

Background: Food and Drug Administration-approved proteasome inhibitors act covalently, which hampers their safety.
Results: Structure activity relationship (SAR) studies, mass spectrometry, and dialysis identified PI-1840 as a noncovalent proteasome inhibitor that sensitizes human cancer cells to p53 and Bcl2 antagonists.
Conclusion: Noncovalent proteasome inhibitors suppress in vivo tumor growth with little toxicity in mouse xenografts.
Significance: Discovery of noncovalent proteasome inhibitors warrants their development as anti-cancer drugs.
Keywords: Anticancer Drug, Apoptosis, Bcl-2 Family Proteins, Cancer Therapy, Cell Proliferation, p53, Proteasome, CT-L, PI-1840, Noncovalent
Abstract
The proteasome inhibitor bortezomib is effective in hematologic malignancies such as multiple myeloma but has little activity against solid tumors, acts covalently, and is associated with undesired side effects. Therefore, noncovalent inhibitors that are less toxic and more effective against solid tumors are desirable. Structure activity relationship studies led to the discovery of PI-1840, a potent and selective inhibitor for chymotrypsin-like (CT-L) (IC50 value = 27 ± 0.14 nm) over trypsin-like and peptidylglutamyl peptide hydrolyzing (IC50 values >100 μm) activities of the proteasome. Furthermore, PI-1840 is over 100-fold more selective for the constitutive proteasome over the immunoproteasome. Mass spectrometry and dialysis studies demonstrate that PI-1840 is a noncovalent and rapidly reversible CT-L inhibitor. In intact cancer cells, PI-1840 inhibits CT-L activity, induces the accumulation of proteasome substrates p27, Bax, and IκB-α, inhibits survival pathways and viability, and induces apoptosis. Furthermore, PI-1840 sensitizes human cancer cells to the mdm2/p53 disruptor, nutlin, and to the pan-Bcl-2 antagonist BH3-M6. Finally, in vivo, PI-1840 but not bortezomib suppresses the growth in nude mice of human breast tumor xenografts. These results warrant further evaluation of a noncovalent and rapidly reversible proteasome inhibitor as potential anticancer agents against solid tumors.
Introduction
Dysregulation of the catalytic processes mediated by the ubiquitin/proteasome system (UPS)2 contributes to the pathogenesis of many diseases, including cancer (1, 2). More than 80% of cellular proteins are degraded by the UPS (3), including proteins that regulate cell cycle progression, DNA repair, and apoptosis (4–6). Deregulation of various components of the UPS resulting in increased degradation of cell cycle inhibitors or pro-apoptotic proteins (e.g. p21Cip1, p27Kip1, p53, and Bax) contributes to malignant transformation (3, 7). The UPS has two distinct steps, recognition/ubiquitination and degradation (5, 8). The ubiquitin-protein ligase system results in the transfer of multiple ubiquitin molecules to the target protein (9). Degradation of such multiubiquitinated proteins occurs on a large 26 S proteaome complex (5, 8) that contains three proteolytic enzymes, peptidylglutamyl peptide hydrolyzing (PGPH), trypsin-like (T-L), and chymotrypsin-like (CT-L) activities, residing in the β1, β2, and β5 catalytic subunits, respectively (3, 7).
In contrast to normal cells, cancer cells generally have higher levels of proteasome activity (3) and have acquired a series of mutations that render them dependent on strong activation of survival pathways (10). One of these is the phosphorylation-dependent recognition and subsequent degradation of cellular proteins by the UPS. Furthermore, compared with normal cells, cancer cells show higher sensitivity toward the pro-apoptotic effects of proteasome inhibition. Therefore, the UPS has become a promising target for anti-cancer strategies (3, 7, 11, 12).
Although two proteasome inhibitors, bortezomib and carfilzomib, are Food and Drug Administration-approved and others are in clinical trials, they are all covalent inhibitors (13, 14). Covalent inhibitors have highly reactive and unstable chemical groups and are therefore less specific (15). This is believed to be a major cause for toxicity to patients. Furthermore, bortezomib is active against liquid but not solid tumors, and its covalent binding, which would limit its widespread tissue distribution, could be a possible reason. In contrast to covalent inhibitors, noncovalent inhibitors have the advantage of rapid binding and dissociation kinetics that would allow broader tissue distribution, reaching both liquid and solid tumors. Only very few noncovalent inhibitors have been identified, and none have entered clinical trials (16, 17). It is important to point out that at present it is not known whether noncovalent inhibitors suffer from the same drawbacks as covalent inhibitors. In this report, we describe the development of a novel noncovalent chemical probe, PI-1840, and provide data that give further support to the notion that noncovalent inhibitors are more effective against solid tumors.
EXPERIMENTAL PROCEDURES
Materials
DMEM, RPMI 1640, DMEM/Ham's F-12, horse serum, penicillin, and streptomycin were purchased from Invitrogen. Fetal bovine serum was from Atlanta Biologicals (Atlanta, GA). Purified 20 S proteasome (rabbit), purified 20 S immunoproteasome (human), fluorogenic peptide substrates N-succinyl-Leu-Leu-Val-Tyr-AMC (for the proteasomal CT-L activity) and benzyloxycarbonyl-Leu-Leu-Glu-AMC (for the proteasomal PGPH activity) were purchased from Boston Biochem (Cambridge, MA). Fluorogenic peptide substrate benzoyl-Val-Gly-Arg-AMC (for the proteasomal T-L activity) was obtained from Biomol International (Plymouth Meeting, PA). Antibodies were obtained from the following suppliers: p27Kip1 (BD Biosciences); β-actin (Sigma); phospho-Akt (Ser-473), phospho-S6 ribosomal protein (Ser-240/244), S6 ribosomal protein (5610), cleaved poly(ADP-ribose) polymerase (Asp-214) (D64E10) XP, cleaved caspase-3 (Asp-175) (5A1E) (Cell Signaling, Danvers, MA); Akt1/2 (N-19), survivin (FL-142), IKB-α (C-21), and Bax (N20) (Santa Cruz Biotechnology, Santa Cruz, CA); MTT (Calbiochem). The pan-Bcl-2 antagonist BH3-M6 and the proteasome inhibitors PI-1833 and PI-1840 were all synthesized in-house as reported previously (18, 19). Bortezomib was purchased from Selleckchem, Houston, TX. Nutlin 1 was purchased from Sigma. All other reagents were from Sigma unless otherwise noted.
Determination of CT-L, T-L, and PGPH Proteolytic Activities
These assays were performed exactly as we described previously (20). Briefly, 1 nm purified 20 S rabbit proteasome or immunoproteasome was incubated with 20 μm N-succinyl-Leu-Leu-Val-Tyr-AMC for the CT-L activity, benzoyl-Val-Gly-Arg-AMC for the T-L activity, and benzyloxycarbonyl-Leu-Leu-Glu-AMC for the PGPH activity for 1 h at 37 °C in 100 μl of assay buffer (50 mm Tris-HCl, pH 7.6) with or without compound, and the hydrolyzed AMC was measured using a Wallac Victor2 counter. To determine proteasome activity in whole cell extracts (5 μg) from cultured cells, lysates were used instead of 20 S rabbit proteasome. Whole cell extracts were prepared by homogenizing the cells in lysis buffer (50 mm Tris-HCl, pH 8.0, 5 mm EDTA, 150 mm NaCl, 0.5% Nonidet P-40), centrifuging the lysates at 12,000 × g, and collecting the supernatants as whole cell extracts as we described previously (20).
Protein Digestion, Peptide Purification, and LC-MS/MS Analysis
These procedures were performed exactly as we described previously (18). Briefly, after purified 20 S proteasome (rabbit) (1 nm) was incubated for 30 min with inhibitors in 50 mm Tris-HCl, pH 7.6, acetonitrile and trypsin were added (4 h, 37 °C). The digest was concentrated, and the peptides were extracted with C18 reversed phase pipette tip columns and injected into a mass spectrometer. To assess LC-MS/MS performance, tryptic peptides from horse apomyoglobin (25 fmol) were added to each LC-MS/MS analysis. LC-MS/MS peptide sequencing experiments were performed using a nanoflow liquid chromatograph (U3000, Dionex, Sunnyvale, CA) interfaced with an electrospray ion trap mass spectrometer to detect and localize modified peptides from the proteasome exactly as we described previously (18).
Database Searching and Data Analysis
Data searching and analysis were performed exactly as we described previously (18). Briefly, the 22 rabbit proteasome protein sequences from UniProt were searched using Sequest (21), and the search results were summarized in Scaffold 3.0. For peptide quantification, the integrated peak areas were calculated from ion chromatograms using QuanBrowser from Xcalibur 2.0 (restriction, m/z (± 0.02); retention time (120 s)). To ensure proper sequence assignment, manual inspection of the accuracy of the m/z values and the fragmentation patterns of the target peptides was performed exactly as we described previously (18).
Dialysis Using Purified Rabbit 20 S Proteasome
We used the same dialysis method that we used in our previous study (18) to determine the effect of dialysis on CT-L activity. Briefly, compounds PI-1840 (1 μm) and lactacystin (2.5 μm) or vehicle (DMSO) were added to 20 S proteasome (rabbit) at a final concentration of 1 nm in proteasome assay buffer (50 mm Tris-HCl, pH 7.6) and incubated at room temperature for 30 min. Then the proteasome/compound mixtures were added to mini dialysis units (3500 MWCO Thermo Scientific Slide-A-Lyzer) (Rockford, IL) and dialyzed against proteasome assay buffer. Immediately (t = 0) and at different time points (20, 60, 120, 240, 480, and 1080 min) of dialysis at 4 °C, samples were taken from the dialysis cassette, and the CT-L activity of 20 S proteasome was determined as we described previously (18). CT-L activity was normalized against CT-L activity of DMSO control.
Cells, Cell Culture, and Extract Preparation
MDA-MB-468 and MDA-MB-231 (human breast cancer cells), HCT-116, HCT-116-p53−/−, and HCT-116-HKH2 (human colon cancer cells), normal foreskin fibroblasts, and PC-3 (human prostate cancer cells) were cultured in DMEM. DU145 and LNCaP (human prostate cancer cells), RPMI-8226 and U266 (human multiple myeloma cells), Colo357 (human pancreatic adenocarcinoma cells), HCA2 normal foreskin fibroblasts, and RXF-397 (human renal carcinoma cells) were cultured in RPMI 1640 medium. All media were supplemented with 10% fetal bovine serum (FBS), and 1% penicillin/streptomycin antibiotics. Normal immortalized MCF-10A breast cells were cultured in DMEM/Ham's F-12 containing 5% horse serum, 20 ng/ml epidermal growth factor (EGF), 100 ng/ml cholera toxin, 500 ng/ml hydrocortisone, and 0.01 mg/ml insulin. Cells were maintained at 37 °C in a humidified incubator in an atmosphere of 5% CO2.
Western Blot Analysis
To prepare whole cell lysates, cells were washed with PBS twice and lysed in 30 mm Hepes, pH 7.5, 10 mm NaCl, 5 mm MgCl2, 25 mm NaF, 1 mm EGTA, 1% Triton-X-100, 10% glycerol, protease inhibitor mixture, 2 mm PMSF, 2 mm Na3VO4, and 6.4 mg/ml p-nitrophenyl phosphate. Lysates were cleared by centrifugation at 12,000 × g for 15 min, and the supernatants were collected as whole cell extracts. The protein concentration was determined by the Bradford assay. Cell lysates (50 μg) were separated by SDS-PAGE, transferred to a nitrocellulose membrane, and probed with specific antibodies, and signals were visualized by enhanced chemiluminescence (ECL, Amersham Biosciences) according to the manufacturer's protocol.
MTT Assay
Cancer cells or normal immortalized MCF-10A breast cells were plated in 96-well plates in 100 μl of above specified medium and allowed to attach overnight. Cells were then incubated for different time points with varying concentrations of drugs or appropriate controls. After that, media were aspirated and replaced with 100 μl of complete media containing 1 mg/ml MTT and incubated for 3 h at 37 °C in a 5% CO2-humidified incubator. After incubation, media were aspirated, and DMSO was added. Cells were then incubated for 10 min at room temperature while shaking, and the absorbance was determined at 540 nm using a μQuant spectrophotometric plate reader (Bio-TEK, Winooski, VT).
Antitumor Study of Human Tumor Xenografts in Nude Mice
Female nude mice (Charles River Laboratories, Wilmington, MA) were maintained and treated in accordance with the Institutional Animal Care and Use Committee procedures and guidelines. Exponentially growing MDA-MB-231 cells were harvested via trypsinization and pelleted at 300 × g for 5 min. Cells were resuspended in 50% Matrigel with DPBS (Invitrogen) at 10 × 106 cells per 200 μl and injected into the right flank of mice. The tumor xenografts were monitored with electronic caliper measurements, and tumor volume (V) was calculated using the following formula: V = (W × L2)/2, where width (W) is the largest diameter, and length (L) is the smallest diameter. When the tumors reached ∼ 250 mm3, the animals were randomized, and treatment schedules were implemented. Treatments consisted of intraperitoneal injections of vehicle control (30% 2-hydroxypropyl-β-cyclodextrin) (n = 5) or PI-1840 (n = 5) at 150 mg/kg (everyday for 14 days) and bortezomib (n = 6) at 1 mg/kg (two times per week for 14 days).
RESULTS
High Throughput Screening and Hit-to-Lead Optimization Identifies PI-1840 as a Potent, Noncovalent, and Rapidly Reversible Proteasome CT-L Inhibitor
Our efforts to develop noncovalent proteasome inhibitors have recently resulted in the identification of the hit PI-1833 from the screening of a ChemBridge 50,000 compound library against the CT-L activity of purified 20 S proteasome (IC50 = 0.6 μm (Fig. 1A)). Through extensive structure activity relationship studies and hit-to-lead optimization, we found that replacing the methyl by a propyl in ring A and replacing ring B tolyl by pyridyl resulted in PI-1840 (IC50 = 27 nm), which is 22-fold more potent than the initial PI-1833 hit (IC50 = 600 nm) (Fig. 1A, right panel). The chemical synthesis of PI-1833, PI-1840, and analogs as well as the details of structure activity relationship and hit-to-lead optimizations studies have recently been published (18). This study describes the biologic characterization of PI-1840 from in vitro and cell culture studies to in vivo anti-tumor efficacy studies.
FIGURE 1.

PI-1833 and its potent analog PI-1840 are selective inhibitors for CT-L activity in vitro. A, chemical structures of PI-1833 and PI-1840 with their IC50 values against different proteasome activities. B, effects of PI-1840 and bortezomib on the CT-L activities of the 20 S constitutive rabbit proteasome and 20 S human immunoproteasome. Data are representative of at least two independent experiments.
PI-1840 was selective for the CT-L over T-L (IC50 value >100 μm) and PGPH-L (IC50 value >100 μm) activities of the proteasome (Fig. 1A). Furthermore, Fig. 1B (left panel) shows that PI-1840 was 121-fold more selective for the constitutive 20 S proteasome over the immunoproteasome (IC50 18 nm versus 2170 nm). In contrast, bortezomib was 2-fold more selective for the immunoproteasome over the constitutive proteasome (IC50 4 nm versus 8 nm) (Fig. 1B, right panel). To determine whether PI-1840 inhibits the CT-L activity of the proteasome in a covalent or a noncovalent manner, we first used LC-MS/MS and then confirmed the results with dialysis as described under “Experimental Procedures.” Fig. 2, A–C, shows the MS spectra of the tryptic digests of the 20 S proteasome after incubation with vehicle, PI-1840, and lactacystin, respectively. Tryptic peptides from the 20 S proteasome treated with vehicle contained unmodified protonated TTTLAFK (m/z 781.4504) (Fig. 2A). TTTLAFK (structure shown in Fig. 3A) corresponds to the N-terminal tryptic peptide of rabbit 20 S proteasome subunit β type-5 with the first Thr in this peptide corresponding to threonine 1 of the active site of CT-L. A similar pattern was observed with tryptic peptides from the 20 S proteosome treated with PI-1840 with the unmodified protonated TTTLAFK (m/z 781.4397) (Fig. 2B). The unmodified Thr-1-containing peptide was confirmed by both the intact mass spectrum and tandem mass spectrum. In contrast, Fig. 2C shows that tryptic peptides from the 20 S proteasome treated with lactacystin (a known covalent and irreversible CT-L inhibitor) (22, 23) contained a doubly charged, lactacystin-modified threonine peptide (m/z 497.7778) (structure shown in Fig. 3B). The observation that only Thr-1 on β-5 was modified by lactacystin was documented by searches matching experimental data to peptides from the database of rabbit 20 S proteasome β-5, β-1, and β-2 subunits. Similarly, searches matching experimental data (from vehicle and PI-1840-treated samples) to peptides from the 22 rabbit 20 S proteasome sequences (UniProt database) show no peptide modifications from the β-5, β-1, and β-2 subunits. Taken together, these results suggest that, unlike lactacystin, PI-1840 does not bind covalently to the proteasome.
FIGURE 2.

Lactacystin but not PI-1840 binds covalently to CT-L of the 20 S proteasome. A–C, LC-MS/MS analysis. Purified rabbit 20 S proteasome was incubated either with vehicle (A), PI-1840 (B), or lactacystin (C), and the tryptic digests were analyzed by LC-MS/MS as described under “Experimental Procedures.” The b ions (red) and y ions (blue) designate the N and C termini of the peptide, respectively. The number next to each ion represents the number of amino acids in that fragment (i.e. y4 = LAFK from C terminus of the peptide).
FIGURE 3.

A, unmodified TTTLAFK peptide; B, clasto-lactacystin-modified TTTLAFK peptide; C, dialysis. Purified rabbit 20 S proteasome was incubated with vehicle control, 1 μm PI-1840, or 2.5 μm lactacystin and was subjected to dialysis at 4 °C for different lengths of time as described under “Experimental Procedures.” Percentage of CT-L activity (relative to vehicle-treated control samples) was then determined at different time points. Data are representative of two (LC/MS-MS) and three (dialysis) independent experiments.
We next determined the reversibility of binding of PI-1840 and lactacystin to CT-L by dialysis as described under “Experimental Procedures.” Fig. 3C shows that the CT-L activity in the dialysis compartment from the sample that was treated with PI-1840 begins to recover within the first few minutes and has recovered fully by 18 h of dialysis. In contrast, the CT-L activity from the lactacystin-treated sample remained potently inhibited even after 18 h of dialysis (Fig. 3C). These results are consistent with the LC-MS/MS results (Fig. 2, B and C) that demonstrated that lactacystin but not PI-1840 binds covalently to the active site Thr-1 of the CT-L subunit of the proteasome.
PI-1840 Is More Potent than PI-1833 at Inhibiting Proteasome Activity, Accumulating Proteasome Substrates, Inhibiting Survival Pathways, and Inducing Apoptosis in Human Cancer Cells
Fig. 1 shows that PI-1840 is more potent than PI-1833 in vitro. We next determined whether PI-1833 and PI-1840 are cell-permeable and whether PI-1840 is more potent than PI-1833 at inhibiting the CT-L activity of the proteasome in intact cells. To this end, we treated MDA-MB-468 cells with PI-1833 and PI-1840 for 2 h and determined the CT-L activity as described under “Experimental Procedures.” We found that both compounds inhibited CT-L activity in a dose-dependent manner, and consistent with our in vitro results, PI-1840 was 10-fold more potent (IC50 = 1.55 μm) than PI-1833 (IC50 = 11.60 μm). We next determined how fast PI-1840 can reach its target and whether its inhibition of CT-L is selective over T-L and PGPH-L activities in intact cells. To this end, we first treated MDA-MB-468 cells with PI-1840 and measured the CT-L, T-L, and PGPH activities over time as described under “Experimental Procedures.” Fig. 4A shows that PI-1840 reached its target within 10 min and inhibited CT-L activity. Consistent with in vitro results, PI-1840 did not inhibit T-L and PGPH activities in intact cells (Fig. 4A).
FIGURE 4.
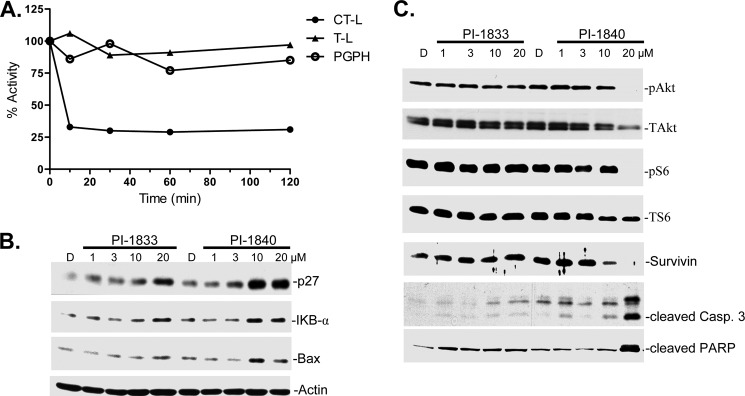
Selective inhibition of the proteasomal CT-L activity in whole cells, accumulation of proteasome substrate proteins, inhibition of cell survival pathways, and induction of apoptosis. A, exponentially growing human breast cancer MDA-MB-468 cells were treated with 5 μm PI-1840 for the indicated time points, followed by measurement of CT-L, T-L, and PGPH activities in whole cell extracts as described under “Experimental Procedures.” B and C, human breast cancer MDA-MB-468 cells were treated with the indicated concentrations of PI-1833 and PI-1840 for 48 h, followed by Western blot assay using the indicated antibodies as described under “Experimental Procedures.” Data are representative of at least two independent experiments. PARP, poly(ADP-ribose) polymerase. Lane D, DMSO.
Inhibition of the CT-L activity of the proteasome is predicted to result in the accumulation of known CT-L substrates. Therefore, we next treated MDA-MB-468 cells with increasing concentrations of PI-1833 and PI-1840 for 48 h and processed the cells for Western blotting as described under “Experimental Procedures.” Fig. 4B shows that both PI-1833 and PI-1840 increased the accumulation of CT-L substrates p27, IκB-α, and Bax but that PI-1840 was more potent than PI-1833, consistent with its more potent activity to inhibit CT-L in vitro and in MDA-MB-468 cells.
Proteasome inhibitors are known to affect many signal transduction pathways that are critical for tumor cell survival (24, 25). We therefore investigated the effects of PI-1833 and PI-1840 on these pathways as well as their ability to induce apoptosis. Fig. 4C shows that PI-1840 was much more potent than PI-1833 at decreasing the levels of p-Akt, Ser(P)-6, and survivin and resulted in induction of apoptosis as apparent from caspase-3 activation and poly(ADP-ribose) polymerase cleavage.
PI-1840 Inhibits the Viability of a Broad Spectrum of Human Cancer Cell Lines
Figs. 1–4 demonstrated that PI-1840 is a potent, selective, noncovalent, and rapidly reversible proteasome inhibitor that induced the accumulation of CT-L substrates, inhibited tumor survival pathways, and induced apoptosis. The cell culture studies were done in one cell line MDA-MB-468; therefore, we next determined the ability of PI-1840 to inhibit CT-L activity and viability in a broad spectrum of human cancer cell lines from different lineages, including breast, colon, prostate, pancreatic, renal, and lung cancers as well as multiple myeloma. To this end, we treated the various cell lines with 20 μm PI-1840 for 2 h and analyzed the CT-L activity. We also treated the various cell lines with increasing concentrations of PI-1840 for 120 h and analyzed viability by MTT assays as described under “Experimental Procedures.” Table 1 shows that PI-1840 reached its target in all the cell lines and inhibited CT-L activity by 45–90% depending on the cell line. Furthermore, PI-1840 also inhibited the viability of all the human cancer cell lines with IC50 values from as low as 2.2 μm in MDA-MB-231 cells to as high as 45.2 μm in RXF-397. Interestingly, in the case of the two HCT-116 isogenic cell lines where the CT-L activities from both HCT-116 (p53+/+) and HCT-116 (p53−/−) were inhibited equally (88.4 ± 4.6 and 89.9 ± 3.0%, respectively) (Table 1), PI-1840 was twice as potent at inhibiting viability in the former than in the latter (IC50 values of 8.7 ± 1.0 and 16.0 ± 1.3 μm, respectively (Table 1), suggesting that p53 may contribute to the anti-proliferative activity of PI-1840. In contrast, in the two HCT-116 isogenic cell lines where the CT-L activities from both HCT-116-HKH-2 (mutant K-Ras) and HCT-116 (wild type K-Ras) were inhibited equally (87.5 ± 5.6 and 88.4 ± 4.6%, respectively) (Table 1), PI-1840 was also as effective in both cell lines at inhibiting viability (IC50 values of 6.3 ± 0.8 and 8.7 ± 1.0 μm, respectively) (Table 1). Moreover, LNCaP cells that are Bax+/+ are 5.7-fold more sensitive to PI-1840 anti-proliferative effects than the Bax−/− DU-145 cells (Table 1). Finally and importantly, PI-1840 inhibited only weakly the viability of “normal” nontransformed breast cells (MCF-10A; IC50 = 314.3 ± 23.9 μm) and normal foreskin fibroblasts (HCA2; IC50 = 86 ± 20 μm) (Table 1).
TABLE 1.
Effects of PI-1840 on CT-L activity and viability of human cancer cells and “normal” cells
Human cancer cells from various lineages and nontransformed MCF-10A and HCA2 cells were treated with 20 μm PI-1840 for 2 h and analyzed for CT-L activity as described under “Experimental Procedures.” In separate experiments, the cells were treated with increasing concentrations of PI-1840 for 120 h and analyzed for viability by MTT assays as described under “Experimental Procedures.”
| Cancer | Cell lines | CT-L, % inhibition at 20 μm | Viability (IC50 (μm)) |
|---|---|---|---|
| Breast | MDA-MB-468 | 63.2 ± 5.4 | 4.8 ± 1.0 |
| MDA-MB-231 | 59.0 ± 4.3 | 2.2 ± 0.5 | |
| Colon | HCT-116 | 88.4 ± 4.6 | 8.7 ± 1.0 |
| HCT-116-p53−/− | 89.9 ± 3.0 | 16.0 ± 1.3 | |
| HCT-116-HKH-2 | 87.5 ± 5.6 | 6.3 ± 0.8 | |
| Prostate | DU-145 | 64.7 ± 5.0 | 28.0 ± 2.8 |
| LNCaP | 88.8 ± 1.0 | 4.9 ± 1.0 | |
| PC3 | 71.4 ± 4.1 | 15.0 ± 1.1 | |
| Multiple myeloma | RPMI 8226 | 49.3 ± 2.5 | 26.0 ± 2.3 |
| U266 | 66.0 ± 58.0 | 15.6 ± 4.2 | |
| Pancreatic adenocarcinoma | Colo 357 | 45.2 ± 25.9 | 15.4 ± 2.4 |
| Renal cell carcinoma | RXF 397 | 53.8 ± 1.9 | 45.2 ± 1.0 |
| Normal foreskin fibroblast | HCA2 | 41.0 ± 2.0 | 86.0 ± 20.0 |
| Normal/immortalized breast cells | MCF-10A | 63.2 ± 5.4 | 314.3 ± 23.9 |
PI-1840 Sensitizes Human Cancer Cells to the mdm2 Antagonist Nutlin
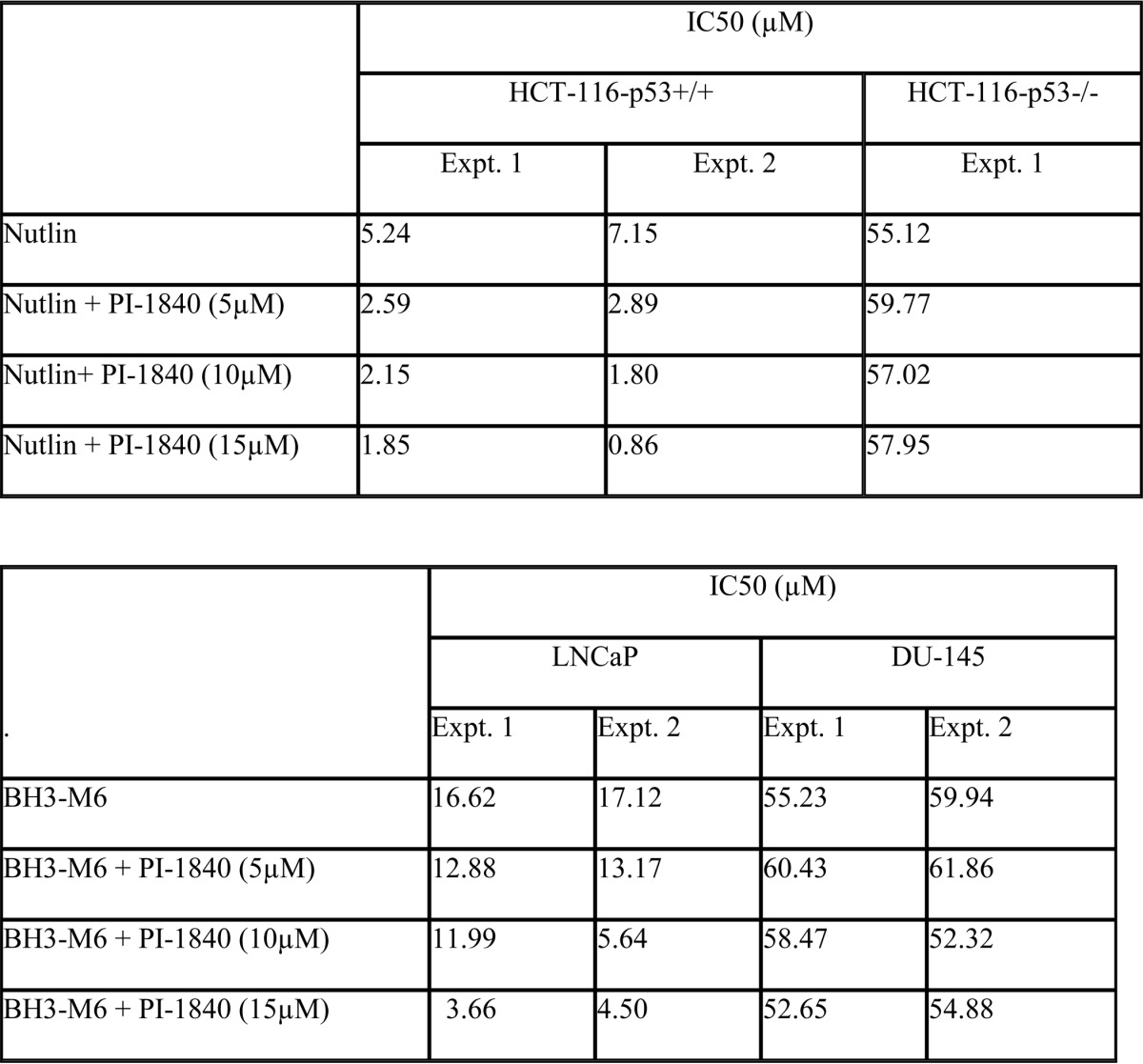
Blocking the degradation of p53 with proteasome inhibitors may not be sufficient to induce apoptosis because the accumulated p53 can be inactivated by binding partners such as mdm2. Therefore, we next evaluated whether nutlin, which disrupts the binding of p53 to mdm2 (26), can sensitize cells to PI-1840. To this end, we treated HCT-116 cells with either nutlin alone or in combination with PI-1840 and determined their effects on viability by MTT as described under “Experimental Procedures.” Table 2 (upper panel) shows that in the absence of PI-1840 nutlin inhibited the viability of HCT-116 cells with an IC50 value of 6.2 μm (average of two experiments). In contrast, in the presence of 5, 10, and 15 μm PI-1840, nutlin IC50 values were 2.7, 2.0, and 1.4 μm. Taken together, these results demonstrate that PI-1840 sensitized HCT-116 to nutlin inhibition of viability by as much as 4.4-fold. To determine whether this sensitization is p53-dependent, we performed similar studies with the isogenic HCT-116 (p53−/−) cells. Table 2 (upper panel) shows that in the absence of PI-1840, nutlin was 8.9-fold less effective at inhibiting the viability of HCT-116 (p53−/−) cells (IC50 value of 55.1 μm) than it is at inhibiting that of its isogenic HCT-116 (p53+/+) counterpart (6.2 μm). Furthermore, PI-1840 did not sensitize HCT-116 (p53−/−) cells to nutlin (IC50 values for vehicle, 5, 10, and 15 μm PI-1840 were 55.1, 59.8, 57.0, and 58.0, respectively).
TABLE 2.
Effects of PI-1840, nutlin, and BH3-M6 on cancer cell viability
Upper panel, PI-1840 sensitizes HCT-116-p53+/+ but not HCT-116-p53−/− cells to nutlin-mediated inhibition of cell viability. The two HCT-116 cell lines were treated with either nutlin alone or in combination with PI-1840, and the effects on viability were determined by MTT as described under “Experimental Procedures.” Lower panel, PI-1840 sensitizes LNCaP but not DU-145 cells to BH3-M6-mediated inhibition of cell viability. LNCaP and DU145 cells were treated with either BH3-M6 alone or in combination with PI-1840, and the effects on viability were determined by MTT as described under “Experimental Procedures.”
PI-1840 Sensitizes Human Cancer Cells to the pan-Bcl-2 Antagonist BH3-M6
Inhibition of the degradation of pro-apoptotic proteins such as Bax by proteasome inhibitors may not be sufficient to induce apoptosis because the accumulated Bax can be inactivated by binding partners such as Mcl-1 or BclxL. Therefore, we next evaluated whether BH3-M6, which disrupts the binding of anti-apoptotic proteins Bcl-xL, Mcl-1, and Bcl-2 to the pro-apoptotic proteins Bax, Bak, and Bim (19), can sensitize cells to PI-1840. To this end, we treated LNCaP cells with either BH3-M6 alone or in combination with PI-1840 and determined their effects on viability by MTT as described under “Experimental Procedures.” Table 2 (lower panel) shows that in the absence of PI-1840, BH3-M6 inhibited the viability of LNCaP with an IC50 value of 16.9 μm (average of two experiments). In contrast, in the presence of 5, 10, and 15 μm PI-1840, BH3-M6 IC50 values were 13.0, 8.8, and 4.1 μm, respectively (Table 2, lower panel), demonstrating that PI-1840 sensitized LNCaP to BH3-M6 inhibition of viability by as much as 4-fold. To determine whether this sensitization is Bax-dependent, we performed similar studies with DU-145 cells that, unlike LNCaP cells, lack Bax. Table 2, lower panel, shows that in the absence of PI-1840, BH3-M6 was 3.4-fold less effective in inhibiting the viability of DU-145 cells (IC50 value = 57.6 μm) than that of LNCaP (16.9 μm). More importantly, unlike in LNCaP cells, PI-1840 did not sensitize DU-145 cells to BH3-M6. Indeed, the BH3-M6 IC50 values for DU-145 treated with vehicle, 5, 10, and 15 μm PI-1840 were 57.6, 61.1, 55.4, and 53.8 μm, respectively (Table 2, lower panel).
PI-1840 but Not Bortezomib Inhibits the Growth of Human Breast Tumor Xenografts in Nude Mice
We next evaluated the anti-tumor activities of PI-1840 and bortezomib in nude mice bearing solid tumors. To this end, we implanted human breast cancer MDA-MB-231 cells s.c. in nude mice, and when tumors reached an average size of about 250 mm3, the mice were treated either with vehicle (30% 2-hydroxypropyl-β-cyclodextrin in H2O), PI-1840 (150 mg/kg/day, i.p, daily), or bortezomib (1 mpk, twice weekly, intraperitoneally). Fig. 5A shows representative examples of mice, treated with either vehicle, PI-1840, or bortezomib. The tumor from the mouse treated with vehicle grew from an initial tumor volume of 279 to 910 mm3. Similarly, the tumor from the bortezomib-treated mouse grew from 239 to 852 mm3. In contrast, the tumor from the PI-1840-treated mouse grew from 280 to only 374 mm3 (Fig. 5A). Thus, compared with vehicle, PI-1840 inhibited tumor growth by 85%, whereas bortezomib had little effect on tumor growth. Fig. 5B shows the average tumor volume change of all mice treated. The volume of tumors from mice treated with vehicle increased on average by 288 ± 91% over the treatment period of 14 days. Similarly, the volume of tumors from mice treated with bortezomib increased on average by 263 ± 28%. In contrast, the volume of tumors from mice treated with PI-1840 increased on average by only 69 ± 17%. Therefore, PI-1840 inhibited the growth of MDA-MB-231 tumor xenografts by 76% ((1 − (69/288)) × 100) (p = 0.04), whereas bortezomib inhibited tumor growth by only 8.7% ((1 − (263/288)) × 100), and this was not a statistically significant effect compared with vehicle-treated mice (p = 0.78). Furthermore, over the 14-day treatment period, the body weight of the vehicle-treated mice increased on average by 4.34%. In contrast, the bortezomib-treated mice lost on average 6.21% of their body weight. The body weight of the PI-1840-treated mice increased on average by 0.12%.
FIGURE 5.
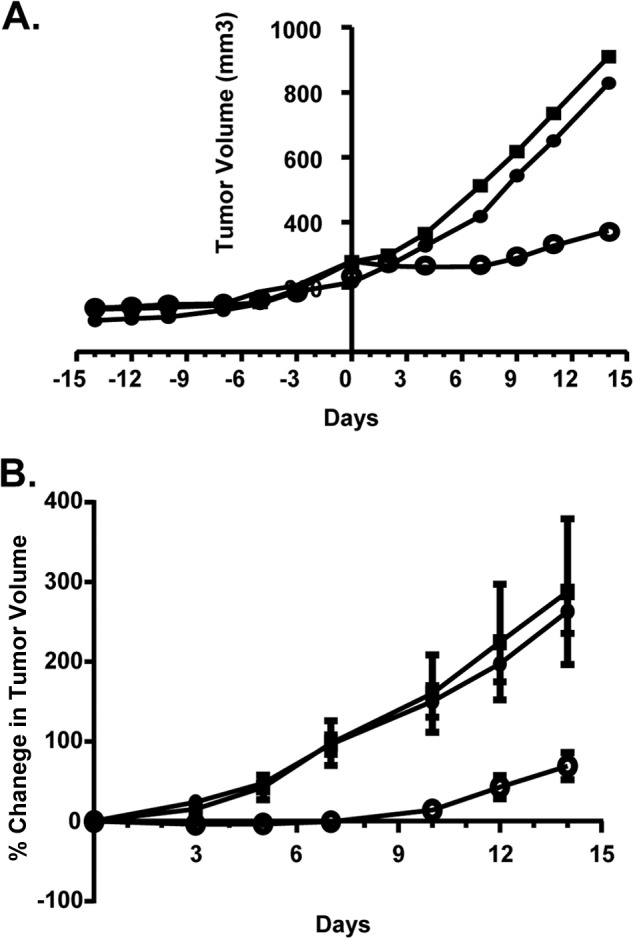
Antitumor efficacy of PI-1840 and bortezomib against human breast cancer MDA-MB-231 xenografts in nude mice. Mice bearing MDA-MB-231 tumors were treated with vehicle (closed squares), PI-1840 (open circles), or bortezomib (filled circles) as described under “Experimental Procedures.” A, representative tumor growth curves form vehicle-, PI-1840-, and bortezomib-treated mice. B, average percent change in tumor volumes from mice treated with vehicle, PI-1840, and bortezomib. There were statistically significant differences between vehicle and PI-1840 in the average percent change in tumor volume on every day of measurement with p values of 0.006, 0.019, 0.009, 0.018, 0.039, and 0.045 on days 3, 5, 7, 10, 12, and 14 of treatment, respectively. In contrast, there were no statistically significant differences between vehicle and bortezomib in the average percent change in volume with p values of 0.243, 0.751, 0.951, 0.842, 0.709, and 0.786 on days 3, 5, 7, 10, 12, and 14 of treatment, respectively.
DISCUSSION
Approval by the Food and Drug Administration of bortezomib further validated targeting the proteasome for the development of anticancer drugs. Although this proteasome inhibitor has benefited patients with multiple myeloma and mantle cell lymphoma, it is ineffective against solid tumors and is associated with undesirable side effects. Carfilzomib, a more recently approved proteasome inhibitor, appears to also be more active against liquid tumors. although it has not been tested as thoroughly as bortezomib against solid tumors. These drawbacks could be due at least in part to the fact that bortezomib inhibits the CT-L activity of the proteasome by binding covalently to threonine 1 of the β-5 subunit of the proteasome. Although this suggestion needs to be further supported with more direct evidence, the development of noncovalent proteasome inhibitors to determine whether they lack these drawbacks is highly desirable. In this study, we describe the discovery of a potent and selective proteasome inhibitor, PI-1840, that blocked selectively the CT-L activity in a noncovalent and rapidly reversible manner. Proteasome inhibitors that are approved by the Food and Drug Administration (bortezomib and carfilzomib) and those that are in clinical trials (i.e. MLN9708, NPI-0052, and CEP18770) all act covalently. There are only a few noncovalent proteasome inhibitors that have been reported, and these include the natural product cyclic peptide TMC-95 (27) and its linear peptide mimics (28) as well as capped peptides (noncyclic and isosteres peptides) (29) and hydroxyurea (30). Furthermore, most of the noncovalent inhibitors previously reported except for hydroxyurea are peptidic in nature, whereas PI-1840 is a nonpeptidic, small organic molecule that is unlikely to suffer from the peptidic compound liabilities such as peptide degradation and poor cellular uptake. Finally, PI-1840 is the only noncovalent proteasome inhibitor that has been evaluated in vivo and shown to have anti-tumor activity against solid tumors. Importantly, in our in vivo studies, PI-1840 was compared head-to-head to bortezomib, and only PI-1840, and not bortezomib, was found to be active against solid tumors.
PI-1840 was able to inhibit the CT-L activity and cell viability in a wide spectrum of tumor types from several lineages, including breast, colon, prostate, pancreatic, and renal cancers as well multiple myeloma (see Table 1). The fact that PI-1840 was two times more potent at inhibiting cell viability in the HCT-116 (p53+/+) than in HCT-116 (p53−/−) suggested that the CT-L substrate p53 contributes at least in part to the ability of PI-1840 to inhibit viability. In contrast, the K-Ras mutation status in the same HCT-116 cell line appears to matter little. Although the two prostate cancer cell lines LNCaP and DU-145 are not isogenic, LNCaP cells that are Bax-positive were severalfold more sensitive to PI-1840 anti-viability effects than the Bax-negative DU-145 suggesting that Bax may be critical. However, other genetic differences between the two cell lines and the fact that PI-1840 inhibited the CT-L activity of LNCaP more potently may also be contributing factors. Finally and importantly, PI-1840 was less active at inhibiting the viability of “normal” cells. Although we do not know why these cells are less sensitive to PI-1840, this is an important finding that is consistent with the finding that in vivo PI-1840 is not toxic as judged grossly by little changes in the body weight of the mice.
Although in about half of human cancers, p53 is inactivated by mutations, and in the other half p53 is wild type and inactivated by various mechanisms (31, 32). One of the major antagonists of p53 is the E3 ligase mdm2 that binds and inactivates p53 at least in part by ubiquitinating p53 and consequently inducing its degradation by the proteasome (33–35). Nutlin is a small molecule that inhibits mdm2/p53 binding freeing up p53 to cause apoptosis (26). However, the ability of the free p53 protein to induce apoptosis could be hampered by its ubiquitination by other E3 ligases and subsequent degradation by the proteasome CT-L. Therefore, inhibiting the CT-L activity with PI-1840 may enhance the ability of nutlin to inhibit survival and induce apoptosis. Indeed, our combination studies demonstrated that PI-1840 sensitized greatly HCT-116 to nutlin inhibition of viability. Furthermore, PI-1840 did not sensitize HCT-116 (p53−/−) cells to nutlin, suggesting that the ability of PI-1840 to sensitize to nutlin is dependent on p53 and that free p53 may be required for PI-1840 to inhibit cell viability. Nutlin itself is much less effective at inhibiting the viability of HCT-116 (p53−/−) cells compared with its isogenic HCT-116 (p53+/+) cells, consistent with previous reports (36, 37). Taken together, these results warrant further evaluation of combination therapy of mdm2 antagonists such nutlin and noncovalent proteasome inhibitors such as PI-1840 in human tumors that express wild type p53.
Recently, we have reported on the development of a novel pan-Bcl-2 antagonist, BH3-M6, which induces apoptosis by inhibiting the binding of the anti-apoptotic proteins Bcl-xL, Mcl-1, and Bcl-2 to the pro-apoptotic proteins Bax, Bak, and Bim, freeing up the latter to cause apoptosis (19). However, the ability of the freed pro-apoptotic proteins to induce apoptosis could be hampered by their degradation by the proteasome CT-L. We found PI-1840 to sensitize greatly LNCaP to BH3-M6 inhibition of viability. In DU-145 cells that lack Bax, BH3-M6 was less effective consistent with our previously published data (19). Furthermore, unlike in LNCaP cells, PI-1840 did not sensitize DU-145 cells to BH3-M6, suggesting that the accumulation of free Bax or other pro-apoptotic proteins may contribute to the mechanism by which PI-1840 inhibits viability. Finally, the fact that LNCaP expresses wild type functional p53 may contribute to its higher sensitivity to PI-1840 because p53 is known to up-regulate Bax.
Our findings that PI-1840 but not bortezomib inhibited the growth in mice of MDA-MB-231 breast tumors coupled with the fact that PI-1840 has little effect on mouse body weight support the suggestion that noncovalent proteasome inhibitors may be less toxic and more active against solid tumors. The fact that PI-1840 demonstrated little toxicity in vivo is consistent with its lack of activity against the normal MCF-10A and HCA2 cells in cell culture studies. Furthermore, selective inhibition of the constitutive proteasome over the immunoproteasome by PI-1840 may be associated with less toxicity to cells of lymphoid origin where the immunoproteasome is selectively expressed (38). Although immunoproteasome-specific inhibitors are believed to have great potential in immune-related diseases such as lupus erythematosus and inflammatory bowel disease, their potential for cancer therapy is not clear (38, 39). Because our interest is mainly in targeting solid tumors, the fact that PI-1840 does not inhibit the immunoproteasome is not a liability.
Finally, although the finding that PI-1840 is active in solid tumors that are resistant to bortezomib is encouraging, further confirmation of this observation with noncovalent proteasome inhibitors other than PI-1840 is important. Furthermore, demonstrating that noncovalent inhibitors lack the drawbacks of covalent inhibitors in other solid tumors as well as in other animal models is also of paramount importance. If confirmed, these findings could be translated into the clinic where noncovalent proteasome inhibitors can be used either as single agents or in combination to treat a wider spectrum of human tumors, including solid tumors.
Acknowledgments
We thank the Chemical Biology Core and the Proteomics Core at the Moffitt Cancer Center for their cooperation and expertise.
This work was supported, in whole or in part, by National Institutes of Health Grant P01-CA118210.
- UPS
- ubiquitin/proteasome system
- CT-L
- chymotrypsin-like
- T-L
- trypsin-like
- PGPH
- peptidylglutamyl peptide hydrolyzing
- AMC
- 7-amido-4-methyl-coumarin
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
REFERENCES
- 1. Pagano M., Benmaamar R. (2003) When protein destruction runs amok, malignancy is on the loose. Cancer Cell 4, 251–256 [DOI] [PubMed] [Google Scholar]
- 2. Schwartz A. L., Ciechanover A. (1999) The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annu. Rev. Med. 50, 57–74 [DOI] [PubMed] [Google Scholar]
- 3. Adams J. (2004) The development of proteasome inhibitors as anticancer drugs. Cancer Cell 5, 417–421 [DOI] [PubMed] [Google Scholar]
- 4. Adams J. (2003) The proteasome: structure, function, and role in the cell. Cancer Treat. Rev. 29, 3–9 [DOI] [PubMed] [Google Scholar]
- 5. Ciechanover A. (1994) The ubiquitin-proteasome proteolytic pathway. Cell 79, 13–21 [DOI] [PubMed] [Google Scholar]
- 6. Ciechanover A. (1998) The ubiquitin-proteasome pathway: On protein death and cell life. EMBO J. 17, 7151–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mani A., Gelmann E. P. (2005) The ubiquitin-proteasome pathway and its role in cancer. J. Clin. Oncol. 23, 4776–4789 [DOI] [PubMed] [Google Scholar]
- 8. Hochstrasser M. (1995) Ubiquitin, proteasomes, and the regulation of intracellular protein degradation. Curr. Opin. Cell Biol. 7, 215–223 [DOI] [PubMed] [Google Scholar]
- 9. Hershko A., Heller H., Elias S., Ciechanover A. (1983) Components of ubiquitin-protein ligase system. Resolution, affinity purification, and role in protein breakdown. J. Biol. Chem. 258, 8206–8214 [PubMed] [Google Scholar]
- 10. Downward J. (2003) Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 3, 11–22 [DOI] [PubMed] [Google Scholar]
- 11. Nalepa G., Rolfe M., Harper J. W. (2006) Drug discovery in the ubiquitin-proteasome system. Nat. Rev. Drug Discov. 5, 596–613 [DOI] [PubMed] [Google Scholar]
- 12. Burger A. M., Seth A. K. (2004) The ubiquitin-mediated protein degradation pathway in cancer: Therapeutic implications. Eur. J. Cancer 40, 2217–2229 [DOI] [PubMed] [Google Scholar]
- 13. Ruschak A. M., Slassi M., Kay L. E., Schimmer A. D. (2011) Novel proteasome inhibitors to overcome bortezomib resistance. J. Natl. Cancer Inst. 103, 1007–1017 [DOI] [PubMed] [Google Scholar]
- 14. Dick L. R., Fleming P. E. (2010) Building on bortezomib: Second-generation proteasome inhibitors as anti-cancer therapy. Drug Discov. Today 15, 243–249 [DOI] [PubMed] [Google Scholar]
- 15. Singh J., Petter R. C., Baillie T. A., Whitty A. (2011) The resurgence of covalent drugs. Nat. Rev. Drug Discov. 10, 307–317 [DOI] [PubMed] [Google Scholar]
- 16. Kisselev A. F., van der Linden W. A., Overkleeft H. S. (2012) Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol. 19, 99–115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Desvergne A., Genin E., Maréchal X., Gallastegui N., Dufau L., Richy N., Groll M., Vidal J., Reboud-Ravaux M. (2013) Dimerized linear mimics of a natural cyclopeptide (TMC-95A) are potent noncovalent inhibitors of the eukaryotic 20 S proteasome. J. Med. Chem. 56, 3367–3378 [DOI] [PubMed] [Google Scholar]
- 18. Ozcan S., Kazi A., Marsilio F., Fang B., Guida W. C., Koomen J., Lawrence H. R., Sebti S. M. (2013) Oxadiazole-isopropylamides as potent and noncovalent proteasome inhibitors. J. Med. Chem. 56, 3783–3805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kazi A., Sun J., Doi K., Sung S. S., Takahashi Y., Yin H., Rodriguez J. M., Becerril J., Berndt N., Hamilton A. D., Wang H. G., Sebti S. M. (2011) The BH3 α-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax- and Bim-dependent manner. J. Biol. Chem. 286, 9382–9392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kazi A., Lawrence H., Guida W. C., McLaughlin M. L., Springett G. M., Berndt N., Yip R. M., Sebti S. M. (2009) Discovery of a novel proteasome inhibitor selective for cancer cells over non-transformed cells. Cell Cycle 8, 1940–1951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yates J. R., 3rd, Eng J. K., McCormack A. L., Schieltz D. (1995) Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal. Chem. 67, 1426–1436 [DOI] [PubMed] [Google Scholar]
- 22. Fenteany G., Schreiber S. L. (1998) Lactacystin, proteasome function, and cell fate. J. Biol. Chem. 273, 8545–8548 [DOI] [PubMed] [Google Scholar]
- 23. Corey E. J., Li W. D. (1999) Total synthesis and biological activity of lactacystin, omuralide and analogs. Chem. Pharm. Bull. 47, 1–10 [DOI] [PubMed] [Google Scholar]
- 24. Hutter G., Zimmermann Y., Rieken M., Hartmann E., Rosenwald A., Hiddemann W., Dreyling M. (2012) Proteasome inhibition leads to dephosphorylation and downregulation of protein expression of members of the Akt/mTOR pathway in MCL. Leukemia 26, 2442–2444 [DOI] [PubMed] [Google Scholar]
- 25. Papandreou C. N., Logothetis C. J. (2004) Bortezomib as a potential treatment for prostate cancer. Cancer Res. 64, 5036–5043 [DOI] [PubMed] [Google Scholar]
- 26. Vassilev L. T., Vu B. T., Graves B., Carvajal D., Podlaski F., Filipovic Z., Kong N., Kammlott U., Lukacs C., Klein C., Fotouhi N., Liu E. A. (2004) In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 303, 844–848 [DOI] [PubMed] [Google Scholar]
- 27. Groll M., Koguchi Y., Huber R., Kohno J. (2001) Crystal structure of the 20 S proteasome:TMC-95A complex: A noncovalent proteasome inhibitor. J. Mol. Biol. 311, 543–548 [DOI] [PubMed] [Google Scholar]
- 28. Groll M., Gallastegui N., Maréchal X., Le Ravalec V., Basse N., Richy N., Genin E., Huber R., Moroder L., Vidal J., Reboud-Ravaux M. (2010) 20 S proteasome inhibition: Designing noncovalent linear peptide mimics of the natural product TMC-95A. ChemMedChem 5, 1701–1705 [DOI] [PubMed] [Google Scholar]
- 29. Blackburn C., Gigstad K. M., Hales P., Garcia K., Jones M., Bruzzese F. J., Barrett C., Liu J. X., Soucy T. A., Sappal D. S., Bump N., Olhava E. J., Fleming P., Dick L. R., Tsu C., Sintchak M. D., Blank J. L. (2010) Characterization of a new series of noncovalent proteasome inhibitors with exquisite potency and selectivity for the 20 S β5-subunit. Biochem. J. 430, 461–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gallastegui N., Beck P., Arciniega M., Huber R., Hillebrand S., Groll M. (2012) Hydroxyureas as noncovalent proteasome inhibitors. Angewandte Chemie 51, 247–249 [DOI] [PubMed] [Google Scholar]
- 31. Favrot M., Coll J. L., Louis N., Negoescu A. (1998) Cell death and cancer: Replacement of apoptotic genes and inactivation of death suppressor genes in therapy. Gene Ther. 5, 728–739 [DOI] [PubMed] [Google Scholar]
- 32. Hupp T. R., Lane D. P., Ball K. L. (2000) Strategies for manipulating the p53 pathway in the treatment of human cancer. Biochem. J. 352, 1–17 [PMC free article] [PubMed] [Google Scholar]
- 33. Kawai H., Wiederschain D., Yuan Z. M. (2003) Critical contribution of the MDM2 acidic domain to p53 ubiquitination. Mol. Cell. Biol. 23, 4939–4947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Love I. M., Shi D., Grossman S. R. (2013) p53 Ubiquitination and proteasomal degradation. Methods Mol. Biol. 962, 63–73 [DOI] [PubMed] [Google Scholar]
- 35. Satija Y. K., Bhardwaj A., Das S. (2013) A portrayal of E3 ubiquitin ligases and deubiquitylases in cancer. Int. J. Cancer 133, 2759–2768 [DOI] [PubMed] [Google Scholar]
- 36. Endo S., Yamato K., Hirai S., Moriwaki T., Fukuda K., Suzuki H., Abei M., Nakagawa I., Hyodo I. (2011) Potent in vitro and in vivo antitumor effects of MDM2 inhibitor nutlin-3 in gastric cancer cells. Cancer Sci. 102, 605–613 [DOI] [PubMed] [Google Scholar]
- 37. Villalonga-Planells R., Coll-Mulet L., Martínez-Soler F., Castaño E., Acebes J. J., Giménez-Bonafé P., Gil J., Tortosa A. (2011) Activation of p53 by nutlin-3a induces apoptosis and cellular senescence in human glioblastoma multiforme. PLoS One 6, e18588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuhn D. J., Orlowski R. Z. (2012) The immunoproteasome as a target in hematologic malignancies. Semin. Hematol. 49, 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bellavista E., Andreoli F., Parenti M. D., Martucci M., Santoro A., Salvioli S., Capri M., Baruzzi A., Del Rio A., Franceschi C., Mishto M. (2013) Immunoproteasome in cancer and neuropathologies: a new therapeutic target? Curr. Pharm. Des. 19, 702–718 [PubMed] [Google Scholar]