounding vessels of the thymi, lungs, and livers (Supplemental Figure 11). These GFP+ cells were identified as CD31+ endothelial cells in thymi and CD31+ LYVE-1+ endothelial cells in lungs and livers (Figure 7). Correspondingly, flow cytometry analysis demonstrated a significant increase in the number of GFP+ CD31+ endothelial cells in lungs and livers of S1P1 GFP signaling mice after LPS treatment (Supplemental Figure 12, A–C).
Figure 7. S1P1 activation in endothelial cells and hepatocytes during systemic inflammation.

LPS (20 mg/kg) or vehicle (PBS) was injected intraperitoneally into S1P1 GFP signaling mice, and the tissues were harvested 3 days after injection. Histological sections were immunostained with antibodies to CD31, LYVE-1, or albumin, and the images were captured using an inverted laser-scanning confocal microscope. The tissues of 4 mice injected with LPS and 3 mice injected with vehicle were examined. Scale bars: 50 μm.
LPS treatment also induced GFP expression in albumin+ hepatocytes (Figure 7). The number of GFP+ hepatocytes increased significantly in S1P1 GFP signaling mice after LPS treatment (Supplemental Figure 12, A and D).
Role of hematopoietically derived S1P during inflammation-induced S1P1 activation in endothelial cells and hepatocytes.
The major source of S1P in plasma is red blood cells (30). Other sources include endothelial cells (35, 36). In order to determine whether hematopoietically derived S1P was a source for S1P1 activation during LPS stimulation, we prepared S1P1 GFP signaling mice with a deficiency of S1P in their plasma. To accomplish this, we transplanted bone marrow cells from pIpC-treated Sphk1fl/fl Sphk2–/– Mx1-cre mice, which have been shown to lack S1P in plasma and are referred to as plasmaS1Pless (pS1Pless) mice (30), into S1P1 GFP signaling mice. Control mice were prepared by transplanting bone marrow cells from pIpC-treated Sphk1fl/fl Sphk2–/– mice, which do not have an S1P deficiency in plasma (30), into S1P1 GFP signaling mice (Figure 8A).
Figure 8. S1P1 activation by hematopoietically derived S1P during systemic inflammation.

(A) Experimental scheme. Bone marrow cells from pS1Pless or control mice were transplanted into S1P1 GFP signaling mice. Ten weeks later, plasma sphingolipid levels were determined; in addition, LPS (20 mg/kg) or vehicle (PBS) was injected intraperitoneally into the mice. After 24 hours, cells were isolated from the lungs and livers, and the percentages of GFP+ cells in CD31+ or CD95+ populations were quantified by flow cytometry. (B) Quantification of dihydrosphingosine (dhSph), sphingosine (Sph), dhS1P, and S1P by HPLC-tandem mass spectrometry in plasma of bone marrow–transplanted mice. Bars represent mean ± SEM (n = 3 for each genotype). (C–E) Flow cytometry analysis was used to determine the percentage of GFP+ cells in the (C) lung CD45– CD31+ population, (D) liver CD31+ population, and (E) CD95+ population 24 hours after LPS or vehicle injection. Bars represent mean ± SEM (n = 7 for vehicle-injected control bone marrow transplant mice and n = 8 for the other 3 groups in C; n = 3 for LPS-injected control bone marrow transplant mice and n = 4 for the other 3 groups in D and E). Student’s t test; *P < 0.05, **P < 0.01, ***P < 0.001.
The levels of S1P and dhS1P in the plasma of these mice were measured by HPLC-tandem mass spectrometry (58) and were found to be reduced by about 80% in the pS1Pless bone marrow–transplanted S1P1 GFP signaling mice compared with those in the control bone marrow–transplanted S1P1 GFP signaling mice. The concentrations of plasma ceramides, sphingosine and dihydrosphingosine, were similar in both groups of mice (Figure 8B and Supplemental Figure 13).
Flow cytometry revealed that, in the control bone marrow–transplanted S1P1 GFP signaling mice, LPS treatment significantly increased the number of GFP+ endothelial cells in the lungs and livers as well as the number of GFP+ hepatocytes, compared with those in vehicle-injected mice (Figure 8, C–E). By comparison, in pS1Pless bone marrow–transplanted S1P1 GFP signaling mice, the number of GFP+ endothelial cells and hepatocytes was significantly lower in the LPS-injected groups compared with that in the LPS-injected control bone marrow–transplanted mice (Figure 8, C–E). Images of frozen tissue sections and the quantitative flow cytometry results indicated a reduced frequency of GFP+ cells in response to LPS in the pS1Pless bone marrow–transplanted S1P1 GFP signaling mice compared with that in the controls (Supplemental Figure 11B).
Overall, the results are consistent with the concept that hematopoietically derived S1P induces S1P1 activation in endothelial cells as well as in parenchymal cells of liver during LPS-induced systemic inflammation.
Discussion
A major limitation in our understanding of the in vivo biology of S1P1 signaling comes from the inability to determine where and when the receptor is activated. We have now established a mouse model that is able to record S1P1 activation events at a cellular level. The strategy that was used is based on an assay (37) that uses GPCR-β-arrestin interactions (38–41) as a means to measure GPCR activation. A unique signaling pathway, separate from endogenous signaling pathways, was formed by the expression of 3 genetic elements: a modified S1P1, a modified β-arrestin, and a transcriptionally activated reporter, H2B-GFP. The reporter gene is activated by the release of a tethered transcription factor as a consequence of the β-arrestin interaction with the activated GPCR. Because the modified receptor and β-arrestin are under control of the endogenous S1P1 promoter, cellular sites of signaling activity should be detectable only in cells that express the endogenous receptor. A similar strategy was been used recently in Drosophila brain to monitor dopamine signaling (59).
The S1P1 GFP signaling mouse model was validated in a number of ways. MEFs containing the modified S1P1 signaling pathway reported S1P1 activation by exogenously added S1P at concentrations that have been reported to induce S1P1 internalization (48). The modified signaling pathway was also induced with the requisite ligand specificity of the endogenous pathway. In addition, the modified signaling pathway was activated in embryos within the vascular system, a developmental site known to require both S1P1 and S1P and presumably rich in S1P1 signaling activity. Finally, the modified S1P1 signaling pathway was induced in several tissues by in vivo administration of FTY720, a well-characterized agonist that is highly active on S1P1.
Because S1P1 is widely expressed (refs. 4–6 and Supplemental Figure 6) and S1P is produced by all cells, S1P1 activation can potentially take place at many locations. However, with the exception of endothelial cells, particularly in lymphoid tissues, activated S1P1 within most tissues was limited under homeostatic conditions. In some parenchymal cell types, such as hepatocytes, which express S1P1, it is likely due to a lack of access to S1P signaling pools, which resulted in substantial receptor reserve. Although liver is known to produce S1P (60), the high levels of degradative enzymes — S1P lyase and phosphatases — likely keep the extracellular S1P levels low enough to prevent receptor activation. Under acute circumstances, such as during systemically induced inflammation, our results are consistent with a scenario in which S1P from the plasma gains access to the interstitial spaces to activate S1P1. This does not rule out the possibility that S1P for S1P1 activation may be produced locally through an upregulation of its synthesis and transport and/or a downregulation of its degradative mechanisms.
Under homeostatic conditions, S1P1 activation in adult mice was most enriched in the endothelial cells of lymphoid organs, with significantly less observed in the endothelial cells of tissues such as liver and heart. Recently, it was shown that S1P released from platelets is crucial to maintain the vascular integrity of high endothelial venules, which support extensive lymphocyte trafficking (61). Our results showing high levels of S1P1 activation on the high endothelial venules imply that S1P1 has a prominent role. The unique features of other lymphoid endothelia that result in enhanced S1P1 activation are not known; however, our observations suggest that S1P signaling pools are able to gain access to S1P1 in these endothelial cells to a greater extent than in nonlymphoid tissues. Indeed, it has been shown that lymphatic endothelial cells are the main source of S1P needed for the egress of lymphocytes from lymph nodes, supporting the idea that the endothelium in lymph nodes, through which lymphocytes exit, has an active role in local S1P production (30, 35). It has also been shown that neural crest–derived pericytes, which underlie the endothelium in thymus, are necessary for the production of S1P to support the emigration of thymocytes into the blood (36). This specialized S1P production may lead to heightened endothelial S1P1 signaling around egress sites. Another factor could be the absence of degradative enzymes, such as the lipid phosphatase, LPP3, which has been shown to be critical for thymocyte egress (62) and may increase local concentrations of S1P in the vicinity of the lymphoid tissue endothelium.
Camerer et al. developed mutant pS1Pless mice engineered to selectively lack S1P in plasma and demonstrated that these pS1Pless mice had increased basal vascular leak and impaired survival after anaphylaxis, exposure to vascular leak–inducing agents, and related inflammatory challenges (12). Increased leak was associated with increased interendothelial cell gaps in venules and was reversed by restoration of plasma S1P levels and by acute treatment with an S1P1 agonist. The results suggested that plasma S1P maintains basal vascular integrity through S1P1 and protects from lethal responses to leak-inducing mediators. These observations on S1P1 function in the endothelium are consistent with our results with the S1P1 GFP signaling mice. LPS intensified S1P1 activation in pulmonary and hepatic endothelial cells as well as in endothelial cells of other tissues. Moreover, we found that the LPS-induced S1P1 activation in endothelial cells was substantially dampened in mice with low circulating S1P levels, suggesting that blood S1P triggers S1P1 activation in endothelial cells during inflammation. Our results are also consistent with findings showing LPS-induced vascular permeability is blocked by induction of S1P receptor signaling on the lung endothelium (63). The question of how S1P from the circulation activates S1P1 on endothelial cells during inflammatory challenge still remains to be answered. It has been suggested that S1P1 may be cryptically expressed on the basal surface of endothelial cells and normally shielded from S1P in the circulation (12). In this case, during conditions of vascular leakage, sufficient S1P may gain access to the basal surface of the endothelium to activate S1P1.
The majority of lymphocytes in the S1P1 GFP signaling mice did not show evidence of S1P1 activation, even though it is believed that some intrinsic S1P1 signaling is required for all lymphocytes to gain access to the circulation from lymphoid tissues (15, 64). It is possible that the lack of a detectable GFP signal may possibly be due to a low level of intrinsic S1P1 activation on lymphocytes that is needed for their egress from lymphoid tissues. Some lymphocytes, however, did show evidence of S1P1 activation. These included B cells in the MZ of the spleen, which are specialized cells that are exposed to high levels of S1P coming from the blood circulation. They shuttle continuously from follicle to the MZ several times a day as directed by S1P1, consistent with the strong activation signal found in these cells (18, 19). This might suggest that continuous cycles of S1P1 activation are required in order to ensure an interaction of the modified receptor and the β-arrestin fusion protein, which is in competition with endogenous β-arrestins, whose levels may be high in lymphocytes. Interestingly, MARCO+ MZ macrophages (52, 53), which presumably undergo extensive exposure to S1P, similar to MZ B cells, also exhibited elevated levels of S1P1 activation.
While FTY720 administration potently activated the modified S1P1 signaling pathway on endothelium and hepatocytes, it had little apparent stimulatory effect on the modified S1P1 signaling pathway lymphocytes. This result is consistent with the observation that FTY720 causes rapid inactivation of lymphocyte S1P1 (15, 16), which may minimize its exposure to the agonist.
The S1P1 signaling pathway has been implicated as a pathway relevant to several diseases. These include multiple sclerosis (25, 26), rheumatoid arthritis (65), and cancer (66). The ability to visualize S1P1 activation in the S1P1 GFP signaling mice is a powerful tool to help delineate the cells that respond to S1P via activated S1P1 during these pathogenic processes. The S1P1 GFP signaling mice can also enable the evaluation of S1P1-active compounds in vivo and how their application would affect S1P1 activation in experimental disease models. Finally, the in vivo design used in the S1P1 GFP signaling mice can in principle be applied to essentially any other GPCR to help determine its functions in physiology and disease.
Methods
S1P1 receptor ligands.
S1P, sphingosine, and LPA were purchased from Avanti Polar Lipids; dhS1P was purchased from Sigma-Aldrich; SEW2871 was purchased from Cayman; and RP-001 was purchased from Tocris.
Generation of S1P1 GFP signaling mice.
A knockin targeting vector (Figure 1B) was prepared containing a bicistronic transcriptional unit, consisting of 2 fusion genes, S1pr1 linked to tTA via a TEV protease cleavage site and mouse β-arrestin 2-TEV protease, which were designed as outlined by Barnea et al. (37). The fusion genes were interconnected by an IRES. The neomycin resistance gene (NeoR) flanked by loxP sites was inserted downstream of the bicistronic transcription unit. Gene targeting in TC1 ES cells and generation of chimeric and heterozygous mice were conducted as described previously (67). One targeted ES clone was used to establish chimeric mice, which were crossed with C57BL/6 mice to obtain heterozygotes. S1pr1 knockin genotypes were determined by Southern blot and PCR analyses of genomic DNA isolated from ES cells and mouse tails. For genotyping by PCR, 3 primers were used: 5′AGAGGAATGTGGGCTGTTGATCCT3′ (primer 1), 5′GGTGAACATCCACCCACTATTCCA 3′ (primer 2), and 5′CCAAATTAAGGGCCAGCTCATTCC3′ (primer 3). Primers 1 and 2 detected the WT allele and amplified a 290-bp fragment. Primers 2 and 3 detected the knockin allele and amplified a 400-bp fragment. Thirty-five cycles of 94°C (30 s), 60°C (30 s), and 72°C (1 min) were used for PCR.
A Cre recombinase transgenic line (EIIa Cre; ref. 68; The Jackson Laboratory, stock no. 003724) was crossed to S1pr1 knockin mice to excise the neomycin resistance gene (NeoR) during embryogenesis. To identify the WT and the S1pr1 knockin (-NeoR) alleles, 3 primers were used. Primers 1 and 2, as described above, detected the WT allele, and primer 2 and 5′GGTTGGCGATTAAATGCTGA3′ (primer 4) detected the S1pr1 knockin (-NeoR) allele and amplified a 630-bp fragment. Thirty-five cycles of 94°C (30 s), 60°C (1 min), and 72°C (1.5 min) were used for PCR.
Both knockin lines (with or without NeoR) were crossed with a histone-EGFP reporter mouse (H2B-GFP; Tg(tetO-HIST1H2BJ/GFP)47Efu/J; The Jackson Laboratory, stock no. 005104), in which human histone 1 protein H2bj and EGFP fusion protein were expressed under the control of a tetracycline-responsive promoter element and cytomegalovirus minimal promoter, to derive the S1P1 GFP signaling mice (Figure 1C and ref. 46). The age-matched offspring from the multiple litters were used for each experiment.
Analysis of S1P1 activation in embryos, tissues, and MEFs.
S1P1 GFP signaling MEFs, which were obtained by breeding H2B-GFP mice with S1pr1 knockin heterozygous mice, were isolated from embryos at E12.5. The MEFs were incubated for 16 hours in DMEM containing 10% charcoal-stripped FBS and received various lipids (S1P, dhS1P, LPA, or sphingosine), selective ligands (SEW2871 or RP-001), or vehicle (4 mg/ml BSA in PBS). GFP expression in the MEFs was observed by an inverted laser-scanning confocal microscope (Carl Zeiss Microscopy) and a BD LSR II flow cytometer (BD Biosciences).
For Western blotting, the MEFs were incubated for 16 hours in DMEM containing 0.1% FBS and received S1P ligands (S1P, SEW2871, or RP-001) or vehicle (4 mg/ml BSA in PBS). After a 10-minute treatment, cells were lysed in a buffer containing 10 mM Tris-HCl (pH 7.2), 150 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 mM sodium orthovanadate, and Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). After centrifugation at 3,000 g for 10 minutes, the supernatants were used for Western blotting. Rabbit monoclonal antibodies for Akt (Cell Signaling, clone 11E7) and phosphor-Akt (Cell Signaling, clone D9E) were used for the primary antibodies, and Alexa Fluor 680 goat anti-rabbit IgG (H+L) (Molecular Probes) and IRDye 800CW goat anti-rabbit IgG (H+L) (LI-COR Biosciences) were used for the secondary antibodies for Western blot. Immunoreactivity was detected by an Odyssey infrared system (LI-COR Biosciences).
S1P1 GFP signaling mouse E9.5 and E10.5 embryos, obtained after timed mating by breeding H2B-GFP with S1pr1 knockin heterozygous mice, were observed with a MZ FLIII stereo-fluorescence microscope (Leica Microsystems) using QCapture imaging software (QImaging).
For histologic analysis, adult mice were anesthetized with tribromoethanol and perfused with normal saline followed by ice-cold 4% PFA solution (in 0.1 M phosphate buffer, pH 7.4). The tissues were harvested, postfixed in 4% PFA for 2 hours at 4°C, and equilibrated in 30% sucrose solution (0.1 M phosphate buffer, pH 7.4) overnight at 4°C. The tissues were embedded in OCT compound, and 12-μm sections were obtained. For immunostaining, nonconjugated primary antibodies for mouse CD31 (dianova, clone SZ31), LYVE-1 (R&D Systems), PNAd (high endothelial venules) (BD Pharmingen, clone MECA-79), albumin (Bethyl Laboratories), B220 (BD Pharmingen, clone RA3-6B2), and MARCO (GeneTex, clone ED31) were used. To detect primary antibodies, fluorescently labeled secondary antibodies were used: donkey anti-rat IgG (Cyanine Cy3) (Jackson ImmunoResearch Laboratories) for CD31, B220, and MARCO; donkey anti-goat IgG (Alexa Fluor 555) (Life Technologies) for albumin; donkey anti-goat IgG (DyLight 405) (Jackson ImmunoResearch Laboratories) for LYVE-1; donkey anti-rat IgG (DyLight 405) (Jackson ImmunoResearch Laboratories) for PNAd. The images were captured with an inverted laser-scanning confocal microscope (Carl Zeiss Microscopy) using the Zeiss LSM software.
For flow cytometry analysis of leukocytes, blood was collected in K2EDTA-coated BD Microtainer tubes. The immune tissues (thymi, spleens, lymph nodes) were harvested, minced with 0.5 ml cold PBS in 60-mm dishes, and filtered through a 40-μm cell strainer (BD Biosciences). The filtrates were centrifuged for 5 minutes at 200 g, and thymus and lymph node pellets were suspended in FACS buffer (5% heat-inactivated FBS, 0.02% sodium azide in PBS). ACK lysis buffer (Lonza) (2 ml) was added to the spleen pellet and blood (100 μl) and incubated for 5 minutes at room temperature. After washing the cells with PBS, they were suspended in FACS buffer.
Lungs were minced and incubated in a digestion buffer (0.05% collagenase IV, 0.01% DNase I, 2% FBS, 10 mM HEPES, pH 7.5) for 30 minutes at 37°C. The reaction was terminated by adding FBS (final concentration 10%) and EDTA (final concentration 5 mM). The cell suspension was forced through a 100-μm cell strainer (BD Biosciences) into a 50-ml Falcon tube, and the filtrates were centrifuged for 5 minutes at 200 g. The pellets were suspended in 2 ml of ACK lysis buffer and incubated for 5 minutes at room temperature. After the cells were washed with PBS, the cells were suspended in FACS buffer.
To isolate liver cells, the left lobes were removed, and warmed (37°C) perfusion medium (Life Technologies) was perfused through the portal vein, followed by liver digestion medium (Life Technologies). The digested liver was torn with forceps in liver digestion medium, and single cells were dispersed in the medium. The reaction was terminated by adding FBS (final concentration 10%) and EDTA (final concentration 5 mM). The cell suspension was forced through a 100-μm cell strainer into a 50-ml Falcon tube, and the filtrates were centrifuged for 5 minutes at 200 g. After the cells were washed with PBS, the cells were suspended in FACS buffer (69).
For immunostaining of leukocytes, the following antibodies were used: phycoerythrin-conjugated (PE-conjugated) rat anti-mouse CD3 (BD Pharmingen, clone 17A2); (allophycocyanin–cyanine 7–conjugated) APC-Cy7–conjugated rat anti-mouse CD4 (BD Pharmingen, clone GK1.5); APC-conjugated rat anti-mouse CD8a (BD Pharmingen, clone 53-6.7); PerCP-Cy5.5–conjugated rat anti-mouse CD11b (BD Pharmingen, clone M1/70); and Pacific Blue–conjugated rat anti-mouse CD45R (B220) (BD Pharmingen, clone RA3-6B2). For immunostaining of lung endothelial cells, APC-conjugated rat anti-mouse CD31 (BioLegend, clone 390) and APC-Cy7–conjugated rat anti-mouse CD45 (BioLegend, clone 30-F11) were used. For immunostaining of liver cells, APC-conjugated rat anti-mouse CD31 (BioLegend, clone 390) and PE-Cy7–conjugated hamster anti-mouse CD95 (BD Pharmingen, clone Jo2) were used. Flow cytometry was performed on a BD LSR II flow cytometer (BD Biosciences).
FTY720 and LPS treatments.
FTY720 (Cayman; 1 mg/kg; dissolved in PBS containing 50% ethanol) or LPS from E. coli 055:B5 (Sigma-Aldrich; 20 mg/kg; dissolved in PBS) was intraperitoneally injected into mice. The tissues were harvested 24 hours (FTY720) or 24–72 hours (LPS) after the injections.
Preparation of plasma S1P-deficient S1P1 GFP signaling mice.
Sphk1fl/fl Sphk2–/– Mx1-cre mice were bred using Sphk1fl/fl mice (30) (MMRRC, stock no. 030038-UCD), Sphk2–/– mice (10), and Mx1-cre mice (70) (The Jackson Laboratory, stock no. 003556). pS1Pless mice were prepared by injection of pIpC into Sphk1fl/fl Sphk2–/– Mx1-cre pups as described previously (30). Controls (Sphk1fl/fl Sphk2–/– mice) were similarly bred and injected. Bone marrow cells were isolated from the femurs and tibias of adult pS1Pless mice or controls 4 weeks after pIpC was injected. A single-cell suspension (1 × 107 cells per mouse) in 0.2 ml saline was injected into the tail veins of irradiated (9 Gy) S1P1 GFP signaling mice. The mice were used 10 weeks after transplantation. Sphingolipid levels were measured by HPLC-tandem mass spectrometry by the Lipidomics Core at the Medical University of South Carolina on a Thermo Finnigan TSQ 7000 triple quadrupole mass spectrometer, operating in a multiple-reaction monitoring positive ionization mode as described previously (58).
Immunohistochemical staining of S1P1 and GFP.
Paraffin-embedded 5-μm tissue sections were deparaffinized in Histo-Clear II (National Diagnostics). Endogenous peroxide activity was quenched with 30-minute incubation in 0.3% hydrogen peroxide solution in methanol, followed by rehydration in a graded ethanol/water series. Antigen retrieval was performed in a 2100 Antigen Retriever according to manufacturer instructions, with slides immersed in Target Retrieval Solution pH 9 (Dako). Blocking was performed with an Avidin/Biotin Blocking Kit (Vector Labs), followed by serum blocking with a solution of 5% goat serum, 5% horse serum, 1% BSA in PBS. For S1P1 staining, nonconjugated primary antibody for EDG-1 H-60 (Santa Cruz Biotechnology, polyclonal) was used, followed by secondary biotinylated horse anti-rabbit IgG (Vector Labs). The VECTASTAIN ABC system was used with VECTOR NovaRED peroxidase substrate (Vector Labs) to produce a red stain. To detect S1P1 activation, GFP staining was performed after avidin/biotin blocking and serum blocking were repeated. Nonconjugated primary antibody for GFP (Abcam, chicken polyclonal antibody) was used, followed by secondary biotinylated goat anti-chicken IgG (Vector Labs). A blue stain was produced using the VECTASTAIN ABC-Alkaline Phosphatase system with Vector Blue phosphatase substrate and Levamisole endogenous phosphatase blocking solution (Vector Labs). Slides were dehydrated in a graded ethanol/water series, cleared with Histo-Clear II (National Diagnostics), and mounted with VectaMount Permanent Mounting Medium (Vector Labs). The images were captured with a Leica DM LB Microscope (Leica Microsystems) using QCapture imaging software (QImaging).
Statistics.
Statistical significance was determined using the Student’s t test. In all cases, P < 0.05 was considered statistically significant.
Study approval.
All animal procedures were approved by the National Institute of Diabetes and Digestive and Kidney Diseases and performed in accordance with the NIH guidelines.
Supplementary Material
Acknowledgments
This research was supported by the Intramural Research Programs of the NIH, National Institute of Diabetes and Digestive and Kidney Diseases, the Lipidomics Shared Resource, Hollings Cancer Center, Medical University of South Carolina (P30 CA138313), and the Lipidomics Core in the SC Lipidomics and Pathobiology COBRE (P20 RR017677). We thank Chuxia Deng and Cuiling Li for helping to generate the chimera mice and Galina Tuymetova for supplying the Sphk mutant mice.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(5):2076–2086. doi:10.1172/JCI71194.
References
- 1.Takabe K, Paugh SW, Milstien S, Spiegel S. “Inside-out” signaling of sphingosine-1-phosphate: therapeutic targets. Pharmacol Rev. 2008;60(2):181–195. doi: 10.1124/pr.107.07113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Blaho VA, Hla T. Regulation of mammalian physiology, development, and disease by the sphingosine 1-phosphate and lysophosphatidic acid receptors. Chem Rev. 2011;111(10):6299–6320. doi: 10.1021/cr200273u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rosen H, Stevens RC, Hanson M, Roberts E, Oldstone MB. Sphingosine-1-phosphate and its receptors: structure, signaling, and influence. Annu Rev Biochem. 2013;82:637–662. doi: 10.1146/annurev-biochem-062411-130916. [DOI] [PubMed] [Google Scholar]
- 4.Chae SS, Proia RL, Hla T. Constitutive expression of the S1P1 receptor in adult tissues. Prostaglandins Other Lipid Mediat. 2004;73(1–2):141–150. doi: 10.1016/j.prostaglandins.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 5.Regard JB, Sato IT, Coughlin SR. Anatomical profiling of G protein-coupled receptor expression. Cell. 2008;135(3):561–571. doi: 10.1016/j.cell.2008.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cahalan SM, et al. Actions of a picomolar short-acting S1P(1) agonist in S1P(1)-eGFP knock-in mice. Nat Chem Biol. 2011;7(5):254–256. doi: 10.1038/nchembio.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, et al. Edg-1, the G protein-coupled receptor for sphingosine-1-phosphate, is essential for vascular maturation. J Clin Invest. 2000;106(8):951–961. doi: 10.1172/JCI10905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Allende ML, Yamashita T, Proia RL. G-protein-coupled receptor S1P1 acts within endothelial cells to regulate vascular maturation. Blood. 2003;102(10):3665–3667. doi: 10.1182/blood-2003-02-0460. [DOI] [PubMed] [Google Scholar]
- 9.Kono M, et al. The sphingosine-1-phosphate receptors S1P1, S1P2, and S1P3 function coordinately during embryonic angiogenesis. J Biol Chem. 2004;279(28):29367–29373. doi: 10.1074/jbc.M403937200. [DOI] [PubMed] [Google Scholar]
- 10.Mizugishi K, Yamashita T, Olivera A, Miller GF, Spiegel S, Proia RL. Essential role for sphingosine kinases in neural and vascular development. Mol Cell Biol. 2005;25(24):11113–11121. doi: 10.1128/MCB.25.24.11113-11121.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng X, et al. Protective effects of sphingosine 1-phosphate in murine endotoxin-induced inflammatory lung injury. Am J Respir Crit Care Med. 2004;169(11):1245–1251. doi: 10.1164/rccm.200309-1258OC. [DOI] [PubMed] [Google Scholar]
- 12.Camerer E, et al. Sphingosine-1-phosphate in the plasma compartment regulates basal and inflammation-induced vascular leak in mice. J Clin Invest. 2009;119(7):1871–1879. doi: 10.1172/JCI38575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Oo ML, et al. Engagement of S1P(1)-degradative mechanisms leads to vascular leak in mice. J Clin Invest. 2011;121(6):2290–2300. doi: 10.1172/JCI45403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Teijaro JR, et al. Endothelial cells are central orchestrators of cytokine amplification during influenza virus infection. Cell. 2011;146(6):980–991. doi: 10.1016/j.cell.2011.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwab SR, Cyster JG. Finding a way out: lymphocyte egress from lymphoid organs. Nat Immunol. 2007;8(12):1295–1301. doi: 10.1038/ni1545. [DOI] [PubMed] [Google Scholar]
- 16.Matloubian M, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427(6972):355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- 17.Allende ML, Dreier JL, Mandala S, Proia RL. Expression of the sphingosine 1-phosphate receptor, S1P1, on T-cells controls thymic emigration. J Biol Chem. 2004;279(15):15396–15401. doi: 10.1074/jbc.M314291200. [DOI] [PubMed] [Google Scholar]
- 18.Cinamon G, Zachariah MA, Lam OM, Foss FW, Jr, Cyster JG. Follicular shuttling of marginal zone B cells facilitates antigen transport. Nat Immunol. 2008;9(1):54–62. doi: 10.1038/ni1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Arnon TI, Horton RM, Grigorova IL, Cyster JG. Visualization of splenic marginal zone B-cell shuttling and follicular B-cell egress. Nature. 2013;493(7434):684–688. doi: 10.1038/nature11738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mandala S, et al. Alteration of lymphocyte trafficking by sphingosine-1-phosphate receptor agonists. Science. 2002;296(5566):346–349. doi: 10.1126/science.1070238. [DOI] [PubMed] [Google Scholar]
- 21.Brinkmann V, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277(24):21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 22.Kharel Y, et al. Sphingosine kinase 2 is required for modulation of lymphocyte traffic by FTY720. J Biol Chem. 2005;280(44):36865–36872. doi: 10.1074/jbc.M506293200. [DOI] [PubMed] [Google Scholar]
- 23.Zemann B, et al. Sphingosine kinase type 2 is essential for lymphopenia induced by the immunomodulatory drug FTY720. Blood. 2006;107(4):1454–1458. doi: 10.1182/blood-2005-07-2628. [DOI] [PubMed] [Google Scholar]
- 24.Thangada S, et al. Cell-surface residence of sphingosine 1-phosphate receptor 1 on lymphocytes determines lymphocyte egress kinetics. J Exp Med. 2010;207(7):1475–1483. doi: 10.1084/jem.20091343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brinkmann V, et al. Fingolimod (FTY720): discovery and development of an oral drug to treat multiple sclerosis. Nat Rev Drug Discov. 2010;9(11):883–897. doi: 10.1038/nrd3248. [DOI] [PubMed] [Google Scholar]
- 26.Choi JW, et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc Natl Acad Sci U S A. 2011;108(2):751–756. doi: 10.1073/pnas.1014154108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4(5):397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Liu Y, Sullards MC, Merrill AH., Jr An introduction to sphingolipid metabolism and analysis by new technologies. Neuromolecular Med. 2010;12(4):306–319. doi: 10.1007/s12017-010-8132-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schwab SR, Pereira JP, Matloubian M, Xu Y, Huang Y, Cyster JG. Lymphocyte sequestration through S1P lyase inhibition and disruption of S1P gradients. Science. 2005;309(5741):1735–1739. doi: 10.1126/science.1113640. [DOI] [PubMed] [Google Scholar]
- 30.Pappu R, et al. Promotion of lymphocyte egress into blood and lymph by distinct sources of sphingosine-1-phosphate. Science. 2007;316(5822):295–298. doi: 10.1126/science.1139221. [DOI] [PubMed] [Google Scholar]
- 31.Olivera A, Allende ML, Proia RL. Shaping the landscape: Metabolic regulation of S1P gradients. Biochim Biophys Acta. 2013;1831(1):193–202. doi: 10.1016/j.bbalip.2012.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brinkmann V, Cyster JG, Hla T. FTY720: sphingosine 1-phosphate receptor-1 in the control of lymphocyte egress and endothelial barrier function. Am J Transplant. 2004;4(7):1019–1025. doi: 10.1111/j.1600-6143.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 33.Rosen H, Sanna MG, Cahalan SM, Gonzalez-Cabrera PJ. Tipping the gatekeeper: S1P regulation of endothelial barrier function. Trends Immunol. 2007;28(3):102–107. doi: 10.1016/j.it.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 34.Cyster JG, Schwab SR. Sphingosine-1-phosphate and lymphocyte egress from lymphoid organs. Annu Rev Immunol. 2012;30:69–94. doi: 10.1146/annurev-immunol-020711-075011. [DOI] [PubMed] [Google Scholar]
- 35.Pham TH, et al. Lymphatic endothelial cell sphingosine kinase activity is required for lymphocyte egress and lymphatic patterning. J Exp Med. 2010;207(1):17–27. doi: 10.1084/jem.20091619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zachariah MA, Cyster JG. Neural crest-derived pericytes promote egress of mature thymocytes at the corticomedullary junction. Science. 2010;328(5982):1129–1135. doi: 10.1126/science.1188222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barnea G, et al. The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci U S A. 2008;105(1):64–69. doi: 10.1073/pnas.0710487105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kohout TA, Lefkowitz RJ. Regulation of G protein-coupled receptor kinases and arrestins during receptor desensitization. Mol Pharmacol. 2003;63(1):9–18. doi: 10.1124/mol.63.1.9. [DOI] [PubMed] [Google Scholar]
- 39.Premont RT, Gainetdinov RR. Physiological roles of G protein-coupled receptor kinases and arrestins. Annu Rev Physiol. 2007;69:511–534. doi: 10.1146/annurev.physiol.69.022405.154731. [DOI] [PubMed] [Google Scholar]
- 40.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. beta-Arrestin: a protein that regulates beta-adrenergic receptor function. Science. 1990;248(4962):1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 41.Shukla AK, et al. Structure of active beta-arrestin-1 bound to a G-protein-coupled receptor phosphopeptide. Nature. 2013;497(7447):137–141. doi: 10.1038/nature12120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci U S A. 1992;89(12):5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baron U, Bujard H. Tet repressor-based system for regulated gene expression in eukaryotic cells: principles and advances. Methods Enzymol. 2000;327:401–421. doi: 10.1016/s0076-6879(00)27292-3. [DOI] [PubMed] [Google Scholar]
- 44.Berens C, Hillen W. Gene regulation by tetracyclines. Constraints of resistance regulation in bacteria shape TetR for application in eukaryotes. Eur J Biochem. 2003;270(15):3109–3121. doi: 10.1046/j.1432-1033.2003.03694.x. [DOI] [PubMed] [Google Scholar]
- 45.Fuchs E, Horsley V. Ferreting out stem cells from their niches. Nat Cell Biol. 2011;13(5):513–518. doi: 10.1038/ncb0511-513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tumbar T, et al. Defining the epithelial stem cell niche in skin. Science. 2004;303(5656):359–363. doi: 10.1126/science.1092436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hochedlinger K, Yamada Y, Beard C, Jaenisch R. Ectopic expression of Oct-4 blocks progenitor-cell differentiation and causes dysplasia in epithelial tissues. Cell. 2005;121(3):465–477. doi: 10.1016/j.cell.2005.02.018. [DOI] [PubMed] [Google Scholar]
- 48.Liu CH, Thangada S, Lee MJ, Van Brocklyn JR, Spiegel S, Hla T. Ligand-induced trafficking of the sphingosine-1-phosphate receptor EDG-1. Mol Biol Cell. 1999;10(4):1179–1190. doi: 10.1091/mbc.10.4.1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jo E, et al. S1P1-selective in vivo-active agonists from high-throughput screening: off-the-shelf chemical probes of receptor interactions, signaling, and fate. Chem Biol. 2005;12(6):703–715. doi: 10.1016/j.chembiol.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 50.Van Brocklyn JR, et al. Dual actions of sphingosine-1-phosphate: extracellular through the Gi-coupled receptor Edg-1 and intracellular to regulate proliferation and survival. J Cell Biol. 1998;142(1):229–240. doi: 10.1083/jcb.142.1.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–354. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 52.Elomaa O, et al. Cloning of a novel bacteria-binding receptor structurally related to scavenger receptors and expressed in a subset of macrophages. Cell. 1995;80(4):603–609. doi: 10.1016/0092-8674(95)90514-6. [DOI] [PubMed] [Google Scholar]
- 53.Elomaa O, et al. Structure of the human macrophage MARCO receptor and characterization of its bacteria-binding region. J Biol Chem. 1998;273(8):4530–4538. doi: 10.1074/jbc.273.8.4530. [DOI] [PubMed] [Google Scholar]
- 54.Newman PJ, et al. PECAM-1 (CD31) cloning and relation to adhesion molecules of the immunoglobulin gene superfamily. Science. 1990;247(4947):1219–1222. doi: 10.1126/science.1690453. [DOI] [PubMed] [Google Scholar]
- 55.Prevo R, Banerji S, Ferguson DJ, Clasper S, Jackson DG. Mouse LYVE-1 is an endocytic receptor for hyaluronan in lymphatic endothelium. J Biol Chem. 2001;276(22):19420–19430. doi: 10.1074/jbc.M011004200. [DOI] [PubMed] [Google Scholar]
- 56.Miyasaka M, Tanaka T. Lymphocyte trafficking across high endothelial venules: dogmas and enigmas. Nat Rev Immunol. 2004;4(5):360–370. doi: 10.1038/nri1354. [DOI] [PubMed] [Google Scholar]
- 57.Akiyama T, Sadahira Y, Matsubara K, Mori M, Igarashi Y. Immunohistochemical detection of sphingosine-1-phosphate receptor 1 in vascular and lymphatic endothelial cells. J Mol Histol. 2008;39(5):527–533. doi: 10.1007/s10735-008-9193-y. [DOI] [PubMed] [Google Scholar]
- 58.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39(2):82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Inagaki HK, et al. Visualizing neuromodulation in vivo: TANGO-mapping of dopamine signaling reveals appetite control of sugar sensing. Cell. 2012;148(3):583–595. doi: 10.1016/j.cell.2011.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bektas M, et al. Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J Biol Chem. 2010;285(14):10880–10889. doi: 10.1074/jbc.M109.081489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Herzog BH, et al. Podoplanin maintains high endothelial venule integrity by interacting with platelet CLEC-2. Nature. 2013;502(7469):105–109. doi: 10.1038/nature12501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Breart B, et al. Lipid phosphate phosphatase 3 enables efficient thymic egress. J Exp Med. 2011;208(6):1267–1278. doi: 10.1084/jem.20102551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McVerry BJ, Garcia JG. Endothelial cell barrier regulation by sphingosine 1-phosphate. J Cell Biochem. 2004;92(6):1075–1085. doi: 10.1002/jcb.20088. [DOI] [PubMed] [Google Scholar]
- 64.Rosen H, Goetzl EJ. Sphingosine 1-phosphate and its receptors: an autocrine and paracrine network. Nat Rev Immunol. 2005;5(7):560–570. doi: 10.1038/nri1650. [DOI] [PubMed] [Google Scholar]
- 65.Kitano M, et al. Sphingosine 1-phosphate/sphingosine 1-phosphate receptor 1 signaling in rheumatoid synovium: regulation of synovial proliferation and inflammatory gene expression. Arthritis Rheum. 2006;54(3):742–753. doi: 10.1002/art.21668. [DOI] [PubMed] [Google Scholar]
- 66.Pyne NJ, Tonelli F, Lim KG, Long JS, Edwards J, Pyne S. Sphingosine 1-phosphate signalling in cancer. Biochem Soc Trans. 2012;40(1):94–100. doi: 10.1042/BST20110602. [DOI] [PubMed] [Google Scholar]
- 67.Deng C, Wynshaw-Boris A, Zhou F, Kuo A, Leder P. Fibroblast growth factor receptor 3 is a negative regulator of bone growth. Cell. 1996;84(6):911–921. doi: 10.1016/S0092-8674(00)81069-7. [DOI] [PubMed] [Google Scholar]
- 68.Lakso M, et al. Efficient in vivo manipulation of mouse genomic sequences at the zygote stage. Proc Natl Acad Sci U S A. 1996;93(12):5860–5865. doi: 10.1073/pnas.93.12.5860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Goncalves LA, Vigario AM, Penha-Goncalves C. Improved isolation of murine hepatocytes for in vitro malaria liver stage studies. Malar J. 2007;6:169. doi: 10.1186/1475-2875-6-169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kuhn R, Schwenk F, Aguet M, Rajewsky K. Inducible gene targeting in mice. Science. 1995;269(5229):1427–1429. doi: 10.1126/science.7660125. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.