
Figure 3.
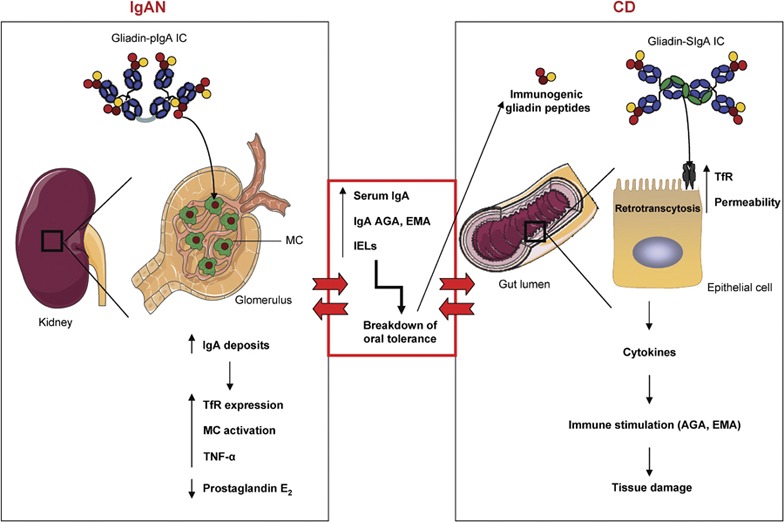
The IgA system constitutes a common link between CD and IgAN. In IgAN patients, gliadins bind pIgA-containing Gal and GalNAc residues, leading to the formation of macromolecular gliadin–IgA ICs. These complexes are deposited on the glomelular mesangium via lectin bonds between gliadin and MCs. This binding induces the overexpression of the mesangial IgA receptor TfR, activates mesangial cells and enhances the in situ IgA deposits. Moreover, binding modulates the production of immunological mediators by mesangial cells (increases in TNF-α and inhibition of prostaglandin E2 production), contributing to IgAN pathogenesis. These patients present with elevated serum IgA, increased production of celiac-specific antibodies (AGA and EMA) and an increase in the number of IELs in the intestine. These dysfunctions are responsible for the breakdown of oral tolerance, which in turn results in perturbations of epithelial cell function and abnormal processing of dietary antigens (immunogenic gliadin peptides). These peptides are retrotranscytosed into the lamina propria via TfR, which is overexpressed on epithelial cells, and induce the release of proinflammatory cytokines. These modifications result in increased intestinal permeability, stimulation of the mucosal immune system and tissue damage, which leads to CD development. Alternatively, the increased intestinal permeability and the presence of circulting IgA-gliadin ICs observed in CD may be a risk factor for the development of IgAN. AGA, antigliadin antibody; CD, celiac disease; EMA, anti-endomysium antibody; Gal, galactose; IgA, immunoglobulin A; IgAN, IgA nephropathy; IC, immune complex; IEL, intraepithelial lymphocyte; MC, mesangial cell; pIgA, polymeric IgA; TfR, transferrin receptor; TNF, tumor necrosis factor. GalNAc, N-acetylgalactosamine.