

Abstract
Human dental tissues consist of inorganic constituents (mainly crystallites of hydroxyapatite, HAp) and organic matrix. In addition, synthetic HAp powders are frequently used in medical and chemical applications. Insights into the ultrastructural alterations of skeletal hard tissues exposed to thermal treatment are crucial for the estimation of temperature of exposure in forensic and archaeological studies. However, at present, only limited data exist on the heat-induced structural alterations of human dental tissues. In this paper, advanced non-destructive small- and wide angle X-ray scattering (SAXS/WAXS) synchrotron techniques were used to investigate the in situ ultrastructural alterations in thermally treated human dental tissues and synthetic HAp powders. The crystallographic properties were probed by WAXS, whereas HAp grain size distribution changes were evaluated by SAXS. The results demonstrate the important role of the organic matrix that binds together the HAp crystallites in responding to heat exposure. This is highlighted by the difference in the thermal behaviour between human dental tissues and synthetic HAp powders. The X-ray analysis results are supported by thermogravimetric analysis. The results concerning the HAp crystalline architecture in natural and synthetic HAp powders provide a reliable basis for deducing the heating history for dental tissues in the forensic and archaeological context, and the foundation for further development and optimization of biomimetic material design.
Keywords: in situ thermal treatment, dental tissue, hydroxyapatite, SAXS/WAXS
1. Introduction
Hydroxyapatite (HAp) crystallites that are closely integrated in the organic matrix at the nanoscale are the main inorganic constituents of the hierarchical mineralized human dental tissues, namely enamel and dentine. Such natural HAp crystallites are understood to be crucial to the physiology and function of dental tissues [1]. In parallel, recent developments in synthetic biomineralization have demonstrated that synthetic HAp powders can play an important role for medical applications, especially for the replacement or treatment of dental tissues [2].
Advanced high-energy treatment techniques are finding increasing use in modern dentistry. In the early 1970s, Kantola et al. [3] showed that laser treatment of dental tissues can be used to increase the mineral content and crystallinity of dentine by preferential removal of the inherent water and protein. With the advent of a variety of new laser systems spanning a range of energy densities and pulse durations, clinical treatments such as laser-assisted caries protection were proposed and developed. Local temperature induced by short pulse laser treatment leads to local heating to temperatures between several hundred to over 1000°C. The alteration of dental tissue remains highly localized, and improved caries prevention is surmised to be associated with increased mineralization and HAp crystallite sintering that leads to the sealing of dentinal tubules, while leaving the remaining tissues intact owing to the confined effect of less than 10 µm in depth (at the fluence of 2 J cm−2) [4,5].
Any change in the local environment of HAp crystallites, for example, through laser illumination or direct heating, is bound to affect the mineral ultrastructure. In the context of archaeological and forensic investigations, while macroscopic alterations (e.g. surface colour) can be used to deduce an approximate temperature range, the investigation of the micro- and ultrastructural alterations of skeletal hard tissue to high temperatures has proved to be crucial to obtain a reliable estimation of the temperature of exposure [6–8]. This, in turn, sheds light on the attendant circumstances, for example, the presence and nature of combustible agent used. Dental tissues, being the most highly mineralized part of the human body, can withstand longer thermal exposure and survive higher temperatures, thus offering the possibility of extending the range of applicability of this technique [9].
The above considerations provide strong motivation for a detailed study into the effect of thermal exposure on the nanostructure (the arrangement of HAp crystallites) of human dental tissue as well as the role of the organic phase. While much research has been carried out into the heat-induced changes that occur in bone, in connection with human remains studied in the context of archaeology, anthropology and palaeontology [10–13], the corresponding range and depth of study of the thermal response of dental tissues remains lacking [6,14].
Multiple techniques have been used to study heat-induced ultrastructural alterations of skeletal hard tissues. Most widely applied techniques are based on the absorption spectra of infrared radiation (Fourier transform infrared spectroscopy, FTIR), or laboratory-based X-ray powder diffraction (XRD) and laboratory-based small angle X-ray scattering (SAXS) [6,10,15–17]. However, there has been ongoing debate regarding the general limitations and validity of the results quantified by FTIR [6,15,16]. Laboratory-based XRD has been shown to be capable of showing the distinct differences in the response of HAp crystallites to heating between animal and human bone [6,16,18]. As a complementary method, laboratory-based SAXS allows the identification of local structural alterations at the nanoscale, and is also capable of a fast throughput of samples [19]. However, both methods usually involve the grinding of a sample into powder to obtain volume-averaged results that do not provide appropriately spatially resolved information on the local changes of skeletal ultrastructure within the tissue [19,20]. In order to understand and quantify more precisely the mechanism of such ultrastructural alterations with temperature, the application of advanced non-destructive techniques offers an appropriate route. One of the most advanced non-destructive techniques is synchrotron-based X-ray diffraction, namely WAXS and SAXS. These methods have been used to characterize human dental tissues [21,22], which enabled the characterization of the ultrastructure of mineralized tissue using intact (not ground) samples, rather than powdered samples used with laboratory-based techniques.
Recently, the non-destructive synchrotron-based techniques (WAXS/SAXS) have been applied to the study of previously thermally treated human dental tissues [9]. However, these tests were conducted ex situ, where it was possible to consider only a limited number of samples after exposure to temperatures in the 400–800°C range. In addition, by necessity, different samples were used for each measurement, and thus the sample-to-sample ultrastructure variation could not be excluded. This aspect may weaken the reliability of the conclusions drawn regarding the mechanism of local ultrastructural changes. Furthermore, the ex situ mode of sample characterization is applied only after heating and cooling the samples to/from different temperatures. Obviously, no information is available on the structure evolution during heating nor cooling. To the best of our knowledge, no results have been reported on monitoring heat-induced ultrastructural changes of HAp crystallites using an in situ experimental protocol spanning the entire relevant range of heating and cooling nor has the comparison been made between the naturally occurring HAp crystallites within human dental tissues and synthetic HAp powders. Thus, in situ thermal measurement will help to understand the internal architecture of complex natural materials (highly mineralized human tissues), and its evolution during thermal exposure.
In this paper, we report the evaluation of the crystallographic properties (d-spacing and crystal perfection and size) and the nanostructure (thickness, orientation and degree of alignment) of HAp crystallites embedded within the dental tissue as well as synthetic HAp powders subjected to in situ thermal treatment. The combination of synchrotron X-ray scattering techniques (WAXS/SAXS) was used. The results were analysed in order to understand how the internal ultrastructure within the human dental tissues and the synthetic HAp powders evolves during the heating and cooling schedule.
2. Material and methods
2.1. Sample preparation
Freshly extracted intact human third molars (ethical approval obtained from the National Research Ethics Committee; NHS-REC reference no. 09.H0405.33/Consortium R&D no. 1465) were washed and cleaned in distilled water to eliminate residues, so that the possibility of contamination or other chemical effects was excluded. The samples were cut into 1 mm-thick cross-sectional slices in the buccolingual orientation including dentine and enamel (figure 1a) using a low-speed diamond saw (Isomet Buehler Ltd, Lake Bluff, IL) and polished using a sequence of grit papers to minimize the induced residual strain. In total, four cross sections were prepared and were kept in distilled water in a commercial fridge at 4°C until the experiment was performed. Low temperature storage conditions ensured relatively low diffusion rates so as to impede or exclude any possibility of superficial demineralization effect of water.
Figure 1.

(a) Schematic of the in situ SAXS/WAXS experimental set-up that ensured that the WAXS and SAXS patterns were collected simultaneously. (b) A representative WAXS pattern showing the Debye–Sherrer ring corresponding to the (002) peak indicated by the red arrow. On the ring, the section along the y-axis in (a) was ‘caked’ (see text) with a 20° range in order to capture the peak shift upon heating and cooling process. (c) A representative SAXS pattern. The dark region in the middle is the beamstop. The ellipsoidal shape of the pattern indicates a partial alignment of HAp crystallites. The pattern for randomly distributed crystals is a round disc with the outline marked with red dashed circle. (d,e) Illustration of the different heating and cooling protocols used for the human dental slice samples and the synthetic HAp powders sample. (Online version in colour.)
A mould was filled with 100 mg of commercially available HAp powder (hydroxyapatite HTP powder, Bio-Rad, Hercules, CA) and compacted with using an Instron 5544 machine (Instron Ltd, Bucks, UK) at 1.8 kN force. The resulting disc was 1.2 mm thick and 8 mm in diameter, with an approximate density of 1150 mg cm−3.
The weight-loss over time was additionally evaluated using thermogravimetry (TGA) at a rate of 12°C per minute using three cube-shaped dental samples (3 × 3 × 3 mm3) extracted from two molars. The weight fraction of the organic component was determined with TGA using one cubic dentine sample that was placed in a rotating 10% formic acid bath for 14 days to eliminate the mineral content, thus producing a purely organic sample.
2.2. In situ scattering measurements
2.2.1. Thermal treatment set-up
In situ thermal treatment of the samples was performed using a remotely operated and monitored furnace that allows collection of SAXS/WAXS data at temperatures up to 1000°C. This furnace is a modification of the design used at Daresbury laboratory [23]. The samples were fixed in specially designed ceramic sample holders. A Kapton window in the furnace allowed a high-energy X-ray transmission set-up to be used, as illustrated in figure 1a. The heating and cooling protocols were controlled using a Eurotherm controller. The protocols incorporated ramps at 12°C min−1, and constant temperature holds of 200 s for each temperature level within dental slice samples studied (enamel and dentine in figure 1d). The protocol for synthetic HAp powders is also shown in figure 1e. Slightly different thermal protocols were used for natural and synthetic materials owing to the limitations of the experimental set-up (furnace heating rates accessible for different samples). However, preliminary measurements carried out showed that, within the range of heating rates considered, the resulting differences were negligible. The temperature output by the furnace controller and thermocouple was of high accuracy (one decimal point, approx. 0.1° nominal precision). Overall, the measurements were conducted at three different points in dentine, one measurement point in enamel and one point in the synthetic HAp sample. Larger number of datasets would improve the quality of data and provide better statistical information on data quality. However, owing to the limited availability of synchrotron beam time, the collection of additional datasets was not possible. The statistical error analysis of dentine has been included in the results, and is reported below.
2.2.2. Beamline experimental set-up
The experiment was carried out on the I22 experimental beamline at Diamond Light Source, Oxford Harwell Campus, Didcot, UK. A monochromatic X-ray beam (photon energy of 18 keV) was used to illuminate the samples with the beam size of 65 × 120 µm2 as illustrated schematically in figure 1a. At each heating or cooling step, WAXS and SAXS patterns were simultaneously collected at consecutive heating and cooling increments at enamel and dentine or synthetic HAp crystallite across the specimens. A lightly compacted disc of NIST standard silicon powder was used for the WAXS data calibration, and a disc of silver behenate powder was used for the SAXS data calibration [24].
WAXS diffraction patterns (figure 1b) were recorded using a Pilatus 100K detector (Dectris, Baden, Switzerland) placed at a sample-to-camera distance of 275.55 mm (figure 1a). Further downstream of the beam a Pilatus 2M detector (Dectris) was positioned at a distance of 2892 mm to collect the SAXS patterns (figure 1c). In order to record simultaneously the WAXS and SAXS patterns at each scanning location, the WAXS detector was titled and offset at 11.5° to the incident beam.
2.3. X-ray scattering data analysis
2.3.1. Wide angle X-ray scattering data analysis
WAXS patterns appear graphically as separated rings with different widths and intensity levels, which contain characteristic information related to the crystalline structure. The analysis of WAXS patterns is performed using Bragg's law, which establishes the relationship between the spacing of atomic planes in crystals and the scattering angle at which these planes produce intense reflections.
| 2.1 |
where λ is the wavelength, 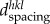 (d-spacing) is the interplanar distance between planes with Miller indices {hkl}, θ is one half of the scattering angle and n is the order of the reflection. Each peak corresponds to a certain family of lattice planes within a certain crystallographic phase. The changes of d-spacing could be used to examine the residual lattice strain and trace the phase transformation. Therefore, diffraction pattern analysis can be used to identify and quantify the crystallographic phases and structure parameters. The typical WAXS pattern is shown in figure 1b can be used to extract and compare the crystallographic properties (e.g. degree of crystal perfection) between different samples or the same samples at different stages of its evolution [20].
(d-spacing) is the interplanar distance between planes with Miller indices {hkl}, θ is one half of the scattering angle and n is the order of the reflection. Each peak corresponds to a certain family of lattice planes within a certain crystallographic phase. The changes of d-spacing could be used to examine the residual lattice strain and trace the phase transformation. Therefore, diffraction pattern analysis can be used to identify and quantify the crystallographic phases and structure parameters. The typical WAXS pattern is shown in figure 1b can be used to extract and compare the crystallographic properties (e.g. degree of crystal perfection) between different samples or the same samples at different stages of its evolution [20].
In figure 1b, the ring with the strongest intensity represents the diffraction from the (002) family of lattice planes. Moreover, the (002) lattice plane reflection is the most distinct ring that is reliably present in all three materials studied (dentine, enamel, synthetic HAp). WAXS data can be interpreted in terms of the shift of the diffraction peak obtained from a cluster of HAp crystallites. The variation of interplanar spacing (d-spacing) with respect to the temperature between the corresponding lattice planes was captured using WAXS. The (002) peak represents the orientation of the scattering vector along the hexagonal c-axis (parallel to the long direction) within the HAp crystallites. It also corresponds to the organic fibril orientation owing to the parallel orientation of fibrils to the long direction in HAp crystallites. The (002) peak is frequently used in determining the crystalline length in the bioapatite and thus was chosen as the principal peak considered in this study [25]. In detail, each two-dimensional diffraction image was firstly pre-processed using Fit2D [26]. The (002) peak of interest from each pattern was ‘caked’ (i.e. binned in the radial–azimuthal coordinates) within the range of 20° in the y-direction (figure 1b). Subsequently, the (002) peak was fitted with the Gaussian curve to obtain the peak centre position of 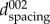 . Finite dimensions of the scattering volumes (crystallites) cause diffraction peak broadening that can be used to deduce the particle size. Diffraction peak width is related to the grain size (e.g. HAp crystallite) by the Scherrer equation that is widely used for the characterization of heat-treated mineralized tissues [9,10].
. Finite dimensions of the scattering volumes (crystallites) cause diffraction peak broadening that can be used to deduce the particle size. Diffraction peak width is related to the grain size (e.g. HAp crystallite) by the Scherrer equation that is widely used for the characterization of heat-treated mineralized tissues [9,10].
| 2.2 |
where L is the particle dimension in the direction of the {hkl} crystallographic plane normal, and 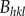 is the full-width at half maximum (FWHM) of the (hkl) peak. The (002) peak width is associated with the crystallite length and thus L deduced from the equation (2.2) of (002) peak could be used to estimate the average length of HAp crystallites [25]. The factor k is a constant related to the crystallite shape, whereas λ and θ have their usual meaning in Bragg's law. By monitoring the variation of FWHM during different heating–cooling procedures, the normalized length of the HAp crystallites was deduced.
is the full-width at half maximum (FWHM) of the (hkl) peak. The (002) peak width is associated with the crystallite length and thus L deduced from the equation (2.2) of (002) peak could be used to estimate the average length of HAp crystallites [25]. The factor k is a constant related to the crystallite shape, whereas λ and θ have their usual meaning in Bragg's law. By monitoring the variation of FWHM during different heating–cooling procedures, the normalized length of the HAp crystallites was deduced.
2.3.2. Small angle X-ray scattering data analysis
Quantitative interpretation of SAXS patterns provides insights into the mean thickness and degree of alignment of HAp crystallites. Two-dimensional diffraction images were initially converted into one-dimensional intensity profiles and processed using Fit2D software package [26].
(a) Mean thickness. In order to determine the mean crystal thickness, the scattering intensity I(q,φ) was radially integrated around the entire range of the azimuthal angle φ to obtain the function I(q), where q is the scattering vector. Based on Porod's law, which is valid in a two-phase system, the Porod chord length T is defined as:
| 2.3 |
where P is the Porod constant given by I(q) = Pq−4 (Porod's law describes the decay of intensity for large values of q), and Q represents the integrated area under the Iq2 ∼ q plot. Here, T is used as the definition of the mean crystal thickness without any assumption of the particle shape. Based on this, the actual thickness of the crystals can be calculated by taking further factors (e.g. volume fraction) into account. The length (period) of the scattering objects forming phase 1 (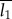 ) and of phase 2 (
) and of phase 2 (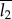 ) in a two-phase system is defined as [27]
) in a two-phase system is defined as [27]
| 2.4 |
where φ1 is the volume fraction of the scattering object. T can be interpreted as an average measure of the thickness and is close to the smaller dimension of the two phases. With the knowledge of φ1, T can be used to deduce the thickness of the scattering objects, with 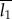 or
or 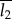 used as the guideline values for the mean thickness of crystallites.
used as the guideline values for the mean thickness of crystallites.
(b) Degree of alignment. The degree of alignment (ρ) of HAp crystallites (percentage of aligned particles) can also be determined from the SAXS patterns. It should be noted that for the enamel, owing to the dense distribution of crystals, the shortest range electron density changes occur in the gaps between crystalline particles, giving rise to the scattering signal [28]. The high volume fraction of HAp crystallites in enamel implies highly aligned tight packing, so that the orientation of the gaps between HAp crystallites corresponds closely to that of the HAp crystallites within the rods [28]. Thus, the information obtained from the value of ρ that describes the degree of alignment of the intercrystallite gaps can be used to deduce the degree of alignment of HAp crystallites in enamel.
Figure 1c gives an example SAXS pattern of enamel at room temperature. In order to quantify the degree of alignment, SAXS patterns were integrated over all spanned scattering vectors q, resulting in a function I(φ) that depends on the azimuthal angle φ [29,30]. The degree of alignment was determined as the ratio of the peak area to the overall area under the curve of I(φ), as has been explained and illustrated in previously published work [21]. The value of the degree of alignment ranges from 0 to 1, where ρ = 0 indicates no predominant orientation within the plane of the section (normal to the beam) and ρ = 1 indicates a perfect alignment of all crystallites [29,30].
(c) Scattering object analysis. The scattering contrast in SAXS arises mainly owing to the fluctuation of electron density. The intensity of SAXS signal is proportional to the square of the difference of electron density between the scattering object(s) as well as the volume fraction of the scattering object(s) [27]
| 2.5 |
where 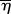 is the mean square fluctuation of electron density, ρ1 and ρ2 are the electron densities of the two scattering phases and φ1 is the volume fraction of the scattering object(s) as defined in equation (2.4). According to the Babinet principle [27], in terms of scattering, a system with a relatively low volume fraction of particles is equivalent to the dense system filled with particles but having the same volume fraction and arrangement of voids (i.e. the low density phase). In the former system, the particles act as scattering objects, whereas in the latter it is the voids.
is the mean square fluctuation of electron density, ρ1 and ρ2 are the electron densities of the two scattering phases and φ1 is the volume fraction of the scattering object(s) as defined in equation (2.4). According to the Babinet principle [27], in terms of scattering, a system with a relatively low volume fraction of particles is equivalent to the dense system filled with particles but having the same volume fraction and arrangement of voids (i.e. the low density phase). In the former system, the particles act as scattering objects, whereas in the latter it is the voids.
3. Results
3.1. Wide angle X-ray scattering data
3.1.1. d-spacing variation
The thermally induced variation of d-spacing of HAp crystallites in human dentine and enamel samples is illustrated in figure 2a,b. This is observed to be almost reversible during heating and cooling, although a visible drop at approximately 200°C is seen in the plot for dentine. By contrast, the synthetic HAp powders display a distinct irreversible change of d-spacing (figure 2c).
Figure 2.

(a–c) The changes in the d002-spacing of HAp crystallites in enamel, dentine and synthetic HAp powders samples, respectively, during heating and cooling stages (heating, red points; cooling, blue points). Intensity variation upon heating and cooling, caused by (d) HAp crystallites in enamel; (e) HAp crystallites in dentine and (f) synthetic HAp powders. The red marks in panel (f) indicate the occurrence of two new peaks. (Online version in colour.)
3.1.2. Crystal perfection
The sharp diffraction peaks of the natural enamel and the synthetic HAp powders show very small changes during the heating process (figure 2d,f). By contrast, the diffraction peaks of natural HAp crystallites in dentine are broad as shown in figure 2e. Furthermore, it is obvious that the peaks start to split and sharpen as the temperature increases, which indicates that the perfection increases remarkably during the heating, similar to the behaviour found in human bone [16,20]. During cooling, the degree of crystal perfection remained unchanged for all three materials. The peaks were indexed by reference to standard HAp patterns (JCPDS 9-432) [31].
3.1.3. Crystalline length
The interpretation of the (002) peak width provides information on the evolution of the crystallite length. Figure 3a–c demonstrates the variation of the normalized crystallite length of HAp crystallites in dentine and enamel and in synthetic HAp powders, with the value at the initial temperature serving as the reference (unity). Overall, not much change is observed in the HAp crystallite length in the enamel, although some oscillation is seen at medium temperatures (from approx. 300°C to approx. 600°C). In dentine, the HAp crystalline length remains almost constant until above approximately 600°C. Upon further heating, a significant length increase occurs, followed by the size remaining almost constant during cooling. In synthetic HAp powders, however, an apparent change of crystallite length occurs above approximately 500°C. The thermal history is different from that in dentine, with the crystallite length continuing to grow even during the cooling process (figure 3c).
Figure 3.

(a–c) The variation of the normalized length of HAp crystallites in human enamel, dentine and synthetic HAp powders (red points for heating and blue points for cooling). (d–f) The normalized mean thickness (with constant volume fraction) of HAp crystallites in enamel, dentine and synthetic HAp powders. (g–i) The normalized mean thickness accounting for the mineral volume fraction variation of HAp crystallites in enamel, dentine and synthetic HAp powders. (Online version in colour.)
3.2. Small angle X-ray scattering data
3.2.1. Scattering intensity and volume fraction variation
In the context of the Babinet principle, initially, the scattering objects in the dentine and synthetic HAp polycrystalline samples are the HAp crystallites. By contrast, in the enamel sample, initially the scattering objects are the gaps between HAp crystallites filled with organic matter. However, owing to the gradual denaturing and disappearance of the organic matrix as well as the accompanying process of crystal growth, the scattering objects in the dentine and HAp polycrystalline samples may also change to gaps. In order to verify the hypothesis that such change in the nature of the scattering objects in human dentine and synthetic HAp polycrystalline samples indeed takes place, the overall intensity variations from the SAXS patterns of the human dentine, enamel and synthetic HAp polycrystalline samples are plotted in figure 4a–c using open markers. The overall low intensity for the enamel sample indicates that the volume fraction of the scattering objects (gaps between crystallites) remains low. As for dentine and synthetic HAp crystallites, the observed large increase in the intensity coincides with the stage at which the HAp crystallites undergo fast growth, as reflected in the crystalline mean thickness plots in figure 3e,f.
Figure 4.

The correlation of the peak intensity variation from SAXS patterns with the mineral volume fraction for (a) human enamel, (b) human dentine and (c) synthetic HAp powders. Open purple markers (heating) and open blue markers (cooling) in panels (a–c) represent the results obtained from constant mineral volume fraction, whereas the red points (heating) and blue points (cooling) in panels (a–c) correspond to the results obtained with the variation of mineral volume fraction taken into account. (Online version in colour.)
Meanwhile, the volume fraction of the scattering objects, the HAp crystallites also increases until the transition temperature (approx. 612°C for dentine and approx. 745°C for synthetic HAp powders). At this point, the increased volume fraction of particles (and the reduced volume fraction of gaps) leads to the gaps between particles becoming the principal scattering objects, resulting in the dramatic decrease of intensity.
3.2.2. Crystalline mean thickness with constant volume fraction
The normalized crystallite mean thickness results for samples were calculated by SAXS interpretation using constant volume fraction of constituent phases (figure 3d–f). During the heating process, a hump is observed in figure 3e for dentine at approximately 300°C. Upon further heating, the mean thickness starts to increase in the range from approx. 500°C to approx. 700°C. This is followed by a slight decrease up to approximately 900°C. The mean thickness remains almost constant during cooling. The variation for the synthetic HAp powders (figure 3f) is similar, but appears smoother compared with that of HAp crystallites in dentine (figure 3e). Crystal growth in dentine appears to be faster than in the synthetic HAp powders. In addition, no obvious hump is observed at low temperature in the graph of the synthetic HAp powders (figure 3f). As for the mean thickness variation of HAp crystallites in the enamel, a drop is visible at low temperature (figure 3d). Beyond this drop, the overall variation is much smaller compared with the results for dentine and synthetic HAp crystallites.
3.2.3. Crystalline mean thickness with varied volume fraction
Based on the above analysis of the scattering objects, we postulate the variation of the volume fraction during heating and cooling as shown in figure 4, starting with the initial volume fraction obtained by TGA analysis. The volume fraction is assumed to remain constant during cooling. Note that the dramatic changes of intensity observed are associated with the change of the principal scattering objects that occurs when the volume fraction passes 50%. By taking the volume fraction variation of HAp crystallites in dentine, enamel and the synthetic HAp polycrystalline sample into account, the updated results of the crystalline mean thickness based on equation (2.4) are shown in figure 3g–i.
3.2.4. Degree of alignment
Because the synthetic HAp powders were produced from powdered HAp crystallites, they show an orientation distribution that is close to random at all temperatures. Thus, only the evolution of the degree of alignment of HAp crystallites in dentine and enamel is presented (figure 5). During heating, the overall degree of alignment of HAp crystallites decreases from 0.75 to 0.29 in the enamel, and from 0.17 to 0.05 in dentine. In more detail, in enamel, the degree of alignment remains almost constant at very low (approx. 0–100°C) and very high temperatures (approx. 800–900°C), whereas an increase occurs above approximately 300–400°C, followed by the decrease up to approximately 800°C. Contrary to the result for the enamel, dentine exhibited a longer low temperature range (approx. 0–300°C) with constant degree of alignment. Afterwards, the evolution follows a similar trend to that in the enamel, with an increase from approximately 300–400°C and a decrease from approximately 400–800°C. However, a more dramatic decrease occurred at approximately 800–900°C compared with that in the enamel at the same temperature range. The degree of alignment remained unchanged during cooling.
Figure 5.
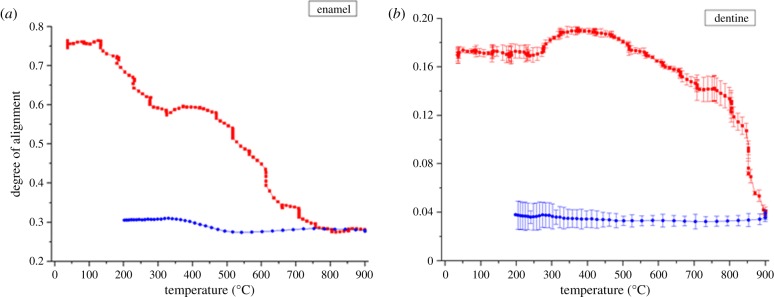
The variation of the degree of alignment of HAp crystallites upon heating/cooling in (a) human enamel and (b) human dentine (red points for heating and blue points for cooling). (Online version in colour.)
3.3. Thermogravimetry
Figure 6a shows the weight loss of dentine, enamel and the organic tooth sample (with most mineral content removed chemically) with increasing temperature. Two dramatic drops are visible in the pure organic tissue, at approximately 300°C and approximately 650°C, respectively. Around 750°C, the organic phase in the tooth almost completely disappears. As for dentine and enamel, the weight loss is observed to be less significant than in the pure organic tooth tissue. A dramatic drop is still apparent at approximately 350°C, and the rate of weight loss continues to decrease slowly at high temperature after approximately 750°C. The initial weight fraction of HAp crystallites in dentine and enamel can be calculated by monitoring the weight remaining at 750°C in dentine (70.3%) and enamel (95.8%), because the weight loss up to this temperature corresponds to that of water and the organic phase. Using the reported HAp density of 3.16 g cm−3 and the methods described in the literature [25], the initial volume fractions of HAp crystallites were calculated to be 45% (dentine) and 90% (enamel).
Figure 6.
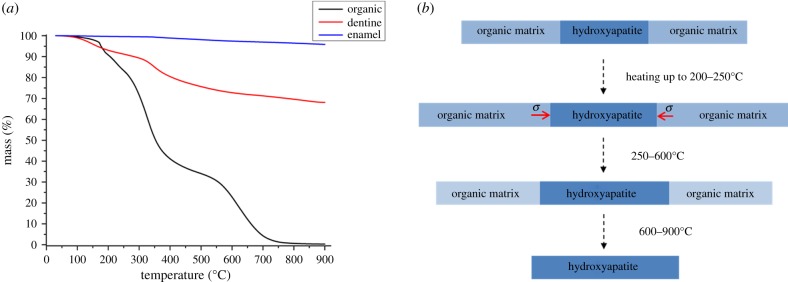
(a) TGA plot of weight loss of dental tissues: pure organic tooth with the inorganic content removed (black line), dentine (red line) and enamel (blue line). (b) A schematic of the ultrastructural alteration of organic matrix and HAp crystallites in human dentine upon heating. The process begins with the superior thermal expansion of the organic matrix compared with the HAp crystallites, resulting in compression of HAp during heating (red arrow). The compression relaxes gradually as the organic matrix begins to denature and burn off, leading to the additional expansion in the HAp crystallites in excess of the pure thermal strain. (Online version in colour.)
4. Discussion
The in situ thermal treatment in this study revealed the complete history of the evolution of the ultrastructure of bioapatite samples during a continuous heating–cooling schedule. In a previous ex situ study [9], only pointwise (in terms of temperature) and post-treatment data could be collected. This led to a fragmented and incomplete picture of the process. In comparison, this study provides reliable information on the sample heating history and ultrastructural response, with the consistency between samples assured by a degree of cross-comparison, full coverage of the entire temperature range and complete observation of the cooling procedure.
In general, SAXS patterns are more sensitive to the nanoscale structure, allowing the determination of the mean thickness of the crystallites by considering the scattering from two-phase systems [19]. The precise nature of the particles involved (e.g. amorphous versus crystalline) is not important in this instance. By contrast, WAXS information is extracted from crystal lattice diffraction, and its clarity depends on the degree of perfection of the crystallites.
The principal outcome of quantitative analysis of SAXS and WAXS patterns was to reveal the temperature-dependent variation of the nanostructure (thickness, orientation and degree of alignment) of HAp crystallites as well as the crystallographic properties (d-spacing, crystal perfection and length) variation during the heating and cooling processes. It is interesting to note how the observed evolution reflects the differences between dentine and enamel, and also the differences between the natural HAp crystallites embedded within organic matrix in human dental tissues, on the one hand, and synthetic HAp powders, on the other hand. The role played by the organic matrix during heating is particularly important, as it turns out to govern the evolution of size and strain of HAp crystallites. It is also important to draw the conclusion that in order to evaluate the exposure temperature precisely (e.g. in forensic cases), enamel and dentine must be investigated separately: the analysis of the entire powdered tooth obscures the sharpness of ultrastructural variations in dentine and enamel [6,32].
4.1. d-spacing variation
In the case of unconsolidated and unconstrained thermal expansion and contraction during heating and cooling, the d-spacing variation curves shown in figure 2a–c would be fully reversible. The observed difference between the heating–cooling curves in figure 2 may arise owing to the existence and relaxation or development of residual microstresses, and in conjunction with structural and compositional (e.g. phase) changes. The reduction in the lattice spacing that occurs in dentine at low temperature (see the curves in figure 2a,b), is likely to be related to the changes that take place in the organic matrix in dentine. The changes that occur in this matrix during heating are illustrated by the schematic diagram in figure 6b. At low temperatures, the organic matrix expands (swells) more than the HAp crystallites owing to its superior thermal expansion coefficient [33]. As a result, the HAp crystallites experience compression (as marked by the red arrow in figure 6b) that reaches a maximum when the protein matrix begins to denature at approximately 200°C. Above that temperature, the protein matrix begins to burn off and gradually disappears with increasing temperature, which is consistent with the TGA plot in figure 6a. In parallel with that, the compression in the HAp crystallites becomes relieved, and the crystallites experience additional expansion over and above the purely thermal strain.
Furthermore, it appears that the synthetic HAp powders experience a larger initial residual stress than HAp powders found in human dental tissue (figure 2a–c). Such differences may be the consequence of two phenomena. First, during sample preparation, a significant compressive stress was applied to the HAp powder to obtain powder discs with a similar density to the dentine tissue. By contrast, in the human dental tissue samples, some residual stresses may have been relaxed during the cutting of slices. Second, in the synthetic HAp powders, the appearance of a new phase is observed above 760°C (tricalcium phosphate, β-TCP). Such decomposition of HAp crystallites to β-TCP has been reported [34]. The phase change leads to large and irreversible changes of the lattice parameters, providing an explanation of the considerable difference in the d-spacing traces during heating and cooling. It is noted that although the β-TCP has also been observed in dentine (900°C) and enamel (800°C) as shown in figure 2d,e, its concentration is very low, and its effect on d-spacing variation can be ignored within the bounds of experimental accuracy, so that the curves appear reversible. The different transformation temperatures indicate an intrinsic difference in the characteristics between the bioapatites (such as the HAp crystallites in dentine and enamel) and the pure synthetic hydroxyapatites.
4.2. Crystal perfection
A high degree of crystal perfection is observed in the enamel and synthetic HAp crystallites compared with dentine, as shown in figure 2d–f. The difference arises from the different micro-morphology of HAp crystallites between the three materials studied. The improved perfection (sharper and stronger peaks) in the dentine with increasing temperature (figure 2e) is due to the sintering of HAp crystallites that takes place at high temperatures (above approx. 800°C). During the heating process, as the organic matrix is gradually burnt off, the interlayers of adjacent HAp crystallites disappear. Further heating leads to crystal growth and annealing of crystal defects, resulting in the significant peak sharpening. Further crystallization of the amorphous part observed in bone alteration [16,20] may also contribute to this effect.
4.3. Degree of alignment
The overall reduction of the degree of alignment in enamel and dentine during heating may be associated with the polydisperse distribution of the particles associated with the changes of crystalline size or shape at high temperature [20]. The initial stage of constant degree of alignment observed both in the enamel (RT-100°C) and dentine (RT-300°C) in figure 5b may correspond to the absence of crystallite rotation or growth. Once the organic matrix begins to denature, a slight increase in the degree of alignment is noted at approximately 300–400°C for both dentine and enamel. This increase corresponds to the gradual disappearance of the organic matrix accompanied by the relaxation of the residual strain. The greater reduction in the degree of alignment observed in dentine compared with enamel in the approximately 800–900°C range reflects the difference in the composite structures. At very high temperatures, the degree of alignment in the enamel remains high, owing to the highly directional structure of long prismatic HAp crystallites that remain aligned even after the organic matrix has been fully burnt off. By contrast, in dentine, the disappearance of the large amount (approx. 45% by volume) of organic matrix is likely to correspond to the loss of the supporting structure, causing the collapse and rearrangement of HAp crystallites that is associated with large rotation and anisotropic sintering.
4.4. Intensity variation
In the analysis of synchrotron-based SAXS data that has so far been reported in the literature, the volume fraction of HAp crystallites was always assumed to remain constant during heating [20]. However, the volume fraction in fact varies owing to the burning off of the protein matrix and the sintering of crystallites during heating (that influences the calculation of the mean crystallite thickness). The overall low intensity change indicates a small ultrastructural alteration which may reflect the limited space for crystal expansion and growth within the enamel sample. The difference between the dentine sample and synthetic HAp powders is also reflected in the scattering behaviour, where the comparatively lower value of intensity for dentine at low temperatures is likely to be associated with the presence of the protein matrix in dentine. Given the similar volume fractions of HAp crystallites in the two materials and close initial density values, the difference in the intensity is ascribed to the electron density contrast between the protein matrix and the crystals, which is smaller than that between gaps and the crystals (see equation (2.5)).
4.5. Crystallite size determined by small angle X-ray scattering/wide angle X-ray scattering
Combined with the WAXS characterization on the length calculation of HAp crystallites, a schematic of how the crystallite dimensions change in the dentine and enamel during heating is depicted in figure 7. Two temperature ranges are identified based on the processes taking place in the organic matrix (the separation temperature is around 500°C). As the organic phase gradually disappears, in ‘temperature range II’, crystal growth becomes unconstrained. Unlike the crystals in enamel, it is also observed that the crystallites in dentine gradually become misaligned, as indicated by the drop in the degree of alignment. Similar phenomena have been observed in ex situ bone characterization [35].
Figure 7.

A schematic of the size variation of HAp crystals within human enamel and dentine upon heating. The crystals in enamel are needle-shaped while those in dentine are platelet-shaped. In temperature range I, the interaction between crystals and the protein matrix occurs, whereas in temperature range II, the protein matrix gradually disappears, and the interaction is weakened. (Online version in colour.)
It is interesting to observe that in the synthetic HAp powders, continued sintering takes place during high temperature hold and even continues during cooling. This is probably due to the fact that sintering is driven by the overall change in the free energy of the system [36].
5. Conclusion
The effects of in situ heat treatment on the hydroxyapatite (HAp) crystallites (human dental tissues) and synthetic HAp crystallites were explored using combined synchrotron-based SAXS/WAXS techniques. From the quantitative analysis of SAXS and WAXS patterns, multi-scale characterization of the hierarchical structure of enamel and dentine establishes the temperature-dependent variation of the nanostructural parameters (thickness, orientation and the degree of alignment) of HAp crystallites from SAXS and crystallographic properties (d-spacing, crystal perfection and length) from WAXS during the heating and cooling processes. The results generally reflect the difference between dentine and enamel, natural and synthetic HAp crystallites. In the analysis presented, the emphasis is placed on understanding the role of the organic matrix during heating. An important observation is made of the fact that the scattering object may change with the volume fraction variation of HAp crystallites during heating.
In conclusion, the synchrotron-based combined SAXS/WAXS analysis has been shown to be a powerful method for the determination of nanostructure variation induced by thermal treatment in human enamel and dentine as well as in synthetic HAp crystallites. The in situ thermal treatment conducted in this study covered the entire relevant temperature range and revealed the complete continuous history of ultrastructure evolution within a single sample during a continuous heating–cooling schedule. It was clearly demonstrated that insignificant structural changes occur during cooling, indicating that relevant conclusions can be drawn when dental remains samples are analysed as part of archaeological or forensic studies. The approach to the study of ultrastructural alteration in skeletal hard tissues exposed to in situ thermal treatment developed in this study provides a better, more reliable basis for deducing the heating history compared with conventional methods based on the monitoring the macro- and microstructural colour [6,7,37]. Furthermore, it allows the interaction between the mineral crystallites and the organic phase during heating to be captured. These advantages show the superior utility of in situ thermal treatment analysis over previous ex situ tests. Statistical error analysis of multiple measurement points in dentine confirmed the reliability of the conclusions drawn. The results of this work will also be beneficial to the optimization of the laser fluence used in dental practice, and in the future design of biomimetic materials.
Acknowledgement
Diamond Light Source is acknowledged for providing the beam time allocation under experiment number SM7981.
Funding statement
A.M.K. acknowledges support of the EPSRC through grants EP/I020691 ‘Multi-disciplinary Centre for In situ Processing Studies (CIPS)’, EP/G004676 ‘Micromechanical Modelling and Experimentation’ and EP/H003215 ‘New Dimensions of Engineering Science at Large Facilities’.
References
- 1.Marshall SJ, Balooch M, Habelitz S, Balooch G, Gallagher R, Marshall GW. 2003. The dentin–enamel junction: a natural, multilevel interface. J. Eur. Ceram. Soc. 23, 2897–2904. ( 10.1016/S0955-2219(03)00301-7) [DOI] [Google Scholar]
- 2.Dorozhkin SV. 2010. Nanosized and nanocrystalline calcium orthophosphates. Acta Biomater. 6, 715–734. ( 10.1016/j.actbio.2009.10.031) [DOI] [PubMed] [Google Scholar]
- 3.Kantola S. 1973. Laser-induced effects on tooth structure 0.8. X-ray-diffraction study of dentin exposed to a CO2-laser. Acta Odontol. Scand. 31, 381–386. ( 10.3109/00016357309002525) [DOI] [PubMed] [Google Scholar]
- 4.Fried D, Zuerlein MJ, Le CQ, Featherstone JDB. 2002. Thermal and chemical modification of dentin by 9–11-μm CO2 laser pulses of 5–100-μs duration. Laser Surg. Med. 31, 275–282. ( 10.1002/Lsm.10100) [DOI] [PubMed] [Google Scholar]
- 5.Zuerlein MJ, Fried D, Featherstone JDB. 1999. Modeling the modification depth of carbon dioxide laser-treated dental enamel. Laser Surg. Med. 25, 335–347. () [DOI] [PubMed] [Google Scholar]
- 6.Piga G, Thompson TJU, Malgosa A, Enzo S. 2009. The potential of X-ray diffraction in the analysis of burned remains from forensic contexts. J. Forensic Sci. 54, 534–539. ( 10.1111/j.1556-4029.2009.01037.x) [DOI] [PubMed] [Google Scholar]
- 7.Thompson TJU. 2005. Heat-induced dimensional changes in bone and their consequences for forensic anthropology. J. Forensic Sci. 50, 1008–1015. ( 10.1520/JFS2004297) [DOI] [PubMed] [Google Scholar]
- 8.Thompson TJU, Islam M, Piduru K, Marcel A. 2011. An investigation into the internal and external variables acting on crystallinity index using Fourier transform infrared spectroscopy on unaltered and burned bone. Palaeogeogr. Palaeoclimatol. 299, 168–174. ( 10.1016/j.palaeo.2010.10.044) [DOI] [Google Scholar]
- 9.Sui T, Sandholzer MA, Le Bourhis E, Baimpas N, Landini G, Korsunsky AM. 2014. Structure–mechanical function relations at nano-scale in heat-affected human dental tissue. J. Mech. Behav. Biomed. Mater. 32, 113–124. ( 10.1016/j.jmbbm.2013.12.014) [DOI] [PubMed] [Google Scholar]
- 10.Shipman P, Foster G, Schoeninger M. 1984. Burnt bones and teeth: an experimental-study of color, morphology, crystal-structure and shrinkage. J. Archaeol. Sci. 11, 307–325. ( 10.1016/0305-4403(84)90013-X) [DOI] [Google Scholar]
- 11.Enzo S, Bazzoni M, Mazzarello V, Piga G, Bandiera P, Melis P. 2007. A study by thermal treatment and X-ray powder diffraction on burnt fragmented bones from tombs II, IV and IX belonging to the hypogeic necropolis of ‘Sa Figu’ near Ittiri, Sassari (Sardinia, Italy). J. Archaeol. Sci. 34, 1731–1737. ( 10.1016/j.jas.2006.12.011) [DOI] [Google Scholar]
- 12.Rogers KD, Daniels P. 2002. An X-ray diffraction study of the effects of heat treatment on bone mineral microstructure. Biomaterials 23, 2577–2585. ( 10.1016/S0142-9612(01)00395-7) [DOI] [PubMed] [Google Scholar]
- 13.Beckett S, Rogers KD, Clement JG. 2011. Inter-species variation in bone mineral behavior upon heating. J. Forensic Sci. 56, 571–579. ( 10.1111/j.1556-4029.2010.01690.x) [DOI] [PubMed] [Google Scholar]
- 14.Reyes-Gasga J, et al. 2008. Structural and thermal behaviour of human tooth and three synthetic hydroxyapatites from 20 to 600 degrees C. J. Phys. D, Appl. Phys. 41, 225407 ( 10.1088/0022-3727/41/22/225407) [DOI] [Google Scholar]
- 15.Hollund HI, Ariese F, Fernandes R, Jans MME, Kars H. 2013. Testing an alternative high-throughput tool for investigating bone diagenesis: FTIR in attenuated total reflection (ATR) mode. Archaeometry 55, 507–532. ( 10.1111/j.1475-4754.2012.00695.x) [DOI] [Google Scholar]
- 16.Rogers K, Beckett S, Kuhn S, Chamberlain A, Clement J. 2010. Contrasting the crystallinity indicators of heated and diagenetically altered bone mineral. Palaeogeogr. Palaeocl. 296, 125–129. ( 10.1016/j.palaeo.2010.06.021) [DOI] [Google Scholar]
- 17.Squires KE, Thompson TJU, Islam M, Chamberlain A. 2011. The application of histomorphometry and Fourier transform infrared spectroscopy to the analysis of early Anglo-Saxon burned bone. J. Archaeol. Sci. 38, 2399–2409. ( 10.1016/j.jas.2011.04.025) [DOI] [Google Scholar]
- 18.Piga G, Malgosa A, Thompson TJU, Enzo S. 2008. A new calibration of the XRD technique for the study of archaeological burned human remains. J. Archaeol. Sci. 35, 2171–2178. ( 10.1016/j.jas.2008.02.003) [DOI] [Google Scholar]
- 19.Hiller JC, Wess TJ. 2006. The use of small-angle X-ray scattering to study archaeological and experimentally altered bone. J. Archaeol. Sci. 33, 560–572. ( 10.1016/j.jas.2005.09.012) [DOI] [Google Scholar]
- 20.Hiller JC, Thompson TJ, Evison MP, Chamberlain AT, Wess TJ. 2003. Bone mineral change during experimental heating: an X-ray scattering investigation. Biomaterials 24, 5091–5097. ( 10.1016/S0142-9612(03)00427-7) [DOI] [PubMed] [Google Scholar]
- 21.Sui T, et al. 2013. Multi-scale modelling and diffraction-based characterization of elastic behaviour of human dentine. Acta Biomater. 9, 7937–7947. ( 10.1016/j.actbio.2013.04.020) [DOI] [PubMed] [Google Scholar]
- 22.Sui T, Sandholzer MA, Baimpas N, Dolbnya IP, Landini G, Korsunsky AM. 2013. Hierarchical modelling of elastic behaviour of human enamel based on synchrotron diffraction characterization. J. Struct. Biol. 184, 136–146. ( 10.1016/j.jsb.2013.09.023) [DOI] [PubMed] [Google Scholar]
- 23.Dent AJ, et al. 1995. A new furnace design for use in combined X-ray-absorption and diffraction of catalysis and ceramics studies: formation from carbonate precursors of Cu, Co, Mn spinels for the oxidation of CO and the formation of Plzt, a piezoelectric ceramic. Nucl. Instrum. Methods B 97, 20–22. ( 10.1016/0168-583X(94)00369-6) [DOI] [Google Scholar]
- 24.Huang TC, Toraya H, Blanton TN, Wu Y. 1993. X-ray-powder diffraction analysis of silver behenate, a possible low-angle diffraction standard. J. Appl. Cryst. 26, 180–184. ( 10.1107/S0021889892009762) [DOI] [Google Scholar]
- 25.Deymier-Black AC, Almer JD, Stock SR, Haeffner DR, Dunand DC. 2010. Synchrotron X-ray diffraction study of load partitioning during elastic deformation of bovine dentin. Acta Biomater. 6, 2172–2180. ( 10.1016/j.actbio.2009.11.017) [DOI] [PubMed] [Google Scholar]
- 26.Hammersley AP. 1997. ‘FIT2D: an introduction and overview’. ESRF Internal Report 1997.
- 27.Glatter O, Kratky O. 1982. Small angle X-ray scattering. London, UK: Academic Press. [Google Scholar]
- 28.Tanaka T, Yagi N, Ohta T, Matsuo Y, Terada H, Kamasaka K, To-o K, Kometani T, Kuriki T. 2010. Evaluation of the distribution and orientation of remineralized enamel crystallites in subsurface lesions by X-ray diffraction. Caries Res. 44, 253–259. ( 10.1159/000314672) [DOI] [PubMed] [Google Scholar]
- 29.Tesch W, Vandenbos T, Roschgr P, Fratzl-Zelman N, Klaushofer K, Beertsen W, Fratzl P. 2003. Orientation of mineral crystallites and mineral density during skeletal development in mice deficient in tissue nonspecific alkaline phosphatase. J. Bone Miner. Res. 18, 117–125. ( 10.1359/jbmr.2003.18.1.117) [DOI] [PubMed] [Google Scholar]
- 30.Tesch W, Eidelman N, Roschger P, Goldenberg F, Klaushofer K, Fratzl P. 2001. Graded microstructure and mechanical properties of human crown dentin. Calcified Tissue Int. 69, 147–157. ( 10.1007/s00223-001-2012-z) [DOI] [PubMed] [Google Scholar]
- 31.Ooi CY, Hamdi M, Ramesh S. 2007. Properties of hydroxyapatite produced by annealing of bovine bone. Ceram. Int. 33, 1171–1177. ( 10.1016/j.ceramint.2006.04.001) [DOI] [Google Scholar]
- 32.Kugler M. 2003. X-ray diffraction analysis in forensic science: the last resort in many criminal cases. Adv. X-ray Anal. 46, 1–16. [Google Scholar]
- 33.Lei HJ, Liu B, Fang DN. 2010. The coefficient of thermal expansion of biomimetic composite. Front. Mater. Sci. China 4, 234–238. ( 10.1007/s11706-010-0088-y) [DOI] [Google Scholar]
- 34.Elliott JC. 1994. Structure and chemistry of the apatites and other calcium orthophosphates. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 35.Pramanik S, Hanif ASM, Pingguan-Murphy B, Abu Osman NA. 2013. Morphological change of heat treated bovine bone: a comparative study. Materials 6, 65–75. ( 10.3390/Ma6010065) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kingery WD, Bowen HK, Uhlmann DR. 1976. Introduction to ceramics, 2nd edn New York, NY: Wiley. [Google Scholar]
- 37.Thompson TJU, Islam M, Bonniere M. 2013. A new statistical approach for determining the crystallinity of heat-altered bone mineral from FTIR spectra. J. Archaeol. Sci. 40, 416–422. ( 10.1016/j.jas.2012.07.008) [DOI] [Google Scholar]