

Abstract
Sphingosine-1-phospate is a lipid mediator that has been implicated in protection from acute kidney injury (AKI) by activation of the sphingosine-1-phosphate receptor S1P1R. A recent study demonstrates that mice with induced deletion of S1P1R on endothelial cells experience increased ischemia-induced AKI. These findings have important translational implications. Indeed, S1P1R agonists have been used for the treatment of patients suffering from autoimmune encephalitis. As such, endothelial S1P1R-signaling could be targeted for AKI prevention in surgical patients.
Keywords: Sphingosine, sphingosine-1-phosphate, adenosine, ischemia, acute kidney injury
Acute kidney injury (AKI) is defined by a rapid loss in kidney function – a disease process that contributes significantly to morbidity and mortality of hospitalized patients1. One of the leading causes of AKI is renal ischemia, such as occurs in patients undergoing major surgery. Particularly, patients undergoing cardiac surgery, thoracic surgery and aortic surgery involving cross-clamping of the aorta are very prone to developing AKI in their perioperative course. The search for novel pharmacologic approaches for AKI treatment or prevention is an area of intense research. Many animal studies show much higher effectiveness for prophylactic treatment approaches of AKI, as compared to treatment studies fully developed AKI. Therefore, surgical patients undergoing surgical procedures with a high risk for developing AKI are an ideal population for the clinical development of preventive AKI treatment approaches. Unfortunately, there are currently no pharmacologic approaches available that are shown to prevent AKI in this patient population1.
Particularly proximal tubular epithelial cells are prone to necrosis and apoptosis induced by renal ischemia. While tubular injury is a critical step for AKI pathogenesis, it is now being appreciated that the intimate interaction between tubular epithelial and other tissue compartments of the kidneys – such as pericytes, resident dendritic cells and vascular endothelial cells - is central to AKI development. Indeed, several studies implicate vascular dysfunction – such as endothelial barrier dysfunction, or persistent reduction in post-ischemic glomerular perfusion - in the pathophysiology of ischemic AKI. For example, the extracellular signaling molecule adenosine has recently been implicated in post-ischemic control of renal perfusion via a crosstalk between renal adenosine transporters and adenosine receptors exclusively expressed on vascular endothelial cells2.
Additional evidence supporting a central role for endothelial dysfunction during AKI comes from a study published in the current edition of Kidney International. The research team of Dr. H. Thomas Lee provides compelling evidence that deletion of the endothelial-expressed sphingosine-1-phosate 1 receptor (S1P1R) is associated with the exacerbation of ischemic AKI3. Sphingolipid metabolites are emerging as an important molecular group of lipid signaling molecules. Sphingolipids form a class of lipids characterized by the aliphatic amino-alcohol sphingosine. Sphingosine-1-phosphate is produced by phosphorylation of sphingosine by sphingosine kinases. Sphingosine-1-phosphate is the natural ligand for a family of five lysophospholipid targeted G-protein coupled receptors.
Previous studies from the laboratory of Dr. Lee showed a critical role for proximal tubule sphingosine kinase-1 in mediating adenosine-dependent kidney protection during AKI4. These studies indicate that sphingosine kinase and sphingosine-1-phosphate synthesis converge as a downstream mechanisms for adenosine-mediated kidney protection, thereby providing a molecular explanation for adenosine-dependent kidney protection during AKI4. During ischemia and reperfusion, the nucleotide adenosine triphosphate (ATP) is released from multiple cellular sources, including for example vascular endothelial cells5, epithelia or activated inflammatory cells6. Once released into the extracellular compartment, ATP is enzymatically converted to adenosine6. This pathway is under the enzymatic control of ecto-nucleotidases – such as the ecto-5’-nucleotidase CD73 – which are induced during conditions of ischemia or hypoxia7, 8. This pathway leads to dramatic increases in the production and signaling effects of extracellular adenosine, and concomitant kidney protection during ischemia1. As such, the current findings from the research team of Dr. Lee of vascular S1P1R signaling in preventing endothelial dysfunction during ischemic AKI are consistent with findings supporting a functional role for adenosine signaling in kidney protection via enhancing vascular functions2.
As next step, Dr. Lee and his colleagues set out to define the tissue-specific contributions for sphingosine-1-phosphate signaling during ischemic AKI. They hypothesized a functional role for endothelial S1P1R signaling to kidney protection during renal ischemia. To address this hypothesis, they took a very elegant and technically highly involving molecular approach. Indeed, they were able to generate a novel mouse line with induced deletion of S1P1R in vascular endothelial cells, including the kidneys3. This was achieved by crossing transgenic mice with a “floxed” S1P1R gene with mice expressing a tamoxifen-inducible form of Cre-recombinase in endothelial cells [Platelet-derived growth factor-b (Pdgfb) Cre+ mice). Indeed, S1P1Rf/f PdgfbiCreER mice have normal S1P1R expression, until they are exposed to tamoxifen. Taken the extra effort for generating an inducible triple-transgenic mouse line was necessary to study the role of endothelial S1P1R signaling during ischemic AKI, as germline global or endothelial S1P1R deletion results in embryonic lethality3. This elegant model revealed that endothelial-specific S1P1R deletion is associated with increased renal tubular necrosis, inflammation, impaired vascular permeability and exacerbated renal tubular apoptosis after ischemic AKI. As mechanism for exacerbated AKI in mice with induced endothelial-specific S1P1R deletion, the authors identified a reduction of heat-shock protein HSP27 expression.
These findings are very interesting in the context of a previous studies showing that S1P1R agonist-elicited kidney protection involves – at least in part - S1P1R signaling on proximal tubules9. This study demonstrated a function of S1P1R in kidney protection independent from the S1P1R-induced lymphopenia. Indeed, the authors demonstrated that kidney protection by S1PR agonists are independent from T and B lymphocytes, Indeed, administration of the nonselective S1PR agonist FTY720 or the selective S1P1R agonist SEW2871 reduced AKI in Rag-1 knockout mice, which are characterized by a lack of T and B lymphocytes. Based on findings showing that SEW2871 significantly attenuated apoptosis in cultured mouse proximal tubule epithelial cells, the authors went on to examine the functional role of S1P1R in proximal tubules in vivo. Indeed, mice deficient in proximal tubule S1P1R experienced a greater decline in renal function after IRI than control mice and were no longer protected by S1P1R agonist treatment9.
The findings of both studies3, 9 are very important from a translational perspective. For example, fingolimod (FTY720) is an immunomodulating drug that is approved for the treatment of patients suffering from multiple sclerosis. Therapeutic effects of fingolimod are related to its function as sphingosine 1-phosphate receptor activator, which leads to the sequenstrcation of lymphocytes in lymph nodes, thereby preventing them from contributing to autoimmune-mediated encephalitis. As discussed above, fingolimod is highly protective in ischemic AKI independent of S1P1R-induced lymphopenia, but functions through activation of S1P1Rs in the kidney9. It seems likely that both mechanisms - tubular epithelial and endothelial S1P1R signaling events - contribute to the kidney protection. On the one hand, tubular epithelial cells are the highly sensitive to ischemia-induced cell death, and their protection will be critical for improving outcomes of ischemic AKI. However, there are many instances where vascular dysfunction precedes tubular injury. As such, the exact conditions of renal ischemia, and the pharmacologic properties of S1P1R agonists will be important in combining tubular and vascular protection. It will be exciting to see these findings going forward from bench to bedside. Indeed, several clinical trials are registered to examine the effects of fingolimod during kidney transplantation (www.clinicaltrial.gov). However, these studies mostly focus on utilizing fingolimod for the prevention of lymphocyte-mediated immune responses to the transplanted kidney10. To move the field of perioperative kidney protection forward, studies of S1P1R agonists in surgical patients will be critical. For example, it is conceivable that fingolimod could prevent AKI in high risk surgical patients.
Figure 1.
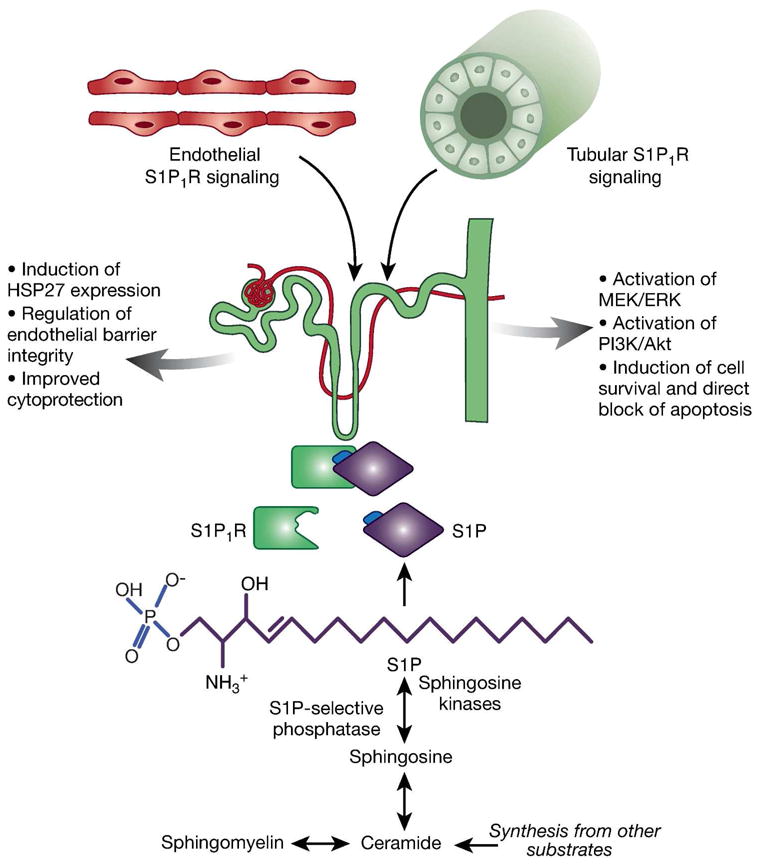
Effects of vascular endothelial and tubular sphingosine-1-phosphate receptor (S1P1R) signaling in ischemic Acute Kidney Injury (AKI). Sphingosine-1-phosphate (S1P) is synthesized from the sphingolipids sphingosine, sphingomyelin, ceramide, and others. S1P synthesis is dependent on the activity of sphingosine kinases and S1P-selective phosphorylase, as they regulate phophorylation of sphingosine to S1P (marked with blue font in figure). As a ligand for S1P1R, S1P initiates various signaling pathways. Endothelial-specific S1P1R deletion is associated with increased renal tubular necrosis, inflammation, impaired vascular permeability and exacerbates renal tubular apoptosis after ischemic AKI. One mechanism for the protective effects of endothelial S1P1R activation lies within the induction of heat-shock protein (HSP)273. In proximal tubule, S1P1R activation leads to direct blockage of apoptosis and induction of cell survival via activation of mitogen-activated protein kinase/extracellular regulated kinase (MEK/ERK) and phosphatidylinositol-3-kinase (PI3K)/Akt induced pathways9.
Acknowledgments
This paper was supported by National Institute of Health Grants R01 DK097075, R01-HL0921, R01-DK083385, R01- HL098294, POIHL114457-01 and a grant by the Crohn’s and Colitis Foundation of America (CCFA) to HKE and a grant by the American Heart Association to A.G.
Disclosure of funding: This work was supported by an American Heart Association (Dallas, TX, USA) grant to Almut Grenz, and National Institutes of Health (Bethesda, MD, USA) grants R01-DK097075, R01-HL0921, R01-DK083385, R01-HL098294, POIHL114457-01, and a grant by the Crohn’s and Colitis Foundation of America (CCFA, New York, NY, USA) to Holger K. Eltzschig.
This is a commentary on article Ham A, Kim M, Kim JY, Brown KM, Fruttiger M, D'Agati VD, Thomas Lee H. Selective deletion of the endothelial sphingosine-1-phosphate 1 receptor exacerbates kidney ischemia-reperfusion injury. Kidney Int.2013 Sep;
Footnotes
Conflicts of interest statement: The authors declare no competing interests.
References
- 1.Bauerle JD, Grenz A, Kim JH, et al. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol. 2011;22:14–20. doi: 10.1681/ASN.2009121217. [DOI] [PubMed] [Google Scholar]
- 2.Grenz A, Bauerle JD, Dalton JH, et al. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. The Journal of clinical investigation. 2012;122:693–710. doi: 10.1172/JCI60214. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 3.Ham A, Kim M, Kim YJ, et al. Selective deletion of endothelial sphingosine-1-phosphate 1 receptor exacerbates kidney ischemia-reperfusion injury. Kidney Int. 2013 doi: 10.1038/ki.2013.345. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Park SW, Kim M, Kim JY, et al. Proximal tubule sphingosine kinase-1 has a critical role in A1 adenosine receptor-mediated renal protection from ischemia. Kidney Int. 2012;82:878–891. doi: 10.1038/ki.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Faigle M, Seessle J, Zug S, et al. ATP release from vascular endothelia occurs across Cx43 hemichannels and is attenuated during hypoxia. PLoS ONE. 2008;3:e2801. doi: 10.1371/journal.pone.0002801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eltzschig HK, Macmanus CF, Colgan SP. Neutrophils as Sources of Extracellular Nucleotides: Functional Consequences at the Vascular Interface. Trends Cardiovasc Med. 2008;18:103–107. doi: 10.1016/j.tcm.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hart ML, Henn M, Kohler D, et al. Role of extracellular nucleotide phosphohydrolysis in intestinal ischemia-reperfusion injury. FASEB J. 2008;22:2784–2797. doi: 10.1096/fj.07-103911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hart ML, Much C, Gorzolla IC, et al. Extracellular adenosine production by ecto-5′-nucleotidase protects during murine hepatic ischemic preconditioning. Gastroenterology. 2008;135:1739–1750. doi: 10.1053/j.gastro.2008.07.064. e1733. [DOI] [PubMed] [Google Scholar]
- 9.Bajwa A, Jo SK, Ye H, et al. Activation of sphingosine-1-phosphate 1 receptor in the proximal tubule protects against ischemia-reperfusion injury. J Am Soc Nephrol. 2010;21:955–965. doi: 10.1681/ASN.2009060662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoitsma AJ, Woodle ES, Abramowicz D, et al. FTY720 combined with tacrolimus in de novo renal transplantation: 1-year, multicenter, open-label randomized study. Nephrol Dial Transplant. 2011;26:3802–3805. doi: 10.1093/ndt/gfr503. [DOI] [PubMed] [Google Scholar]