

Abstract
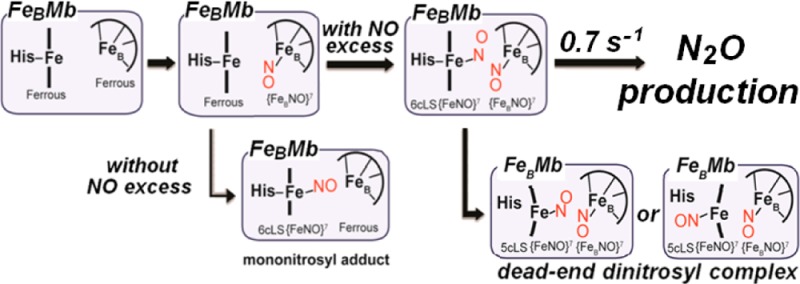
Denitrifying NO reductases are transmembrane protein complexes that are evolutionarily related to heme/copper terminal oxidases. They utilize a heme/nonheme diiron center to reduce two NO molecules to N2O. Engineering a nonheme FeB site within the heme distal pocket of sperm whale myoglobin has offered well-defined diiron clusters for the investigation of the mechanism of NO reduction in these unique active sites. In this study, we use FTIR spectroscopy to monitor the production of N2O in solution and to show that the presence of a distal FeBII is not sufficient to produce the expected product. However, the addition of a glutamate side chain peripheral to the diiron site allows for 50% of a productive single-turnover reaction. Unproductive reactions are characterized by resonance Raman spectroscopy as dinitrosyl complexes, where one NO molecule is bound to the heme iron to form a five-coordinate low-spin {FeNO}7 species with ν(FeNO)heme and ν(NO)heme at 522 and 1660 cm–1, and a second NO molecule is bound to the nonheme FeB site with a ν(NO)FeB at 1755 cm–1. Stopped-flow UV–vis absorption coupled with rapid-freeze-quench resonance Raman spectroscopy provide a detailed map of the reaction coordinates leading to the unproductive iron-nitrosyl dimer. Unexpectedly, NO binding to FeB is kinetically favored and occurs prior to the binding of a second NO to the heme iron, leading to a (six-coordinate low-spin heme-nitrosyl/FeB-nitrosyl) transient dinitrosyl complex with characteristic ν(FeNO)heme at 570 ± 2 cm–1 and ν(NO)FeB at 1755 cm–1. Without the addition of a peripheral glutamate, the dinitrosyl complex is converted to a dead-end product after the dissociation of the proximal histidine of the heme iron, but the added peripheral glutamate side chain in FeBMb2 lowers the rate of dissociation of the promixal histidine which in turn allows the (six-coordinate low-spin heme-nitrosyl/FeB-nitrosyl) transient dinitrosyl complex to decay with production of N2O at a rate of 0.7 s–1 at 4 °C. Taken together, our results support the proposed trans mechanism of NO reduction in NORs.
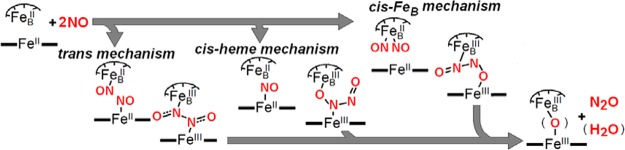
Nitric oxide reductases (NORs) from denitrifying bacteria catalyze the two-electron reduction of nitric oxide (NO) to nitrous oxide (N2O) as part of the conversion of nitrite (NO2–) and nitrate (NO3–) to dinitrogen gas (N2).1−3 The NOR reaction has important significance to human health because it provides many pathogenic organisms with resistance to high NO concentration exposure, and as a whole, to the mammalian immune response.4−6 Cytochrome c-dependent NORs (cNORs) are integral membrane proteins evolutionarily related to the heme/copper oxidases (HCOs). They have a catalytic subunit NorB composed of 12 central transmembrane helices that anchor a low-spin heme b and a heme/nonheme diiron active site (heme b3/nonheme FeB), analogous to the heme a3/CuB center of HCO.7,8 Despite significant advances in the structural characterization of NORs, mechanistic studies continue to be limited by multiple experimental hurdles. For example, although purified cNOR exhibits a reasonable steady-state turnover rate, the reduced enzyme adopts an ill-defined resting form that reacts very sluggishly with NO (on the minute time scale) in pre-steady-state conditions. Flow-flash experiments with the carbonyl complex of cNOR can circumvent this inactive reduced form to provide access to fast kinetics,9−11 but the approach is typically limited to measuring UV–vis absorption changes that reflect reactions at the heme sites without revealing direct insights about events occurring at the nonheme FeB site. Additional complications arise because oxidized cNOR exists in multiple configurations, including a μ-oxo bridged diferric structure where a five-coordinate high-spin heme iron(III) is magnetically coupled to the high-spin nonheme FeB(III),12 and alternative forms where only weak magnetic coupling occurs between the two iron(III) centers.13,14 Because of these hurdles, the mechanism of NORs remains unclear, and one can envision many possible reaction routes that allow for N–N bond formation and N–O bond cleavage, as shown in Scheme 1.15 The so-called “trans mechanism” proposes that one NO binds per iron(II) to form two facing {FeNO}7 species with reductive activation of the nitrosyl moieties to form a hyponitrite complex. Differences in the number and location of NO metal coordination provide multiple alternatives to these first steps leading to N–N bond formation. Theoretical studies by Siegbahn and co-workers16,17 favor binding of a first NO molecule to the heme iron(II) with subsequent reductive activation facilitated by the electrostatic interaction of the nitrosyl moieties with FeB(II). In this “cis-heme mechanism”, side-on attack by a second NO produces a heme iron(III)-hyponitrite dianion complex that is stabilized by its electrostatic interaction with the FeBIII site.16,17 Finally, a “cis-FeB mechanism” where NO coordination occurs only at the FeB site can also be envisioned.14,18 Whether N–O bond cleavage and N2O production requires protonation of the coordinated hyponitrite, or if the hyponitrite species decays through rearrangement to form a bridging μ-oxo and N2O, also remains an open question.15
Scheme 1. Possible Mechanisms for the Binding of NO at the Diiron Site and the Formation of Putative Hyponitrite Intermediate before N2O Production.
Clearly, simpler and better-behaved NOR models would be helpful to test these various mechanistic routes, and engineered proteins are an attractive alternative to synthetic biomimetic inorganic complexes. One such protein is the L29H/F43H/V68E triple variant of myoglobin (FeBMb1) which provides side-chain ligands for an FeB site within its distal heme pocket. Specifically, X-ray crystallography has confirmed that after loading with iron(II), the new side chains and the native distal histidine H64 are recruited to reproduce the (3His,1Glu) coordination sphere of FeB in cNOR.19 In a second construct (FeBMb2), an additional glutamate (I107E) that is peripheral to the two metal ions’ open-coordination sites interacts with a water molecule and forms a hydrogen bond within the diiron active site.20 The crystal structures of the reduced proteins loaded with FeII confirm successful coordination of the nonheme iron(II) by three histidines, one glutamate, and a solvent molecule, as observed for the FeB center of cNOR (Figure 1).
Figure 1.
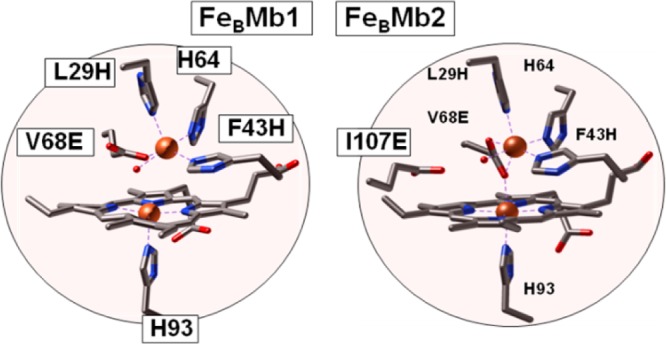
Heme/nonheme diiron centers of reduced FeBMb1 (PDB entry 3K9Z), and reduced FeBMb2 (PDB entry 3M39).19,20
Using resonance Raman (RR) spectroscopy, we previously showed that the heme iron(II) is five-coordinate high-spin (5cHS) in both reduced FeBMb1 and FeBMb2, with an invariant ν(Fe–NHis) regardless of whether or not metal ions have been loaded at their FeB sites.21 We also showed that exposure of these reduced proteins, loaded with Fe(II) or Zn(II) at their FeB site, to 1 equiv of NO produces stable six-coordinate low-spin (6cLS) heme {FeNO}7 complexes with exceptionally low ν(NO) stretching frequencies.21 We assigned the observed deviations in ν(FeNO) and ν(NO) frequencies to the stabilization of a heme Fe(III)-NO– electronic configuration by electrostatic interaction of the NO– group with FeBII. In addition, we noted how the nitroxyl-like electronic structure of the [heme-NO/FeB] complex coincided with the initial mononitrosyl complex predicted by theoretical studies.16,17,21 These results demonstrated the potential of these engineered myoglobin proteins as investigative tools. In the current study, we examine the reaction of the iron(II)-loaded reduced proteins (simply described within as reduced FeBMb1 and FeBMb2) with excess NO using time-resolved techniques to probe for the formation of both heme and nonheme iron-nitrosyl species and to identify productive NOR reaction routes. A major finding is that both constructs proceed through a [{FeNO}7]2 6cLS heme/nonheme iron-nitrosyl dimer prior to N2O production, thus supporting the catalytic validity of the "trans mechanism" in denitrifying NORs.
Materials and Methods
Loading of FeII in E-FeBMbs Preparations
E-FeBMb1 (swMb L29H/F43H/V68E) and E-FeBMb2 (swMb L29H/F43H/V68E/I107E) were expressed and purified with empty FeB sites as described previously,19,20 and protein concentrations were calculated using a 406-nm extinction coefficient, ε406, of 175 mM–1cm–1 in the oxidized form. One millimolar solutions of E-FeBMbs in 50 mM Bis-Tris buffer (pH 7) were transferred to an anaerobic glovebox containing less than 1 ppm O2 (Omnilab System, Vacuum Atmospheres Co.). The proteins were reduced by addition of ∼5 mM sodium dithionite followed by the removal of excess reducting agent using desalting spin columns (7K MWCO, Zeba, Thermo Scientific). The loading of FeII into E-FeBMbs was performed using previously described methods.21 Briefly, an FeIICl2 solution (0.01 M HCl) containing 1.3 equiv of iron was added to 1 mM reduced E-FeBMbs in 50 mM Bis-Tris buffer (pH 7.0) at a rate of 0.5 μL/min with gentle stirring. After the addition of Fe(II), the protein solution was incubated at room temperature for 20 min, and the excess iron was removed with desalting spin columns (7K MWCO, Zeba, Thermo Scientific). Iron incorporation into the FeB site was confirmed by UV–vis spectroscopy using a Cary 50 spectrophotometer (Varian Inc.).
Preparation of NO Solution and Reaction Conditions
NO solutions were prepared using NO (99.5%, Airgas) and 15NO gas (>98% 15N, Cambridge Isotope Laboratory), which were further purified by bubbling through 1 M NaOH solutions to remove the degradation products N2O3 and NO2. All procedures were performed inside an anaerobic glovebox containing less than 1 ppm O2 (Omnilab System, Vacuum Atmospheres Co.). The purified gases were drawn with gastight Hamilton syringes and bubbled through Bis-Tris buffer solutions in serum bottles fitted with septa. After 5 min of mixing, the NO concentration in the buffered solutions reached the expected NO saturation concentration of ∼2 mM. The exact NO concentration was determined each time by titration against deoxymyoglobin.
To prepare FeBMb samples reacted with excess NO, the sample headspace in UV–vis cuvettes, Raman capillaries, or NMR tubes was thoroughly exchanged with purified NO gas. For low temperature FTIR experiments, the NO exposure was performed in Eppendorf tubes prior to transfer of the aqueous samples to FTIR cells. The success of the reaction was confirmed by UV–vis spectra collected directly in Raman capillaries, NMR tubes, or FTIR cells. Preparations of the mononitrosyl complexes of FeBMbs were performed by stoichiometric addition of NO to fully reduced FeBMbs as described previously.21
FTIR Detection of N2O
Production of N2O was monitored by FTIR spectroscopy with the ν(N–N) mode of N2O at 2231 cm–1, as described previously.22,23 Reduced FeBMb solutions were mixed with the NO-donor diethylammonium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate (diethylamine NONOate, t1/2 ∼ 16 min at 22 °C, Cayman Chemical, Ann Arbor, MI) before recording the FTIR spectra. Diethylamine-NONOate stock solutions were prepared using an ε250 of 6500 M–1 cm–1 in 0.01 M NaOH, and aliquots were added to deoxymyoglobin to confirm the concentration of NO produced. Approximately 30-μL aliquots of 1 mM FeBMbs solutions were deposited as a droplet on a CaF2 window (International Crystal Laboratories, Garfield, NJ) before adding diethylamine-NONOate solution to produce 2 equiv of NO per iron. Immediately after mixing, a second CaF2 window was gently dropped on the sample to form an optical cell whose path length was controlled by a 100-μm Teflon spacer (International Crystal Laboratories, Garfield, NJ). Room temperature FTIR spectra were obtained on a Bruker Tensor 27 equipped with a liquid-N2-cooled MCT detector, purged with compressed air, dried, and depleted of CO2 (purge gas generator, Puregas LLC). Sets of 100-scan accumulations were acquired every 2 min until no further growth of the 2231 cm–1 band was observed. Buffer blank spectra were recorded under the same conditions. Intensity standards for the 2231 cm–1 band of N2O in aqueous solution were obtained from dilutions of N2O saturated solutions.
RR Experiments
Typical protein concentrations for RR experiments ranged from 100 μM for room temperature measurements to 300 μM for data collected at 110 K. RR spectra were recorded using a custom McPherson 2061/207 spectrograph (set at 0.67-m or 1-m focal length with 2400 grooves per millimeter of holographic gratings) equipped with a liquid-N2-cooled CCD detector (LN-1100PB, Princeton Instruments). The 406-nm excitation laser was derived from a Kr laser (Innova 302C, Coherent). A Kaiser Optical supernotch filter or a long-pass filter (RazorEdge, Semrock) was used to attenuate Rayleigh scattering. Room temperature RR spectra were collected using in a 90° scattering geometry on samples mounted on a reciprocating translation stage. Frequencies were calibrated relative to indene, which are accurate to ±1 cm–1. Polarization conditions for room temperature RR spectra were optimized using CCl4 and indene. The integrity of the RR samples was confirmed by direct monitoring of their UV–vis absorption spectra in Raman capillaries or NMR tubes before and after laser exposure. Low-temperature spectra were recorded in a backscattering geometry on samples maintained at 110 K in a liquid nitrogen coldfinger. Frequencies were calibrated relative to aspirin and are accurate to ±1 cm–1. To assess the photosensitivity of the NO adducts, rapid acquisitions within a range of laser powers and continuous sample translation or spinning were compared with longer data acquisitions on static samples.
FTIR Photolysis Experiments
Low-temperature FTIR photolysis was conducted using a previously described method.24−26 FTIR sandwich films were prepared as described above by loading approximately 15 μL of a 1 mM protein solution into an FTIR cell with a 15-μm Teflon spacer. After the formation of the NO adduct was confirmed by UV–vis absorption spectroscopy, the FTIR cell was mounted to a sample rod which was then flash-frozen in liquid N2 and inserted into the sample compartment of a closed-cycle cryogenic system (Omniplex, Advanced Research Systems). The cryostat was placed inside the sample compartment of the FTIR, and the sample was kept in the dark during cooling to 15 K. The temperature of the sample was monitored and controlled with a Cryo-Con 32 or a Lake Shore Model 331 unit. FTIR spectra were recorded with sets of 1000-scan accumulations at 4-cm–1 resolution. Photolysis of nitrosyl complexes was performed by continuous illumination of the sample directly in the FTIR sample chamber using a 300-W arc lamp after filtering out heat and NIR emissions. The photolysis process was completed within a few minutes of illumination at 15 K and was confirmed to be fully reversible by reproducing the same photoinduced difference spectra after raising the sample temperature to 80 K.
Rapid-Freeze-Quench (RFQ) Experiments
We recently described in detail our protocols for the preparation of RFQ samples.27 Briefly, glass syringes (1 or 2 mL) were loaded with protein solutions (0.6 mM reduced FeBMbs in 50 mM Bis-Tris, pH 7.0) and NO solutions (2 mM 14NO and 15NO in 50 mM Bis-Tris, pH 7.0) inside the anaerobic glovebox before mounting them to the System 1000 Chemical/Freeze Quench Apparatus (Update Instruments). A water bath maintained the sample temperature inside the apparatus at 4 °C. Reaction times were controlled by varying the syringe displacement rate from 1 to 8 cm/s or by varying the length of the reactor hose after the mixer. Five milliseconds were added to the calculated reaction times to account for the time-of-flight and the freezing time in liquid ethane. Mixed volumes of 250 μL were ejected into a glass funnel attached to NMR tubes filled with liquid ethane at or below −120 °C. The frozen samples were packed into the tube as the assembly sat within a Teflon block cooled to −120 °C. Liquid ethane was subsequently removed by incubation of samples at −80 °C for 2 h. RR spectra obtained before and after the cryosolvent removal showed no change except for the loss of ethane vibrations. In previous RFQ-RR studies,28,29 sodium selenate or cacodylic acid were used as intensity standards to determine the relative resonance enhancement of porphyrin vibrations in the starting material and reaction product. In the current study, reduced FeBMbs and end-point products of the reaction with excess NO where prepared at 15 μM concentration in the presence of 150 mM sodium selenate. Before these samples were frozen in NMR tubes, the integrity of the reduced FeBMbs and end-point products was confirmed by UV–vis spectroscopy. Using the ν1 symmetric stretch of selenate at 838 cm–1 to compare the intensities of RR bands in the reduced and end products of FeBMb1 and FeBMb2 showed that the closest match is observed near 1615 cm–1 where contributions from porphyrin vinyl stretching modes predominate (data not shown). On these bases, all RFQ-RR spectra were normalized relative to their maximum intensity at ∼1615 cm–1. Without direct measurement of the intensity standard in RFQ samples, this choice of normalization procedure is arbitrary, but alternative intensity normalizations, for example using integrated areas in the porphyrin ν4 region, does not affect the identification of intermediate species.
Stopped-Flow UV–vis Spectroscopy
Stopped-flow experiments were performed with an SX20 apparatus (Applied Photophysics) with a 1-cm path length cell equilibrated at 4 °C inside an anaerobic glovebox containing less than 1 ppm O2 (Nexus System, Vacuum Atmospheres Co.). FeBMb stock solutions were diluted to obtain a final protein concentration of 10 μM in 50 mM Bis-Tris buffer, pH 7.0. NO solutions ranging from ∼0.1 to 1.8 mM in the same buffer were prepared in 1.2 mL glass vials capped with tight septa and were used immediately. After each experiment, remaining premixed solutions were recovered from the stopped-flow apparatus to confirm the concentration of the reactants. Controls using 1:1 mixtures of fully reduced FeBMb solution and buffer were run to ensure that the UV–vis spectrum of FeBMb alone did not change during the course of a stopped-flow experiment. The single-wavelength data at 388, 420, and 440 nm were fit using Pro-Data software by Applied Photophysics or Origin 9.0 software by Origin Lab Corporation. Initial estimates of rate constants and amplitudes were derived from either single- or multiple-exponential fits to these single wavelength data sets. The kinetics data were better fit to triple exponential leading to pseudo-first-order rate constants k1obs, k2obs, and k3obs. Additionally, complete sets of time-resolved spectra were examined by global analysis using a Marquardt–Levenberg algorithm (Pro-K software, Applied Photophysics). As expected, the single wavelength and global analyses provided equivalent kinetic parameters. The reported rate constants are from global analyses and are the average of at least three different rapid mixing experiments. Second-order rate constants were obtained from linear regression analyses of kobs versus NO concentration plots.
Results
Monitoring N2O Production Using FTIR Spectroscopy: No Evidence of N2O Production by FeBMb1 but at Least 50% Productive Single Turnover Reaction in FeBMb2
Prior measurements of N2O production by FeBMb proteins were based on GCMS analyses of reaction headspaces, which showed that under identical conditions, FeBMb2 produced twice as much N2O than FeBMb1.19,20 However, the GCMS approach requires long incubation times (∼20 h) to allow for the equilibration of NO and N2O partition between the aqueous phase and the headspace.
To obtain a more direct measurement of N2O produced under single turnover conditions, we used FTIR spectroscopy and monitored the appearance of N2O in solution upon exposure of reduced FeBMb to excess NO. The ν1 mode of N2O which primarily corresponds to an N–N stretch motion is observed at 2231 cm–1 in aqueous solution. Although the sensitivity of these transmittance measurements in aqueous films is relatively poor, a 1 mM solution of reduced protein provides an effective estimate of the yield of productive single turnover because the IR cell is sealed immediately after mixing the protein with a diethylamine-NONOate stock solution. FTIR spectra were acquired during the release of NO by the NO donor, and while samples prepared with FeBMb1 do not show significant absorption at 2231 cm–1, the ν1 mode of N2O is clearly observed in spectra of samples prepared with FeBMb2 (Figure 2). In addition, the growth rate of the 2231-cm–1 band matches the lifetime of the diethylamine-NONOate (t1/2 ∼ 16 min, at 22 °C), suggesting that, in these measurements, NO-release is the rate-limiting step. Comparison of the 2231 cm–1 peak intensity with calibration curves from aqueous solutions prepared with N2O-saturated stock solution shows that 0.55 ± 0.05 equiv of N2O is produced per reduced FeBMb2 protein.
Figure 2.
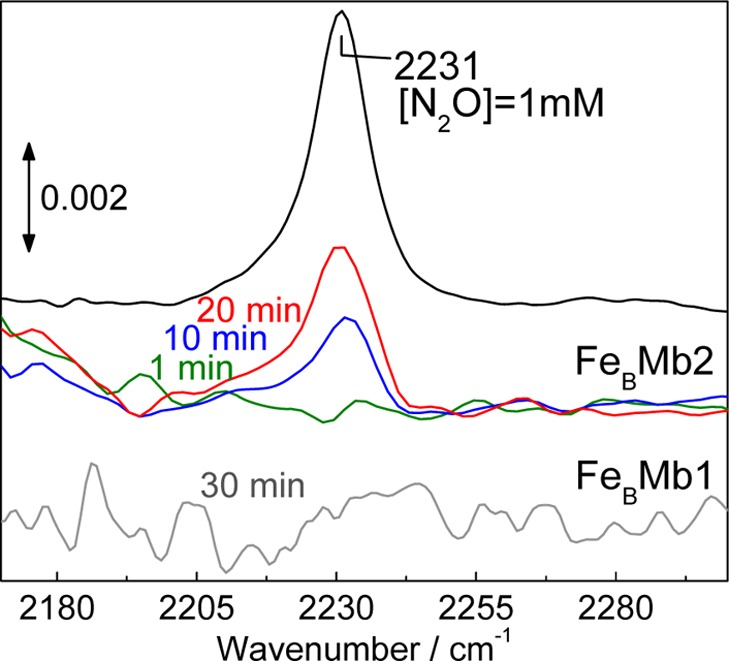
Room temperature FTIR detection of N2O produced by 1 mM reduced FeBMbs following exposure to diethylamine-NONOate. The NO released after 1, 10, and 20 min is 0.1, 0.75, and 1.5 mM, respectively. For the same time points, peak heights at 2231 cm–1 in the FTIR spectra of FeBMb2 correspond to [N2O] <0.1, 0.3, and 0.5 mM, respectively. One millimolar N2O solution was used as an intensity standard.
Reaction Product of FeBMb1 with Excess NO: Evidence for a [5cLS heme-NO/FeB-NO] Dinitrosyl Dead-End Complex
Before investigating the kinetic parameters of the reaction of reduced FeBMb1 with excess NO, we used RR spectroscopy to characterize the end point of this reaction. High-frequency RR spectra obtained with Soret excitation are dominated by porphyrin skeletal modes which report on the oxidation, coordination, and spin states of the iron.30,31 Furthermore, RR modes involving nitrosyl groups are identifiable by their observed frequencies and sensitivity to 15NO and 15N18O isotopic substitutions. After reaction with excess NO, FeBMb1 displays a high frequency RR spectrum with ν4, ν3, ν2, and ν10 modes at 1375, 1505, 1583, and 1641 cm–1, respectively (Figure S1, Supporting Information). Also observed are RR bands at 1666 and 520 cm–1 that shift to lower frequencies with 15NO and 15N18O (Figure S1), thus supporting their assignment to ν(NO) and ν(FeNO) modes, respectively. These frequencies are very different from that of the 6cLS heme {FeNO}7 complex formed upon exposure of reduced FeBMb1 to 1 equiv of NO; instead they are characteristic of a 5cLS heme {FeNO}7 species (Figure 3 and Table 1).
Figure 3.
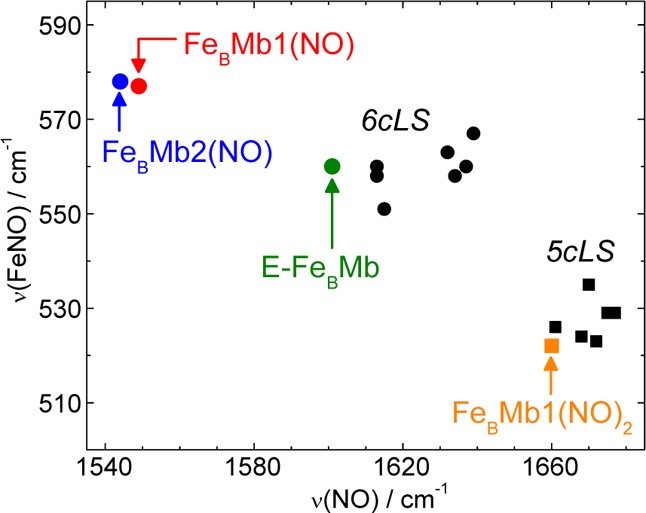
ν(FeNO) vs ν(NO) plot of 5cLS and 6cLS heme {FeNO}7 complexes and proteins (see Table 1 for frequencies and references).
Table 1. Vibrational Frequencies (cm–1) of Heme and Nonheme {FeNO}7 Speciesa.
| {FeNO}7 species | ν(FeNO) (Δ15N) | ν(N–O) (Δ15N) | reference | |
|---|---|---|---|---|
| 6CLS heme(NO) | E-FeBMb1(NO) | 560 (−24) | 1601 (−31) | (21) |
| FeBMb1(NO) | 577 (−25) | 1549 (−22) | (21) | |
| FeBMb2(NO) | 578 (−25) | 1544 (−25) | (21) | |
| FeBMb1(NO)2 intermediate | 572(−20) | – | this work | |
| FeBMb2(NO)2 intermediate | 568(−20) | – | this work | |
| sperm whale Mb(NO) | 560 | 1613 | (56) | |
| horse heart Mb(NO) | 558 | 1613 | (56) | |
| Hb(NO) | 551 | 1615 | (57) | |
| FixL | 558 | 1634 | (58) | |
| DosH E. coli | 563 | 1632 | (59) | |
| DosH M. thermoautotrophicum | 567 | 1639 | (59) | |
| PDEA1H | 560 | 1637 | (59) | |
| 5cLS heme(NO) | FeBMb1(NO)2 end-point | 522 (−12) | 1660 (−30) | this work |
| Horse Mb(NO), pH4 | 524 | 1668 | (56) | |
| sperm whale H93G-Mb(NO) | 535 | 1670 | (60) | |
| CooA(NO) | 523 | 1672 | (51) | |
| cytochrome c′(NO) | 526 | 1661 | (42) | |
| sGC(NO) | 529 | 1677 | (61) | |
| FixL(NO) | 529 | 1675 | (58) | |
| Nonheme(NO) | FeBMb1(NO)2 end-point | – | 1755 (−32) | this work |
| FeBMb1(NO)2 intermediate | – | 1755 (−32) | this work | |
| FeBMb2(NO)2 intermediate | – | 1759 (−33) | this work | |
| Deflavo-FDP(NO)2 | 459 | 1749 | (23) | |
| R2(NO)2 | 445 | 1742 | (35) | |
| SOR(NO) | 475 | 1721 | (34) |
When available, low-temperature values are listed.
In the high frequency region, beyond the range of porphyrin skeletal modes, a very weak signal at 1756 cm–1 that shifts to 1724 cm–1 with 15NO is suggestive of a ν(NO) from a nonheme {FeNO}7 species.23,32−36 Alternative assignments of these RR signals to heme combination bands with contributions from NO-isotope sensitive modes were considered, but none could match the observed frequencies; moreover, RR spectra of RFQ samples show that the time-dependence of the 1756 cm–1 band does not coincide with that of heme vibrations (see below). Considering that the extinction coefficient of nonheme {FeNO}7 ligand-to-metal charge-transfer transition between 400 and 450 nm are ∼100 fold lower than that of Soret absorption from hemes, the resonance enhancement of vibrational modes from the {FeBNO}7 complex are expected to be very low, as observed. Attempts were made to find an excitation wavelength more favorable to the nonheme {FeNO}7 species, but the best results were obtained with a 406 nm excitation. While room temperature RR spectra must be obtained at low laser power and for limited data acquisition, RR spectra with higher signal-to-noise ratios can be obtained on samples at 110 K (Figure 4). At this temperature, the heme ν(FeNO) is 2 cm–1 higher and the heme ν(NO) is 6 cm–1 lower than at room temperature, which may reflect a decrease in Fe–N–O bond angle. In contrast, the nonheme ν(NO) is not significantly affected by the change in temperature and remains at 1755 cm–1.
Figure 4.
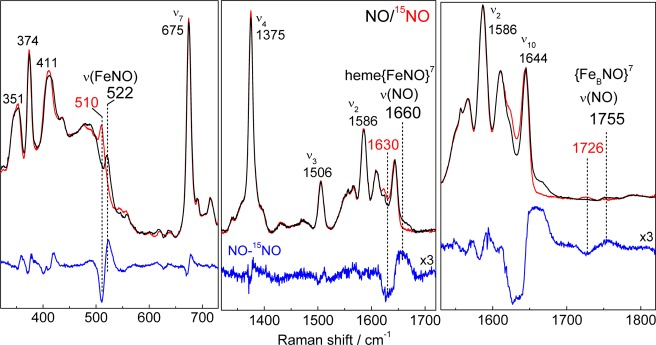
RR spectra (110 K) of the reaction end product of reduced FeBMb1 with excess NO (protein concentration, 300 μM; samples frozen ∼2 min after the addition of NO; excitation wavelength: 406 nm).
Low-temperature FTIR photolysis difference spectra were also able to isolate the ν(NO) from the {FeBNO}7 complex in FeBMb1. Nonheme {FeNO}7 species can be dissociated upon illumination and trapped as photolyzed species at cryogenic temperatures.23,35 Accordingly, a photosensitive band observed at 1751 cm–1 with 14NO and 1719 cm–1 with 15NO is assigned to the ν(NO) of the {FeBNO}7 complex (Figure 5). A significantly weaker 14NO minus 15NO differential signal at 1669 (−31) cm–1 is assigned to the ν(NO) of the 5cLS heme {FeNO}7 species. This small differential signal may reflect a minor population of heme-nitrosyl complex that can be stabilized in the dissociated state at 15 K. However, photolysis of 5cLS heme {FeNO}7 species forms highly unstable four-coordinate hemes which efficiently recombine even at cryogenic temperatures.37 Thus, the differential signal at 1669 cm–1 may reflect an indirect perturbation of the heme-nitrosyl ν(NO) upon dissociation of the nonheme {FeBNO}7 species; indeed, we observed such indirect perturbations between ligands across binuclear centers in dicarbonyl complexes.24 Other FTIR differential signals at 1460 and 1537 cm–1 are induced by illumination but are insensitive to NO-isotope substitutions and are likely to reflect a minor reorganization of coordinating imidazole side chains upon dissociation of the NO group at the FeB site, as seen in redox-based FTIR difference spectra of Cu,Zn-superoxide dismutase.38
Figure 5.
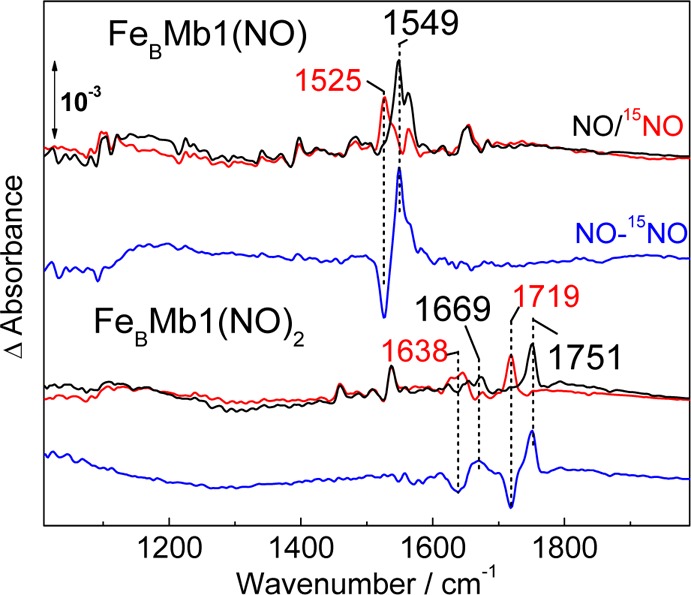
Low temperature FTIR spectra of the mononitrosyl complex formed in FeBMb1 after reaction with 1 equiv of NO (top three traces) and of the end product of the reaction of FeBMb1 with excess NO (the samples were frozen ∼4 min after the addition of NO; bottom three traces). Black and red traces correspond to reactions performed with 14NO and 15NO, respectively. Also shown, are 14NO – 15NO double-difference spectra (blue traces).
Reaction Product of FeBMb2 with Excess NO: Evidence for a Mixture of Oxidized Protein and [Heme-NO/FeB-NO] Dinitrosyl Dead-End Complex
The 110 K high frequency RR spectrum of FeBMb2 with excess NO shows equivalent bands to those assigned to the 5cLS heme {FeNO}7 species formed in FeBMb1, but it also reveals ν4 and ν3 components at 1370 and 1512 cm–1, respectively, which we assign to oxidized heme (Figure 6). Without knowledge of the respective resonance enhancement for these two end products, the relative populations of these states cannot be deduced from the RR intensities; however, the spectra suggest that both states are present in significant concentrations, thus corroborating the 1:1 oxidized:dead-end product ratio predicted by the production of 0.5 equiv of N2O.
Figure 6.
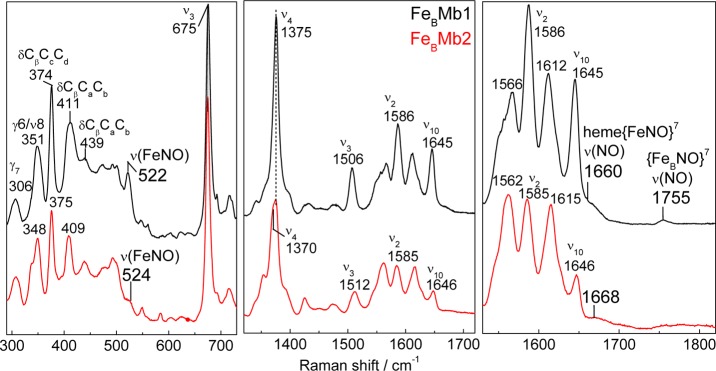
110 K RR spectra of the end product of the reaction of reduced FeBMb1 (black) and FeBMb2 (red) with excess NO (protein concentration, 300 μM; samples frozen ∼2 min after the addition of NO; excitation wavelength: 406 nm).
RFQ-RR Characterization of the Reaction of FeBMb1 with Excess NO: A First NO Binds to FeB before Binding of a Second NO to the Heme Iron(II)
To explore the reaction pathway leading to the dead-end [5cLS heme-NO/FeB-NO] dinitrosyl complex, we prepared RFQ samples of the reaction of reduced FeBMb1 with a 3-fold excess of NO at 6, 40, and 140 ms. RFQ samples of reduced FeBMb1 and of its [5cLS heme-NO/FeB-NO] dinitrosyl dead-end complex were also prepared. High-frequency RR spectra obtained with Soret excitation are shown in Figure 7 (middle panel). The RR spectrum of the 6-ms RFQ sample retains significant contributions from the reduced HS heme with porphyrin skeletal modes ν4 and ν3 at 1352 and 1471 cm–1, respectively. At 40- and 140-ms, however, RFQ samples exhibit growing RR bands at 1375 and 1506 cm–1 which are associated with low-spin heme-nitrosyl. Spectral changes in the 1550 to 1650 cm–1 region are more complex to analyze because of the overlap of multiple porphyrin vibrational modes, but along with the changes in ν4 and ν3 modes, they support the notion of a relatively slow conversion from ferrous heme to a heme-nitrosyl complex. Importantly, the characteristic ν10 of the 5cLS heme-nitrosyl at 1645 cm–1 only begins to gain intensity in the spectrum of the 140-ms RFQ sample. In the low-frequency region (Figure 7, left panel), a RR band at 572 cm–1 that shifts to 552 cm–1 with 15NO is characteristic of a ν(FeNO) from a 6cLS heme {FeNO}7 species (Table 1). This ν(FeNO) mode is weak in the RR spectra of the 6-ms RFQ sample and becomes most prominent in the RR spectra of the 40 ms RFQ sample before decaying in the 140 ms RR spectra, presumably in favor of the 5cLS heme {FeNO}7 species. Accordingly, a 522 cm–1 band assignable to the ν(FeNO) of the 5cLS heme-nitrosyl complex is observed in the RR spectra of the 140-ms RFQ sample (Figure 7, left panel).
Figure 7.
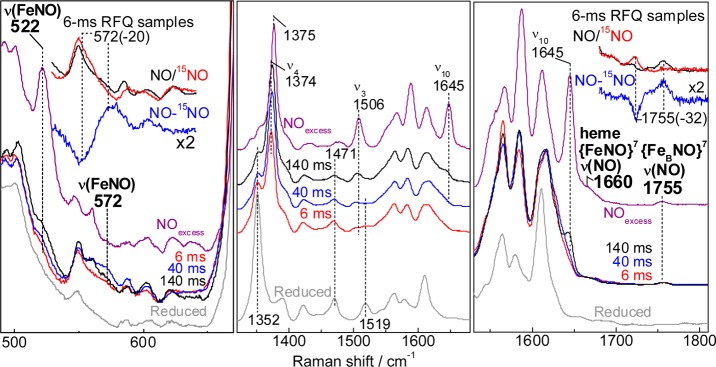
RR spectra of RFQ samples of the reaction of reduced FeBMb1 with excess NO. Also shown for comparison are the RR spectra of resting reduced FeBMb1 (bottom gray traces) and the dinitrosyl end product (purple traces) (protein concentration, 300 μM; excitation wavelength 406 nm; sample temperature 110 K). Porphyrin skeletal modes highlight the ferrous 5cHS to 6cLS conversion (middle panel). The low frequency region shows the growth of heme-nitrosyl ν(FeNO) modes (left panel), and the high frequency region is where ν(NO) modes are observed (right panel). Upper insets focus on the RR data for 6-ms RFQ samples prepared with excess 14NO (black), excess 15NO (red trace), and 14NO – 15NO difference spectra (blue trace).
The RR spectra of the 6-ms RFQ samples prepared with 14NO also reveal a mode at 1755 cm–1 that shifts to 1723 cm–1 with 15NO (Figure 7, right panel). These frequencies are beyond the range of porphyrin vibrational frequencies and instead match those assigned to the ν(NO) from the {FeBNO}7 species in the resting dead-end product (Figure 4). Importantly, the intensity of these bands relative to the porphyrin modes are unchanged in the RFQ samples, supporting the notion that this species is formed within the millisecond mixing time of these experiments and that it does not evolve further before freezing. Thus, these RFQ-RR data suggest that one NO molecule binds to the nonheme FeB site before a second NO molecule binds to the heme iron(II) to form a [6cLS heme-NO/FeB-NO] dinitrosyl intermediate (Scheme 2). Scheme 2 is also supported by the lack of photosensitivity of the nonequilibrium 6cLS heme-nitrosyl complex trapped in the RFQ samples, which we assign to molecular crowding at the heme distal pocket by the {FeBNO}7 complex. Indeed, the 6cLS heme-nitrosyl complex formed after exposure of reduced FeBMb1 to 1 equiv of NO is extremely photolabile, with its dissociated state pumped by the 406-nm Raman excitation, even at laser power <0.1 mW (data not shown). In contrast, the 6cLS heme-nitrosyl complex trapped in the RFQ samples shows no evidence of photolysis even with 50 mW laser power.
Scheme 2. Reaction Steps Leading to the [5cLS heme-NO/FeB-NO] Dinitrosyl Dead-End Complex in FeBMb1.
RFQ-RR Characterization of the Reaction of FeBMb2 with Excess NO: FeBMb1 and FeBMb2 Share Common Intermediate States
As with FeBMb1, 6-, 40-, and 140-ms RFQ samples of the reaction of reduced FeBMb2 with a 3-fold excess of NO were prepared along with samples of reduced FeBMb2 and of the reaction end product. Once again, the RR spectra reveal porphyrin skeletal modes supporting a relatively slow conversion of the reduced HS heme to a low-spin heme-nitrosyl species (Figure 8), as evidenced by the growth of a ν(FeNO) from a 6cLS heme {FeNO}7 species at 568 cm–1, a value within 4-cm–1 of that observed in FeBMb1. Importantly, while this mode decreases in intensity after 140 ms in FeBMb1, the ν(FeNO) of the 6cLS heme-nitrosyl species continues to gain intensity after 140 ms in FeBMb2, without significant growth in RR modes expected for a 5cLS heme {FeNO}7 species. In the high-frequency region, beyond the range of porphyrin vibrational frequencies, the RR spectra of FeBMb2 presents a background more complex than that of FeBMb1 (the same RR background was observed for three independent sets of RFQ samples prepared from different batches of FeBMb2 proteins), but difference spectra between RR spectra of 6-ms RFQ samples prepared with 14NO and 15NO isolate a ν(NO) stretch at 1759 cm–1 of comparable intensity to the ν(NO) of the {FeBNO}7 complex in FeBMb1 (Figure 8, right panel). The relatively weak resonance enhancement of the nonheme ν(NO) precludes us from evaluating the evolution of its intensity at different RFQ time points with full confidence (Figure S2, Supporting Information), but the data are sufficient to demonstrate that a [6cLS heme-NO/FeB-NO] dinitrosyl intermediate forms in FeBMb2. Overall, the RFQ-RR data provide evidence supporting a common reaction pathway for NO in FeBMb1 and FeBMb2 and they further suggest that it may be differences in their kinetic rates that cause the difference in N2O production versus dead-end [5cLS heme-NO/FeB-NO] dinitrosyl complex.
Figure 8.
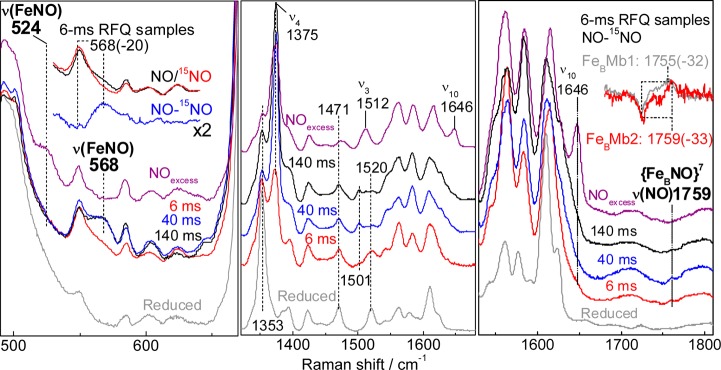
RR spectra of RFQ samples of the reaction of reduced FeBMb2 with excess NO compared to those of resting reduced FeBMb2 (gray traces) and the end product of the reaction (purple traces) (excitation wavelength 406 nm, sample temperature 110 K). Also shown in the upper right inset is an overlay of the 14NO – 15NO differential signal for the nonheme ν(NO) modes in the 6-ms RFQ samples of FeBMb1 (gray) and FeBMb2 (red).
Stopped-Flow Absorption Analysis of the Reaction of FeBMb1 with Excess NO
In an effort to gather individual second-order rate constants, the reaction of reduced FeBMb1 with NO was monitored at 4 °C, with a fixed protein concentration of 5 μM and varying NO concentrations of ∼50 μM to 900 μM under pseudo-first-order conditions. This series of reactions supports the notion that the ferrous high-spin heme, with a Soret absorption at 434 nm, is converted to a 6cLS heme-nitrosyl species with Soret maxima at 421 nm (Figure 9); furthermore, the conversion occurs on a millisecond time scale which indicates a much lower association rate than in wild-type deoxymyoglobin (Table 2). Subsequent conversion to a 5cLS heme-nitrosyl species with a Soret maximum below 410 nm occurs on a second time scale (Figure 9). The first blue shift of the Soret maximum from 434 to 421 nm is multiphasic and complex. Single wavelength analyses lead to observed rates with linear dependence on NO concentration and second-order rate constants k1 ∼ 580 ± 46 mM–1 s–1 and k2 = 6.1 ± 0.3 mM–1 s–1 (Figure S3, Supporting Information). In addition to these two millisecond phases, comparing the spectrum of reduced FeBMb1 with the first few spectra obtained by stopped-flow UV–vis at low NO concentrations (i.e., 54 and 180 μM) reveals a significant hypochromic effect on the Soret absorption. On the basis of the RFQ-RR data which indicate that the first event upon exposure to NO is the formation of a {FeBNO}7 complex, we tentatively assign the drop in Soret absorbance to a [ferrous heme/FeB-NO] complex. Equally satisfactory fits of the stopped-flow data can be achieved with (i) a linear reaction scheme involving two subsequent 6cLS heme-nitrosyl conformers or (ii) branching mechanisms involving two forms of the reduced protein or two diverging heme-nitrosyl species. Despite these mechanistic uncertainties, the RFQ-RR and stopped-flow data convincingly show that a [6cLS heme-NO/FeB-NO] dinitrosyl complex accumulates to near stoichiometry prior to the breakage of the heme iron histidine bond and the formation of the dead-end [5cLS heme-NO/FeB-NO] dinitrosyl complex. This last kinetic phase corresponds to a blue shift of the Soret absorption from 421 to 408 nm with a second-order rate k3 = 2.1 ± 0.1 mM–1 s–1 (Figure 9). NO-dependent dissociation of a proximal histidine from a 6cLS heme-nitrosyl complex has been observed previously in soluble guanylate cyclase (sGC) and in cytochrome c′39−41 and has been assigned to the displacement of the proximal histidine ligand by a second NO molecule.42,43 Table 2 summarizes the kinetic information obtained here along with relevant published rates for other heme proteins. For completeness, the dissociation rate constant for NO in the heme-nitrosyl adduct formed with 1 equiv of NO was also determined optically with dithionite as NO scavenger and was found to be ∼0.1 × 10–3 s–1 (data not shown).
Figure 9.
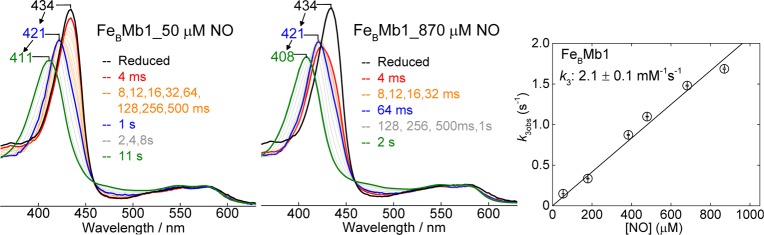
Stopped-flow UV–vis absorption spectra of the reaction of FeBMb1 with 50 and 870 μM NO at 4.0 °C. Also shown is the dependence of the last rate constant (k3obs) on NO concentration.
Table 2. Rate Constants (mM–1 s–1) for NO Binding to Heme Cofactor.
Stopped-Flow Analysis of the Reaction of FeBMb2 with Excess NO
As with FeBMb1, the reaction of reduced FeBMb2 with NO was monitored at 4 °C, with a fixed protein concentration of 5 μM and varying NO concentrations of ∼50 μM to 900 μM under pseudo-first-order conditions. Once again, the conversion of the ferrous heme to a 6cLS heme-nitrosyl is multiphasic, with a dependence on NO concentrations leading to second-order rate constants k1 = 377 ± 22 mM–1 s–1 and k2 = 18 ± 1.5 mM–1 s–1 (Figures 10, and Figure S3, Supporting Information). Although the RFQ-RR data indicate that FeBMb2 also binds NO at the FeB(II) site first, the hypochromic effect seen on the Soret absorption of the ferrous heme in FeBMb1 is not observed in FeBMb2. In the latter case, the build-up of a [6cLS heme-NO/FeB-NO] dinitrosyl complex with a Soret maximum at 420 nm is followed by a decrease in the intensity of the Soret absorption and a blue shift to 412 nm with no significant dependence on NO concentrations beyond 200 μM, resulting in an observed rate k3 ∼ 0.6 s–1 (Figure 10). The hyperbolic dependence of the observed rate on NO concentration can be modeled with the inclusion of a rapid dissociative equilibrium,
and
Figure 10.
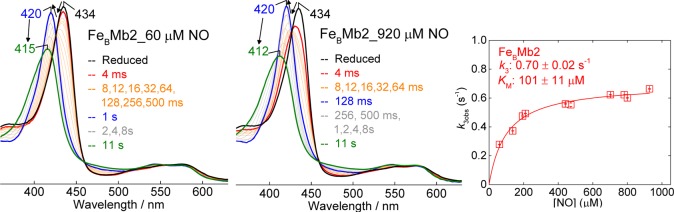
Stopped-flow UV–vis absorption spectra of the reaction of FeBMb2 with 60 and 920 μM NO at 4.0 °C. Also shown is the dependence of the last rate constant (k3obs) on NO concentration.
This analysis yields values of k3 = 0.7 ± 0.02 s–1 and Kd = 101 ± 11 μM (Figure 10 and Scheme 3). It is unclear if the latter value reports on the dissociation of the heme-nitrosyl or the {FeBNO}7 species in the [6cLS heme-NO/FeB-NO] dinitrosyl intermediate. For completeness, we measured the dissociation rate constant for the heme-nitrosyl adduct formed with 1 equiv of NO as koff ∼ 0.5 × 10–3 s–1 (data not shown), but this value may not apply to the heme-nitrosyl species in the dinitrosyl intermediate.
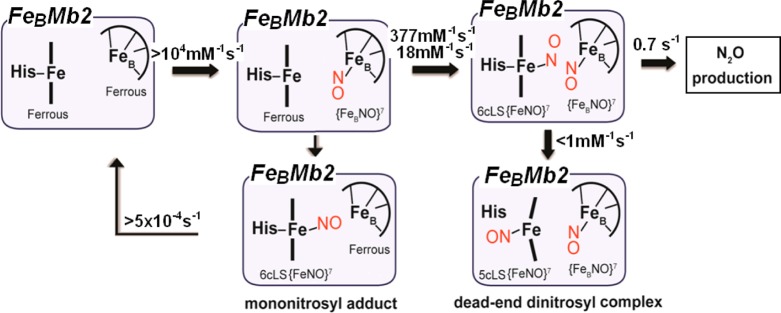
Scheme 3. Reaction Steps Leading to the Production of N2O in FeBMb2.
The reactivity to excess NO of the [6cLS heme-NO/FeB(II)] complex, formed at equilibrium after exposure of fully reduced FeBMbs to 1 equiv of NO, was also evaluated by stopped-flow absorption spectroscopy. As seen for the reaction of FeBMb2 with excess NO, this reaction revealed the same 419 to 412 nm kinetic phase (Figure S4, Supporting Information), demonstrating that the polarized heme-nitrosyl complex does not react more quickly with NO than reduced FeBMb2, which transits through a [6cLS heme-NO/FeB-NO] dinitrosyl complex before production of N2O.
Discussion
Our time-resolved analyses of the reactions of reduced FeBMb1 and FeBMb2 with excess NO reveal that NO first binds to the FeB site rather than to the heme iron. This conclusion is drawn from the RFQ-RR spectra which show a characteristic nonheme ν(NO) at 1755 cm–1 at the earliest quenching time of 6 ms. Without internal intensity standard(s), a truly quantitative evolution of the {FeNO}7 complex cannot be deduced from these RFQ-RR data (see Materials and Methods), but by matching background levels and normalizing all RR spectra to the cluster of porphyrin skeletal modes peaking at 1610 cm–1, it appears that the 1755 cm–1 band remains unchanged in all the RFQ samples and the dead-end complex of FeBMb1. Thus, the NO association rate to the FeB site is likely to be beyond the millisecond time resolution of our RFQ and stopped-flow experiments. Multiple studies have shown that deoxymyoglobin offers several accessibility routes for exogenous diatomic ligands to reach the distal side of the heme iron where the FeB site is situated in FeBMbs.44−47 Once an NO group is bound to the FeB site, molecular crowding at the distal pocket could explain why the NO association rate to the heme iron can be as much as 2 orders of magnitude smaller than in wild-type deoxymyoglobin (Table 2).48
Our previous work has shown that exposing reduced FeBMbs to 1 equiv of NO leads to stable six-coordinate low-spin (6cLS) heme-nitrosyl complexes,21 but these equilibrium complexes are not populated under pre-steady-state conditions, and the build-up of these resting heme-nitrosyl complexes reflects their low dissociation rates in the 2–4 h–1 range. Because observed rates for the formation of the heme-nitrosyl species are always first rather than zero order with respect to NO concentrations, we rule out a gating mechanism where NO would be required to bind to the FeB site before it can migrate to the heme iron. Both FeBMbs exhibit two second-order kinetic rates for the formation of the 6cLS heme-nitrosyl species. Single wavelength and global analyses are unambiguous in identifying two kinetic phases of comparable amplitude, but the data do not distinguish whether these two distinct phases correspond to a branching reaction pathway or if they reflect the occurrence of two consecutive intermediates. Despite the complex kinetics, a clear result of this work has been to show that, upon exposure to excess NO, both FeBMbs transit through a [6cLS heme-NO/FeB-NO] dinitrosyl intermediate complex which accumulates to near stoichiometry after 0.1 to 1.0 s depending on the NO concentration.
In FeBMb1, the [6cLS heme-NO/FeB-NO] dinitrosyl intermediate decays to a dead-end [5cLS heme-NO/FeB-NO] dinitrosyl complex with a second-order rate constant dependent on the NO concentration, k3 = 2.1 mM–1 s–1. The formation of a 5cLS heme-NO caused by the dissociation of the proximal histidine is well documented in many hemoproteins including hemoglobin, guanylate cyclase, NO and CO sensory proteins, cytochrome c, and cytochrome c′.39,41,43,49−52 The trans ligand effect of the distal nitrosyl on the Fe-His bond facilitates the dissociation of the proximal histidine, but the dependence of this process on NO concentration indicates that a second NO molecule is involved in the generation of the 5cLS heme-nitrosyl species. This second NO may bind to an allosteric site, or it may directly participate in the displacement of the proximal histidine through the formation of a transient heme-dinitrosyl complex before dissociation of the distal NO occurs, as recently documented in sGC by Martin and co-workers.43 An equivalent process in FeBMb1 would leave the [5cLS heme-NO/FeB-NO] dinitrosyl complex with each nitrosyl group separated from one another by the porphyrin cofactor. Such arrangement provides a straightforward explanation for the complete lack of reactivity of this [{FeNO}7]2 complex.
The hypothesis of a proximal NO as the driving force for the 6cLS to 5cLS conversion of the heme iron nitrosyl is further supported by the impact the I107E substitution in FeBMb2 has on this reaction step. FTIR-photolysis studies of the carbonyl complex of myoglobin have shown that a bulky I107F substitution severely restricts the migration of CO from the distal pocket to the proximal cavity.53 Although the I107E substitution does not significantly affect the van der Waals volume of the side chain, the replacement of the aliphatic chain with a carboxylate group is likely to affect the migration of NO around the porphyrin cofactor. Accordingly, the substitution already affects the NO association rate with the ferrous heme in FeBMb2 relative to FeBMb1. A 3-fold decrease in the 6cLS to 5cLS heme-nitrosyl conversion rate would be sufficient to allow the intramolecular reaction between the two iron-nitrosyl groups and N2O production to proceed. Our data show that the reaction of NO with FeBMb2 branches into an ∼1:1 mixture of oxidized protein and dead-end [5cLS heme-NO/FeB-NO] dinitrosyl complex, thus suggesting that the I107E substitution may not be optimized.54 We expect that further engineering of the myoglobin scaffold should be able to fully inhibit the 6cLS to 5cLS heme-nitrosyl conversion to achieve stoichiometric production of N2O. Specifically, mutations that impact the accessibility of diatomic ligands to the heme proximal pocket should be explored.
While the I107E substitution partially protects the 6cLS heme-nitrosyl against a proximal attack by NO, it does not appear to affect the electronic structure of the [6cLS heme-NO/FeB-NO] dinitrosyl intermediate because the ν(FeNO)heme and ν(NO)FeB vibrations are nearly unchanged in FeBMb1 and FeBMb2. In addition, the same dinitrosyl intermediate state is formed when the equilibrium heme-nitrosyl complex, formed upon exposure to 1 equiv of NO, is further exposed to excess NO. Therefore, contrary to expectation from theoretical studies,16,17 the polarized [6cLS heme-NO/FeB] complex is not primed for electrophilic attack by a second NO as in the cis-heme mechanism. The decay of the [6cLS heme-NO/FeB-NO] dinitrosyl intermediate provides no evidence for a build-up of a hyponitrite intermediate, and because the turnover in FeBMb2 is substochiometric, we cannot precisely characterize the subsequent step(s) of the reaction. Nevertheless, the kinetic data reveals a turnover rate of ∼0.7 s–1 at 4 °C which can be used as a valuable benchmark for the evaluation of future NOR models.
Our present study of FeBMb1 and FeBMb2 shows that FeBII is the initial site of NO binding, prior to the coordination of a second NO molecule to the heme iron to produce a [6cLS heme-NO/FeB-NO] trans-dinitrosyl intermediate. In FeBMb1, this transient species is converted to a nonproductive [5cLS heme-NO/FeB-NO] trans-dinitrosyl via the dissociation of the proximal histidine. By contrast, the I107E mutation in FeBMb2 lowers the rate of this inhibitory step and allows the trans-iron-nitrosyl dimer to proceed toward N–N bond formation and the production of N2O. At this time, the exact molecular basis for these changes in reactivity remains uncertain. Mutations of I107 in swMb are known to influence the access of diatomic molecules to the heme proximal site, and in reduced FeBMb2, Glu107 stabilizes a hydrogen bond network with a water molecule and the FeBII site. Thus, the impact of the I107E substitution on the reactivity of the [6cLS heme-NO/FeB-NO] trans-dinitrosyl intermediate may have proximal as well as distal origins. New FeBMb constructs such as swMb L29H/F43H/V68E/I107F should provide further insights on this issue.
Acknowledgments
This work was supported by grants GM074785 (P.M.-L.) and GM062211 (Y.L.) from the National Institutes of Health, A JSPS fellowship for H.M., and a Vertex pharmaceutical scholarship for T.H. We thank Dr. Colin Andrew for helpful discussions.
Supporting Information Available
Room temperature RR spectra of the end product of the reaction of reduced FeBMb1 with excess NO, low temperature RR spectra of RFQ samples of FeBMb2, the dependence of observed rate constants k1 and k2 on NO concentration for the reaction of reduced FeBMbs with excess NO, stopped-flow UV–vis absorption spectra, and the dependence of the reaction of FeBMb2(NO) with excess NO on NO concentration at 4 °C. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
§ Department of Synthetic Chemistry and Biological Chemistry, Graduate School of Engineering, Kyoto University, Katsura, Kyoto 615-8510, Japan.
The authors declare no competing financial interest.
A very recent report describes the EPR and Mössbauer spectra of the {FeBNO}7 complex in a Zn-porphyrin substituted form of FeBMb1 {Chakraborty et al. Angew. Chem. Int. Ed.2014, 53, in press}. Reduction of NO to N2O through a trans bisnitrosyl intermediate was also shown to occur in a heme/nonheme diiron synthetic model {Collman et al. J. Am. Chem. Soc.2008, 130, 16498}. For a review of efforts with heme/nonheme model compounds see {Schopfer et al. Inorg. Chem.2010, 49, 6267}.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Wasser I. M.; de Vries S.; Moënne-Loccoz P.; Schröder I.; Karlin K. D. Chem. Rev. 2002, 102, 1201. [DOI] [PubMed] [Google Scholar]
- Zumft W. G. J. Inorg. Biochem. 2005, 99, 194. [DOI] [PubMed] [Google Scholar]
- Watmough N. J.; Field S. J.; Hughes R. J.; Richardson D. J. Biochem. Soc. Trans. 2009, 37, 392. [DOI] [PubMed] [Google Scholar]
- Barraud N.; Hassett D. J.; Hwang S. H.; Rice S. A.; Kjelleberg S.; Webb J. S. J. Bacteriol. 2006, 188, 7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevanin T. M.; Read R. C.; Poole R. K. Gene 2007, 398, 62. [DOI] [PubMed] [Google Scholar]
- Arkenberg A.; Runkel S.; Richardson D. J.; Rowley G. Biochem. Soc. Trans. 2011, 39, 1876. [DOI] [PubMed] [Google Scholar]
- Hino T.; Matsumoto Y.; Nagano S.; Sugimoto H.; Fukumori Y.; Murata T.; Iwata S.; Shiro Y. Science 2010, 330, 1666. [DOI] [PubMed] [Google Scholar]
- Tosha T.; Shiro Y. IUBMB Life 2013, 65, 217. [DOI] [PubMed] [Google Scholar]
- Hendriks J. H.; Jasaitis A.; Saraste M.; Verkhovsky M. I. Biochemistry 2002, 41, 2331. [DOI] [PubMed] [Google Scholar]
- Flock U.; Thorndycroft F. H.; Matorin A. D.; Richardson D. J.; Watmough N. J.; Adelroth P. J. Biol. Chem. 2008, 283, 3839. [DOI] [PubMed] [Google Scholar]
- Lachmann P.; Huang Y.; Reimann J.; Flock U.; Adelroth P. J. Biol. Chem. 2010, 285, 25531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moënne-Loccoz P.; Richter O.-M. H.; Huang H. W.; Wasser I. M.; Ghiladi R. A.; Karlin K. D.; de Vries S. J. Am. Chem. Soc. 2000, 122, 9344. [Google Scholar]
- de Vries S.; Strampraad M. J.; Lu S.; Moënne-Loccoz P.; Schröder I. J. Biol. Chem. 2003, 278, 35861. [DOI] [PubMed] [Google Scholar]
- Timoteo C. G.; Pereira A. S.; Martins C. E.; Naik S. G.; Duarte A. G.; Moura J. J.; Tavares P.; Huynh B. H.; Moura I. Biochemistry 2011, 50, 4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moënne-Loccoz P. Nat. Prod. Rep. 2007, 24, 610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg L. M.; Blomberg M. R.; Siegbahn P. E. Biochim. Biophys. Acta 2006, 1757, 240. [DOI] [PubMed] [Google Scholar]
- Blomberg M. R.; Siegbahn P. E. Biochemistry 2012, 51, 5173. [DOI] [PubMed] [Google Scholar]
- Cordas C. M.; Duarte A. G.; Moura J. J.; Moura I. Biochim. Biophys. Acta 2013, 1827, 233. [DOI] [PubMed] [Google Scholar]
- Yeung N.; Lin Y. W.; Gao Y. G.; Zhao X.; Russell B. S.; Lei L.; Miner K. D.; Robinson H.; Lu Y. Nature 2009, 462, 1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. W.; Yeung N.; Gao Y. G.; Miner K. D.; Tian S.; Robinson H.; Lu Y. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 8581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Miner K. D.; Yeung N.; Lin Y. W.; Lu Y.; Moënne-Loccoz P. Biochemistry 2011, 50, 5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Lin M. T.; Ganesan K.; Chen Y.; Fee J. A.; Gennis R. B.; Moënne-Loccoz P. Biochemistry 2009, 48, 883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi T.; Caranto J. D.; Wampler D. A.; Kurtz D. M.; Moënne-Loccoz P. Biochemistry 2010, 49, 7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Suharti; de Vries S.; Moënne-Loccoz P. J. Am. Chem. Soc. 2004, 126, 15332. [DOI] [PubMed] [Google Scholar]
- Lu S.; Sazinsky M. H.; Whittaker J. W.; Lippard S. J.; Moënne-Loccoz P. J. Am. Chem. Soc. 2005, 127, 4148. [DOI] [PubMed] [Google Scholar]
- Hayashi T.; Lin I. J.; Chen Y.; Fee J. A.; Moënne-Loccoz P. J. Am. Chem. Soc. 2007, 129, 14952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumura H.; Moënne-Loccoz P. In Metalloproteins: Methods and Protocols; Fontecilla-Camps J. C., Nicolet Y., Eds.; Springer Science+Business Media: New York, 2014; Vol. 1122, in press. [Google Scholar]
- Lu S.; Wiertz F. G. M.; de Vries S.; Moënne-Loccoz P. J. Raman Spectrosc. 2005, 36, 359. [Google Scholar]
- Yukl E. T.; de Vries S.; Moënne-Loccoz P. J. Am. Chem. Soc. 2009, 131, 7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro T. G.; Li X. Y. In Biological applications of Raman spectroscopy; Spiro T. G., Ed.; Wiley: New York, 1988; p 1. [Google Scholar]
- Spiro T. G.; Czeruszewicz R. S. In Physical methods in bioinorganic chemistry; Que L., Jr., Ed.; University Science Books: Sausalito, CA, 2000, p 59. [Google Scholar]
- Nocek J. M.; Kurtz D. M. Jr.; Sage J. T.; Xia Y. M.; Debrunner P.; Shiemke A. K.; Sanders-Loehr J.; Loehr T. M. Biochemistry 1988, 27, 1014. [DOI] [PubMed] [Google Scholar]
- Feig A. L.; Bautista M. T.; Lippard S. J. Inorg. Chem. 1996, 35, 6892. [DOI] [PubMed] [Google Scholar]
- Clay M. D.; Cosper C. A.; Jenney F. E. Jr.; Adams M. W.; Johnson M. K. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu S.; Libby E.; Saleh L.; Xing G.; Bollinger J. M. Jr.; Moënne-Loccoz P. J. Biol. Inorg. Chem. 2004, 9, 818. [DOI] [PubMed] [Google Scholar]
- Zheng S.; Berto T. C.; Dahl E. W.; Hoffman M. B.; Speelman A. L.; Lehnert N.. J. Am. Chem. Soc. 2013, 135(13), 4902–4905. [DOI] [PubMed] [Google Scholar]
- Yoo B. K.; Lamarre I.; Martin J. L.; Andrew C. R.; Negrerie M. J. Am. Chem. Soc. 2013, 135, 3248. [DOI] [PubMed] [Google Scholar]
- Dupeyrat F.; Vidaud C.; Lorphelin A.; Berthomieu C. J. Biol. Chem. 2004, 279, 48091. [DOI] [PubMed] [Google Scholar]
- Stone J. R.; Marletta M. A. Biochemistry 1996, 35, 1093. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Brandish P. E.; Ballou D. P.; Marletta M. A. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 14753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D. M.; Stevenson C. E.; Andrew C. R.; Eady R. R. EMBO J. 2000, 19, 5661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew C. R.; George S. J.; Lawson D. M.; Eady R. R. Biochemistry 2002, 41, 2353. [DOI] [PubMed] [Google Scholar]
- Martin E.; Berka V.; Sharina I.; Tsai A. L. Biochemistry 2012, 51, 2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver T. E.; Rohlfs R. J.; Olson J. S.; Gibson Q. H.; Blackmore R. S.; Springer B. A.; Sligar S. G. J. Biol. Chem. 1990, 265, 20007. [PubMed] [Google Scholar]
- Schotte F.; Lim M.; Jackson T. A.; Smirnov A. V.; Soman J.; Olson J. S.; Phillips G. N. Jr.; Wulff M.; Anfinrud P. A. Science 2003, 300, 1944. [DOI] [PubMed] [Google Scholar]
- Bourgeois D.; Vallone B.; Schotte F.; Arcovito A.; Miele A. E.; Sciara G.; Wulff M.; Anfinrud P.; Brunori M. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 8704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maragliano L.; Cottone G.; Ciccotti G.; Vanden-Eijnden E. J. Am. Chem. Soc. 2009, 132, 1010. [DOI] [PubMed] [Google Scholar]
- Moore E. G.; Gibson Q. H. J. Biol. Chem. 1976, 251, 2788. [PubMed] [Google Scholar]
- Chan N. L.; Kavanaugh J. S.; Rogers P. H.; Arnone A. Biochemistry 2004, 43, 118. [DOI] [PubMed] [Google Scholar]
- Nioche P.; Berka V.; Vipond J.; Minton N.; Tsai A. L.; Raman C. S. Science 2004, 306, 1550. [DOI] [PubMed] [Google Scholar]
- Reynolds M. F.; Parks R. B.; Burstyn J. N.; Shelver D.; Thorsteinsson M. V.; Kerby R. L.; Roberts G. P.; Vogel K. M.; Spiro T. G. Biochemistry 2000, 39, 388. [DOI] [PubMed] [Google Scholar]
- Silkstone G.; Kapetanaki S. M.; Husu I.; Vos M. H.; Wilson M. T. Biochemistry 2012, 51, 6760. [DOI] [PubMed] [Google Scholar]
- Lamb D. C.; Nienhaus K.; Arcovito A.; Draghi F.; Miele A. E.; Brunori M.; Nienhaus G. U. J. Biol. Chem. 2002, 277, 11636. [DOI] [PubMed] [Google Scholar]
- EPR experiments aimed at quantifying iron-nitrosyl species in FeBMb1 and FeBMb2 are hindered by magnetic exchange coupling between the two iron centers. Our earlier study of the equilibrium NO adducts formed after exposure of reduced FeBMbs to 1 equiv of NO showed that magnetic coupling between the 6cLS heme-nitrosyl (i.e., 6cLS {FeNO}7S = 1/2) and FeBII (HS FeIIS = 2) leads to an S = 3/2 or 5/2 magnetic system with weak EPR resonances at g = 6.2 and 6.1 observable only below 30 K.21 Similarly, the (5cLS heme-nitrosyl/FeB-nitrosyl) dead-end dinitrosyl complex formed in FeBMb1 after exposure to excess NO suffers from exchange coupling and is not detectable by EPR above 30 K; weak and broad signals observed below 30 K are poor quantitative tools. EPR was also used by Lin et al. to follow the reaction of reduced FeBMb2 with excess NO and revealed a g = 2 signal characteristic of a 5cLS heme-nitrosyl species that increases in intensity within the 1 to 15 min incubation period at room temperature. Because this EPR-active 5cLS heme-nitrosyl species was not observed in equivalent experiments carried out with FeBMb1, it was suggested that it contributes to the higher NOR activity seen with FeBMb2.20 However, our current stopped-flow and RFQ-RR data reveal reaction rates for FeBMb2 with excess NO that approach 1 s–1 at 4 °C, and we now believe that the growth of the EPR-active 5cLS heme-nitrosyl species in the minute time scale corresponds to the reaction of oxidized FeBMb2 with excess NO, after the production of N2O. Autoreduction of ferric heme-nitrosyl complexes in myoglobins has been extensively studied,55 and although this reaction is typically much slower, occurring on the hour time scale, distal mutations, NO, nitrite, and free iron may accelerate this reaction in FeBMb constructs. More detailed EPR studies of the reaction of FeBMbs with excess NO will be carried out in our laboratories.
- a Fernandez B. O.; Ford P. C. J. Am. Chem. Soc. 2003, 125, 10510. [DOI] [PubMed] [Google Scholar]; b Addison A. W.; Stephanos J. J. Biochemistry 1986, 25, 4104. [DOI] [PubMed] [Google Scholar]
- Tomita T.; Hirota S.; Ogura T.; Olson J. S.; Kitagawa T. J. Phys. Chem. B 1999, 103, 7044. [Google Scholar]
- Tsubaki M.; Yu N. T. Biochemistry 1982, 21, 1140. [DOI] [PubMed] [Google Scholar]
- Lukat-Rodgers G. S.; Rodgers K. R. Biochemistry 1997, 36, 4178. [DOI] [PubMed] [Google Scholar]
- Tomita T.; Gonzalez G.; Chang A. L.; Ikeda-Saito M.; Gilles-Gonzalez M. A. Biochemistry 2002, 41, 4819. [DOI] [PubMed] [Google Scholar]
- Thomas M. R.; Brown D.; Franzen S.; Boxer S. G. Biochemistry 2001, 40, 15047. [DOI] [PubMed] [Google Scholar]
- Deinum G.; Stone J. R.; Babcock G. T.; Marletta M. A. Biochemistry 1996, 35, 1540. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.