

Abstract
Children with systemic Juvenile Idiopathic Arthritis (sJIA), the most severe subtype of JIA, are at risk from destructive polyarthritis and growth failure, and corticosteroids as part of conventional treatment can result in osteoporosis and growth delay. In children where there is failure or toxicity from drug therapies, disease has been successfully controlled by T-cell-depleted autologous stem cell transplantation (ASCT). At present, the immunological basis underlying remission after ASCT is unknown. Immune reconstitution of T cells, B cells, natural killer cells, natural killer T cells and monocytes, in parallel with T-cell receptor (TCR) diversity by analysis of the β variable region (TCRVb) complementarity determining region-3 (CDR3) using spectratyping and sequencing, were studied in five children with sJIA before and after ASCT. At time of follow up (mean 11·5 years), four patients remain in complete remission, while one child relapsed within 1 month of transplant. The CD8+ TCRVb repertoire was highly oligoclonal early in immune reconstitution and re-emergence of pre-transplant TCRVb CDR3 dominant peaks was observed after transplant in certain TCRVb families. Further, re-emergence of pre-ASCT clonal sequences in addition to new sequences was identified after transplant. These results suggest that a chimeric TCR repertoire, comprising T-cell clones developed before and after transplant, can be associated with clinical remission from severe arthritis.
Keywords: autologous stem cell transplantation, childhood arthritis, immune reconstitution, T-cell receptor repertoire clonal diversity
Introduction
Juvenile idiopathic arthritis (JIA) is a common childhood autoimmune disease, and encompasses a heterogeneous group of disorders classified according to clinical features observed during the first 6 months of disease.1 Systemic JIA (sJIA), still currently classified as a subtype of JIA, is characterized by arthritis, quotidian fever, erythematous rash, lymphadenopathy, hepatosplenomegaly and serositis and has a risk of associated mortality if not well controlled.2 However, given its features of systemic inflammation, sJIA is now considered to be more similar to an autoinflammatory disorder and may be considered to have separate pathogenesis from other JIA subtypes.3
Despite significant advances in the treatment of sJIA, there remain children who suffer from active disease that is refractory to novel anti-rheumatic drugs. These children can endure considerable complications secondary to chronic inflammation and cumulative drug toxicity, including joint erosions, osteoporosis and growth failure.4
A number of children with very severe and refractory disease have been successfully treated by autologous stem cell transplantation (ASCT).5–9 Those who achieve drug-free remission after transplant benefit significantly from a catch up of growth and experience a considerable improvement in psychosocial morbidity and quality of life.
However, although outcomes can be highly successful, the risks associated with ASCT are also significant: intense immunosuppression and prolonged T-cell lymphopenia can lead to serious infection and exacerbation of macrophage activation syndrome, and these complications are associated with a high mortality.5,7,10 Therefore, it is crucial that only children who are least expected to suffer from complications and have a good prospect of remission from disease are identified for transplantation.
A number of other autoimmune disorders, including multiple sclerosis (MS), rheumatoid arthritis and systemic lupus erythematosus, have also been managed successfully by ASCT.11–14 However, despite encouraging results, similar to those challenges faced in sJIA, stem cell mobilization, immune ablation and stem cell infusion in some patients are associated with multiple adverse events, including hepatic toxicity, cardiac failure and infection. Furthermore, the response to ASCT varies considerably between patients and it is challenging to determine which patients will have disease remission. Interestingly, it has been observed that younger patients suffering from MS had significantly higher remission probabilities in comparison to older patients.11
It has been suggested that high-dose immunosuppression administered as part of the ASCT protocol might contribute towards the elimination of autoreactive lymphocytes mediating the pathological autoimmune response.15,16 However, to date, the immunological basis of remission is poorly understood. Numerous studies investigating ASCT for the management of severe autoimmune diseases report that remission is associated with a marked expansion of recent thymic emigrants and the emergence of a new and more diverse T-cell receptor (TCR) repertoire, consistent with the generation of a ‘new’ T-cell compartment of the immune system.17,18
However, one recent study investigating ASCT in MS identified the re-emergence of autoreactive T cells, by expanding T cells on known antigens, in the context of remission. It was suggested that these antigens might have played a role in pathology before transplant.19 Another study exploring immune reconstitution in JIA showed an increased regulatory activity in autoreactive T-cell clones post transplant.6 Furthermore, a preferential reconstitution of CD4+ CD25bright T regulatory cells in the thymus was observed. Together, these might suggest that induction of self-tolerance and remission from disease could be achieved through the ‘resetting’ of immune balance.
Of note, a predominance of CD45RO+ memory T cells is observed very early post transplant in the treatment of various autoimmune and malignant conditions.7,17,20–22 It is yet to be confirmed whether this can be attributed to the persistence of pre-primed, pre-transplant T cells with greater capacity for expansion compared with de novo generated cells, acquisition of CD45RO expression by rapidly proliferating naive T cells, or both.
To further understand the immunological mechanisms underlying remission from sJIA after ASCT, in this study we investigated the immune reconstitution and T-cell repertoire of children with sJIA undergoing transplantation. Results indicate that remission from severe arthritis can be associated with an immune system comprising of re-emerging T-cell clones that were previously identified before transplant as well as de novo generated clones. In addition, preliminary data from one patient who relapsed shortly post transplant suggest that the presence of full-length TCR complementarity determining region-3 (CDR3) diversity early during immune reconstitution might be associated with an inadequate conditioning regimen, inadequate immune depletion and relapse of disease.
These results, together with past and future studies, may help to elucidate which patients are most likely to benefit from ASCT, and may help to determine optimal conditioning regimens for induction of remission while minimizing risks associated with intense immunosuppressive therapy.
Materials and methods
Patient samples and cell preparation
Peripheral venous blood samples were obtained from five children with sJIA before, 1 month, 3–12 months and 2–3 years after ASCT, with fully informed parental consent and age appropriate child assent. The study had full ethical approval. Peripheral blood mononuclear cells were prepared by density gradient centrifugation using lymphoprep (Axis-Shield, Dundee, UK).
Immunophenotypic analysis
Peripheral blood mononuclear cells were assessed for expression of T-cell, B-cell, natural killer (NK) cell, NK-T-cell and monocyte surface markers by flow cytometry [CD3, CD19, CD16, CD14, CD4, CD8, CD45RA and CD45RO, using the following fluorochromes: FITC, phycoerythrin, PC7, allophycocyanin, Peridinin chlorophyll protein-Cy5.5, Qdot605, allophycocyanin-Cy7 and V450, purchased from: BD (Franklin Lakes, NJ), Life Technologies (Carlsbad, CA), eBioscience (San Diego, CA) or Beckman Coulter (Brea, CA)]. TCRVb staining was performed using the iotest® Beta Mark Kit (Beckman Coulter) according to the manufacturer's instructions. LIVE/DEAD Fixable blue Dead Cell Stain (Life Technologies) was used to exclude dead cells. Cytometric analysis was performed using LSRII or FACScan (both BD) and flowjo (Treestar Inc., Ashland, OR).
TCR repertoire analysis
CD4+ and CD8+ T-cell populations were separated using CD4+-positive selection magnetic bead sorting (Miltenyi Biotec, Bergisch Gladbach, Germany) and the CD4− fraction was used as the source of CD8+ T cells. Sort purity for CD4+ sorted cells was typically ≥ 90%. Messenger RNA was extracted from sorted cells using RNAzol (Biogenesis, Westminster, CO) and cDNA was synthesized using SuperScript reverse transcriptase and Oligo-dT (both Life Technologies). The TCR β variable regions (TCRVb) were amplified from cDNA using Vb family primers (see Supporting information, Table S1). The following cycling conditions were used: 95° for 25 seconds, 35 cycles of 95° for 25 seconds, 60° for 45 seconds, 72° for 45 seconds, then 72° for 5 min.
For TCRVb CDR3 spectratyping, PCR products from each TCRVb were used in primer-extension reactions with 5′ FAM-labelled Vb primer (Table S1). The following cycling conditions were used: 95° for 25 seconds, 40 cycles of 95° for 25 seconds, 60° for 45 seconds and 72° for 45 seconds, then 72° for 5 min. Spectrayping was performed on an AB3130 Genetic Analyzer (Applied Biosystems, Carlsbad, CA).
For TCRVb sequencing, PCR products from each TCRVb were cloned using a TOPO kit (Life Technologies). Between 16 and 25 clones per TCRVb family per time-point were amplified using M13 primers (Table S1). The following cycling conditions were used: 94° for 5 min, 30 cycles of 94° for 30 seconds, 55° for 30 seconds and 72° for 45 seconds, then 72° for 5 min. Sequencing was performed on a 3730xl capillary sequencer (UCL Genomics, London, UK).
Results
Patient clinical features and outcomes of ASCT
To study immune reconstitution after ASCT, peripheral venous blood was obtained from five children with sJIA before and at several time-points after transplant. Clinical features and outcomes for these patients are summarized in Table 1. We have previously reported on the clinical details of transplant and early follow up, for these patients:5 longer-term outcome data are now available and presented in Table 1. Patients included in this study were predominantly female (4/5), with a mean age of 7 years (range 6–12 years) at the time of transplant. The mean disease duration before ASCT was 5·3 years (range 2·5–8 years). Patients had a variety of treatment approaches before ASCT including corticosteroids, methotrexate and biological agents although for several of these patients ASCT was carried out at a time when biological agents were less available for JIA than they are currently.
Table 1.
Clinical features and outcomes of patients studied
| Patient | Transplant year | Age at transplant/gender | Treatment before ASCT | Conditioning regimen | Long term follow up |
|---|---|---|---|---|---|
| 1 | 2000 | 6/F | S, M, CsA | ATG 20, Cy 200, TBI, (P) | CR 13 years |
| 2 | 2000 | 10/M | S, M, Ig | ATG 40, Cy 200, (P) | CR 13 years |
| 3 | 2004 | 7/F | S, M, CsA, TNF, IL-6 | ATG 20, Cy 200, (P) | Early relapse |
| 4 | 2003 | 10/F | S, M, Ig, TNF, IL-6 | ATG 20, Cy 120, Flu 150, (P) | CR 10 years |
| 5 | 2003 | 12/F | S, M, Ig, CsA, Cy | ATG 20, Cy 100, Flu 150, (P) | CR 10 years |
ASCT, autologous stem cell transplantation; S, corticosteroids; M, methotrexate; CsA, cyclosporine A; Ig, intravenous immunoglobulin (high dose, immunomodulatory); TNF, tumour necrosis factor-α inhibitors; IL-6, interleukin-6 receptor inhibitor; Cy, cyclophosphamide in mg/kg/total dose; ATG, anti-T-cell globulin (rabbit) in mg/kg/total dose; TBI, total body irradiation (400 cGy); P, prednisolone; Flu, fludarabine in mg/kg/total dose; CR, complete remission.
Some data adapted from Abinun et al.5
Conditioning regimens for ASCT included anti-T-cell globulin, cyclophosphamide, prednisolone with fludarabine or total body irradiation in some cases (Table 1). Four of the five patients achieved complete remission after 10 or more years of follow up (mean 11·5, range 10–13 years), whereas one patient (patient 3) relapsed shortly after transplant.
Frequency of peripheral blood cell subsets are highly skewed early after ASCT
Peripheral blood mononuclear cells were isolated from venous blood samples taken before transplant, shortly after transplant (1 month), at intermediate follow up (3–12 months) and late follow up (2–3 years) and assessed for expression of leucocyte and NK cell markers by flow cytometry (Fig. 1). In Fig. 1(a) representative CD4 and CD8 staining on CD3+ T cells is shown; apparent differences in staining intensities are the result of technical differences in flow cytometer settings over time, and not a biological effect. Immediately after ASCT, mononuclear cell subsets were highly skewed compared with cell numbers before transplant. Considerable decreases in CD3+ T-cell and CD19+ B-cell frequencies were observed, while an increase in frequency of CD14+ monocytes was seen (Fig. 1b). However, the apparent increase in one cell subset reflects the decrease in another subset, and actual counts in all cell subsets during this period were low, as illustrated in the Supporting information, Table S2 for one representative patient. By 12 months post transplant, no significant differences compared with before ASCT were observed in frequency of T and B cells, monocytes, NK cells as defined by CD16+ CD3− and NK-T cells as defined by CD16+ CD3+; however, relative cell proportions were still variable at this time-point.
Figure 1.
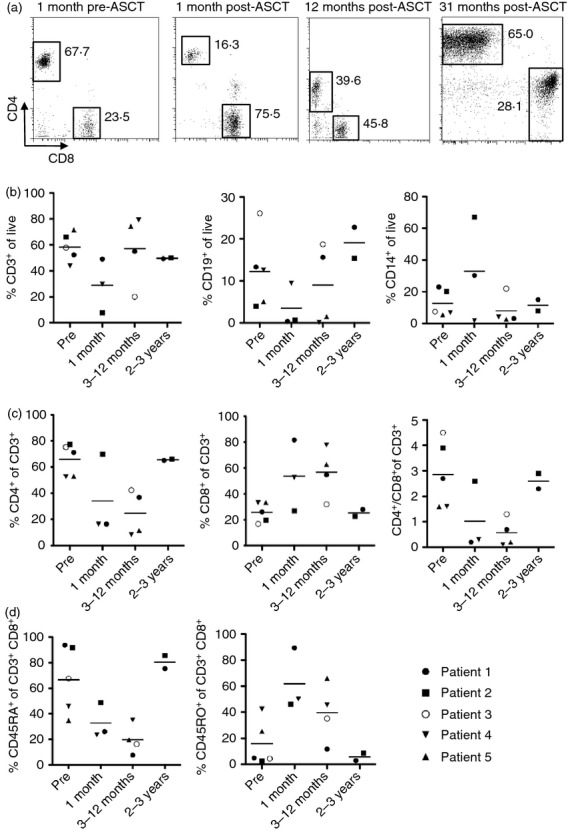
Highly skewed frequency of peripheral blood cell subsets early after autologous stem cell transplantation (ASCT). Expression of cell surface markers examined before and after ASCT by flow cytometry. (a) Representative flow cytometry plots, showing CD4 (y-axis) versus CD8 (x-axis) expression on cells gated on live CD3+ T cells 1 month before and 1, 12 and 31 months after ASCT in patient 1. Percentages of events in each gate are shown. (b) Total T (CD3+, left) and B (CD19+, middle) cell and monocyte (CD14+, right) proportions before ASCT and at 1 month, 3–12 months and 2–3 years after ASCT. All gated on live cells according to forward and side scatter, some using live–dead discrimination. (c) CD4+ (left) and CD8+ (middle) T-cell proportions, gated on live CD3+ cells, and the CD4/CD8 ratio (right). (d) Naive (CD45RA+, left) and memory (CD45RO+, middle) T-cell proportions, gated on live CD3+ CD8+ T cells. Throughout, horizontal bars in summary plots represent the mean, n = 2–5 (where samples were available).
To further investigate the immunological features associated with remission from severe childhood arthritis, expression of T-cell sub-population markers was examined at the same time-points as above. The CD4+ to CD8+ T-cell ratio reversed immediately post stem cell transplantation, and had recovered to the baseline ratio by 2–3 years (Fig. 1c), indicating an early expansion of CD8+ T cells relative to CD4+ T cells. In the CD8+ T-cell population, relative frequency of CD45RA+ naive T cells decreased while frequency of CD45RO+ memory T cells increased, normalizing to baseline by 2–3 years post transplant (Fig. 1d). A similar, but less striking, pattern of CD45RA and CD45RO expression was observed in the CD4+ T-cell population (data not shown).
Oligoclonal TCR repertoire in CD8+ T-cell subset early in immune reconstitution
To assess the reconstitution of the T-cell immune repertoire after immune depletion and stem cell infusion, the clonality of the T-cell repertoire was studied before and after ASCT by TCRVb CDR3 length spectratyping. Spectratyping is a multiplex T-cell assay that can separate TCRVb CDR3 by length, and can be a useful technique to study overall TCR diversity.23,24 Where a large clonal population is present, the amplification of the CDR3 region from this clone produces a ‘spike’ in the typically Gaussian distribution of CDR3 amplification products.
At the same time-points as investigated above, TCRVb CDR3 spectratyping from sorted CD4+ and CD4− T-cell subsets was performed. The CD4-negative sorted fraction was used as a source of CD8+ T-cell TCR mRNA analysis, because other cells in this fraction (monocytes, B cells, NK cells, γδ T cells) do not express a TCR-αβ, and NK-T cells represent a very low percentage of cells.
The TCR diversity of three patients (patients 1, 2, 3) was analysed in detail. In the CD4+ T-cell subset, the TCRVb CDR3 distribution was normally diverse in all Vb families before transplant. One month post ASCT, CDR3 length distribution was predominantly oligoclonal. By 12 months post ASCT, full CDR3 length diversity was restored in all Vb families tested and with quality reads. Data from one individual (patient 1), in whom ASCT led to remission of severe arthritis, are shown in Fig. 2, with five representative TCRVb.
Figure 2.
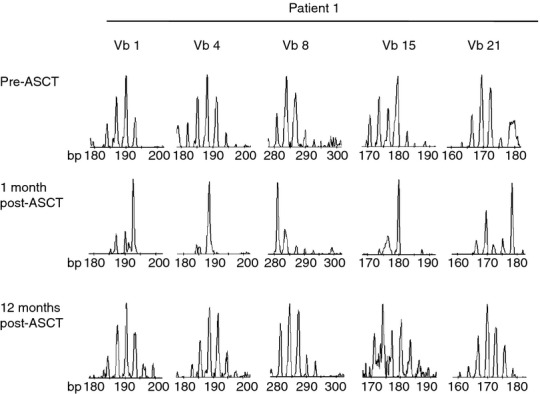
Highly oligoclonal CD4+ T-cell receptor β variable region (TCRVb) repertoire early in immune reconstitution in patient 1, who obtained remission after autologous stem cell transplantation (ASCT). TCRVb CDR3 length spectratyping for Vb1, Vb8 Vb4, Vb15 and Vb21; samples tested from before and 1 and 12 months after ASCT (top to bottom as shown). Representative TCRVb CDR3 spectratypes are shown from patient 1.
We observed different patterns in the clonality of TCRVb, when comparing diversity before, immediately after, and at later time-points after ASCT. In Fig. 3, TCRVb diversity of the CD4-negative T-cell subset (representing CD8+ T cells) is shown for three patients before and after transplant; patient 1 (Fig. 3a) and patient 2 (Fig. 3b) had successful ASCT, whereas patient 3 (Fig. 3c) relapsed after ASCT. For patient 1, at baseline, in all Vb families other than Vb15 and Vb21, TCRVb CDR3 distribution was normally diverse (representative examples shown in Fig. 3a, top). One month after transplant, TCRVb CDR3 distribution was highly skewed in every Vb family, suggesting that a highly oligoclonal T-cell repertoire was generated early in immune reconstitution. Twelve months post transplant, full CDR3 length diversity was re-established in five out of 24 families, namely TCRVb Vb1, 5, 8, 17 and 23 (Fig. 3a, pattern A). Increased T-cell clonal diversity in these instances might suggest that de novo T-cell development and TCR rearrangement occurs after ASCT and this may contribute to remission from autoimmune disease. In comparison, however, CDR3 length distribution remained skewed at 12 months post transplant in 13 of 24 families (Vb2, 3, 4, 6a, 6b, 9, 11, 13a, 13b, 14, 15, 18 and 21). Of interest, in some families, in addition to dominant peaks at different lengths, a re-emergence of some dominant peaks from pre-ASCT was observed (Fig. 3a, pattern B). This might suggest that in certain instances, some T cells, bearing specific TCRVb CDR3 expression, could persist through immunosuppressive conditioning and have a proliferative advantage over T-cell clones that are newly generated post transplant.
Figure 3.

Highly oligoclonal CD8+ T-cell receptor β variable region (TCRVb) repertoire early in immune reconstitution in patients, who were in remission after autologous stem cell transplantation (ASCT), (Patterns A and B) compared with high TCRVb diversity in the patient who relapsed (Pattern C). Patient 1 (a) and patient 2 (b) TCRVb CDR3 length spectratyping for respective TCRVb falling into pattern A (left) and pattern B (right); TCRVb as labelled; samples tested for patient 1: before and 1 and 12 months after ASCT and for patient 2: before and 23·5 months after ASCT. (c) TCRVb CDR3 length spectratyping in same Vb families before and 3 months after ASCT in patient 3 (pattern C).
TCRVb diversity analysis of the CD4-negative T-cell subset (representing CD8+ T cells) in patient 2 (Fig. 3b) showed similar patterns to patient 1. Some TCRVb showed oligoclonal distribution before and later after transplant (namely TCRVb 1, 4, 7, 16, 21, Fig. 3b, pattern A). Pattern B, re-emergence of dominant clones of particular CDR3 lengths, was also observed in patient 2 (Fig. 3b, pattern B, TCRVb3 11, 12, 13b and 15). To investigate the relative frequency of TCRVb family usage TCR family expression was analysed by flow cytometry. No major change in TCRVb family usage was observed in CD8+ T cells before and at later time-points after transplant (see Supporting information, Fig. S1). However, we observed skewing in TCRVb use early after transplant, which correlated well with the oligoclonality seen in the spectratyping analysis.
Spectratyping of TCR species from the CD8+ T-cell population was also studied in the one child who relapsed after ASCT. At baseline, TCRVb CDR3 distribution was normally diverse in the majority of Vb families (representative examples shown in Fig. 3c, pattern C). However, in striking contrast to the spectrayping pattern observed in patient 1 and 2, full TCRVb CDR3 length diversity was observed early after transplant in every Vb family tested (Fig. 3c, pattern C). Of note, this child received cyclophosphamide and anti-thymocyte globulin in standard dose pre-transplant, compared with patient 2 who received a higher dose of anti-thymocyte globulin, or other patients who received another agent or treatment (total body irradiation, fludarabine). It has been suggested previously that the conditioning regimen with lower ATG dose or no third agent, may not be sufficient for full immunodepletion.5
A chimeric TCR repertoire, comprising clones from before and after transplant, can be associated with remission from severe arthritis
The TCR spectratyping data presented above suggested that some T-cell clones might persist through immunosuppressive conditioning, surviving either in the patient or possibly being transferred in the grafted stem cells themselves, and expand after transplant. To further investigate this possibility, clonality of T cells was studied at the level of TCR sequences. TCR sequencing analysis provides a semi-quantitative analysis of clonal frequency within a population of T cells, can identify clones that have expanded from a single T-cell clone, and may provide evidence for survival of specific T-cell clones, after this treatment strategy.
Vb4, 15 and 21 family CD4− T-cell subset PCR products from patient 1, represented in Fig. 3, pattern B, and who achieved complete remission from severe arthritis after ASCT, were chosen for sub-cloning and TCR sequencing. As predicted, in the Vb15 family, specific TCR sequences that were identified from the sample obtained before ASCT were also identified post-ASCT (Table 2 and Fig. 4a). Interestingly, the relative clonal sizes of re-emerging clones were comparable before, and 12 months after, ASCT (clone with N region sequence GVGG 4% before and 4·8% after transplant, DYEN 48% before and 81% after transplant). In contrast, in Vb4 and Vb21 families, new dominant clones were identified 12 months after ASCT, with the clone with N region sequence GTGE representing 54% of total sequences in Vb4 and the clone with N region sequence HGTG representing 100% of Vb21 (Fig. 4b and c, respectively). Furthermore, the dominant clones before transplant (Vb4: clone with N region sequence RHIP 11%, Vb15: AAGA 48%) were not found post transplant (Fig. 4b and c, respectively). However, because only between 16–24 clones were sequenced in these Vb families at each time-point, overlapping clones could have been missed.
Table 2.
T-cell receptor β variable region (TCRVb) sequences across CDR3 region in patient 1
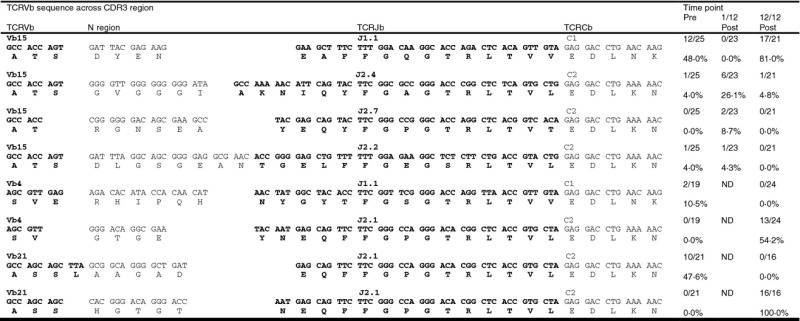 |
Figure 4.
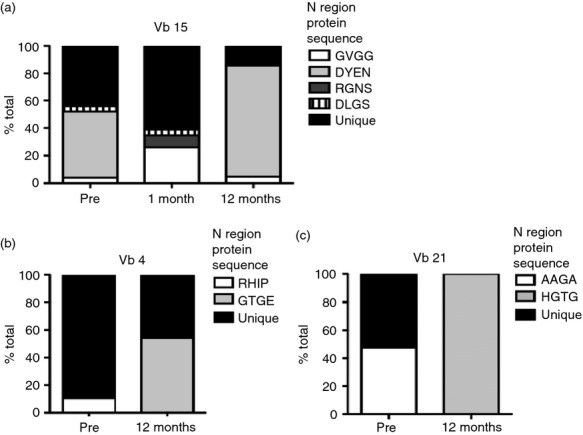
Two patterns of T-cell receptor (TCR) β repertoire: re-emergence of old and establishment of new T-cell clones after autologous stem cell transplantation (ASCT). Sub-cloning of the CDR3 of Vb15 (a), Vb4 (b) and Vb21 (c) in CD4− T-cell subset (representing CD8+ T cells). (a) Vb15 with N region protein sequence: GVGG: white; DYEN: light grey; RGNS: dark grey and DLGS: black stripes. (b) Vb4 with N region protein sequence: RHIP: white and GTGE: light grey. (c) Vb21 with N region protein sequence: AAGA: white and HGTG: light grey. Throughout, unique N region sequences in black. 16–24 sequences per Vb analysed.
Discussion
Systemic juvenile idiopathic arthritis is the most severe form of JIA and has the potential to be highly debilitating or even fatal. Where therapies fail due to drug toxicity or ineffectiveness, autologous stem cell transplantation is a valuable treatment option. When successful, ASCT can lead to full sustained remission; however, pre-transplant immunosuppressive conditioning and parallel treatment can have severe side effects with high mortality. To date we do not understand how ASCT works or who is likely to respond. It has been suggested that ASCT might reset the immune system or give rise to a new immune repertoire.16
The data presented here suggest that, although cell subsets are skewed and the T-cell repertoire is oligoclonal early after ASCT, long-term alterations in frequency of cell subsets and the generation of an entirely new T-cell repertoire might not be necessary for remission from severe arthritis. Remission from severe arthritis can be associated with an immune system comprising T-cell clones found before transplant as well as de novo generated T-cell clones. In addition, on the basis of data from one patient who relapsed rapidly, it could be speculated that full TCRVb CDR3 length diversity early after transplant may be associated with relapse of arthritis after ASCT, and could be indicative of unsatisfactory conditioning.
It has been suggested that in MS, ASCT works through the generation of a mainly de novo T-cell compartment with higher diversity. However, in a study of seven adult MS patients treated with CD34+ enriched autologous haematopoietic stem cells, a small but detectable number of pre-transplant clones were clearly picked up after transplant. The re-emergence of peaks was also seen on spectratyping in some TCRVb families.17 Furthermore, clones reactive to known disease-associated antigens were found post transplant in another study of ASCT in MS.19 Interestingly these had decreased (pathological) T helper type 17 (Th17) responses, but equivalent Th1 responses compared with cells isolated before transplant. The self-protein heat-shock protein 60 (hsp60) has been implicated in regulating arthritis,25–27 and T cells from patients with JIA after ASCT had increased interleukin-10 and Th2 responses with reduced Th1/interferon-γ responses when re-stimulated with hsp60-loaded antigen presenters.6 These results might suggest that induction of self-tolerance and remission from disease may be achieved through the resetting of immune balance. In this study we were not able to compare cytokine responses of T cells before and after transplant. However, demonstrating the re-emergence of pre-transplant T-cell receptor clones after transplant in the context of remission enriches and corroborates the evidence of the above studies, by showing that the same clones re-emerge; compared with reactivity to the same antigens described by the studies above. Thus, these findings contribute towards our understanding of the immunological basis of remission after ASCT.
In agreement with previous studies,17,20,21 we found that relative frequencies of cell subsets were highly skewed early after transplant, with a reversal of the CD4/CD8 T-cell ratio, low CD45RA and high CD45RO frequencies among both CD4+ and CD8+ T cells. It is known that early in immune reconstitution CD8+ T cells have proliferative advantage over CD4+ T cells.28 In this study by Mackal et al., this was due to proliferation of CD8+ T cells outside the thymus, probably from non-depleted memory CD8+ T cells, whereas CD4+ T cells mainly re-generated through thymic selection. Thymic reconstitution of the periphery takes time. It has been shown that normal peripheral CD4+ T-cell numbers are typically reached after approximately 1 year post transplant.10 This may explain the reversal of CD4+ and CD8+ T cells early after transplant.
This study could not investigate regulatory T-cell frequencies, however, another study on JIA ASCT showed that after transplant there is a rebound in the numbers of regulatory T cells.6 In an animal arthritis model of autologous bone marrow transplantation regulatory T cells increased early after transplant and actively controlled arthritis to a low level later after transplant.29
It has been speculated that immune depletion may reset ‘immunological memory’. However, interestingly, results of this study may suggest that although some T-cell depletion is needed, complete T-cell depletion might not be essential for remission and re-emergence of dominant clones is a feature of successful ASCT. The presence of identical N region (V-D-J) nucleotide sequences of individual TCR clones found at varying time points, as we have demonstrated here, is a strong indicator that these are cells from the same clone, i.e that such clones have in fact persisted (in vivo or in transplanted material) from before transplant to post transplant.
Patient 3 relapsed early after transplant and showed polyclonal TCRVb spectra, potentially indicating that the conditioning dose may have been unsatisfactory.5 It could be speculated that full TCRVb CDR3 length diversity seen early after transplant in patient 3 might be associated with the failure of ASCT as treatment. However, this is only one patient and further investigations would be necessary to confirm this. Other limitations of this study include the low number of sequences analysed for TCRVb sequencing, meaning that further reoccurring but low-frequency clones might have been missed. However, even in a small sequence pool, as analysed here, the overlap of pre- and post-transplant clonality strongly indicates reoccurrence of T-cell clones post transplant as a real phenomenon in ASCT. A larger sample size for each time-point with phenotyping and TCR diversity analysis would have been desirable; however, available samples and patients undergoing ASCT were limited.
It is important to note that all patients in this study were treated at a time when disease-modifying anti-rheumatic drugs such as methotrexate were the mainstay of treatment but before many of the biological agents were available. The management of this disease has changed considerably since the advent of these novel biological agents in recent years, such as Tocilizumab (humanized anti-interleukin-6 receptor antibody) and Anakinra (interleukin-1 receptor antagonist) or other interleukin-1β blockade agents. With the introduction of more effective biological agents, ASCT is likely to become a later treatment step in JIA. However, there are many uncertainties about the success of ASCT after intense immunosuppressive effects associated with long-term use of powerful novel biological agents. At present, it is unclear at what time-point and for which children ASCT may be a valuable and appropriate treatment. Therefore, submission of outcomes to the national (UK) and European blood and marrow transplant database (http://www.ebmt.org) may be a vital contribution in informing future management decisions.
Understanding mechanisms underlying disease and treatment is important, to achieve the future goal to successfully treat patients with JIA and other autoimmune conditions, both through ASCT and other immunomodulatory methods. There is a need for treatments with fewer adverse events and minimal drug toxicity. Further research is needed to help define patients most suited for ASCT, and it may help to determine optimal conditioning regimens for induction of remission, while minimizing risks associated with intense immunosuppressive therapy. The results presented here indicate that development of an entirely new immune system with complete re-generation of de novo T cells might not be necessary for remission from severe arthritis. Indeed, it appears that a chimeric TCR repertoire, comprising clones generated before and after transplant, can be associated with long-term remission from severe arthritis.
Acknowledgments
The authors are grateful to patients, parents and staff for contribution of samples. Wedderburn and Brogan group members are thanked for their help, advice and donation of antibodies. All authors read and approved the final version to be published and had input in revising it for intellectual content and style. QW, AMP and LRW had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Experimental design: AMP, QW, AS, DK and LRW. Acquisition of data: QW, AMP, AS and DK. Analysis and interpretation of the data: QW, AMP, AS and LRW. Drafting of the manuscript: QW, AMP and LRW. This work was supported by Arthritis Research UK (QW held an Arthritis Research UK student fellowship), Nuffield Oliver Bird Programme (AMP held an OB Fellowship), and SPARKS UK (AS), ref 01ICH02.
Glossary
- ASCT
autologous stem cell transplantation
- TCRVb
T-cell receptor diversity by analysis of β variable region
- CDR3
complementarity determining region-3
- sJIA
systemic juvenile idiopathic arthritis
Disclosure
The authors have no conflicts of interest to disclose.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Table S1. Primer sequences.
Table S2. Absolute cell counts for patient 1.
Figure S1. T-cell receptor β variable region (TCRVb) expression before and after autologous stem cell transplantation (ASCT). Frequency of expression of 24 TCRVb families was assessed using flow cytometry before (top left), and 1 month (top centre), 19 months (top right), 23·5 months (bottom left) and 37·5 months (bottom centre) after transplant in the CD8+ T-cell subset in patient 2. Gated on live CD8+ T cells and then each TCRVb population. Pie charts show relative proportion of Frequency of total cells positive for each TCRVb shown (in greyscale), with other (in white) representing frequency of CD8+ T cells not accounted for by any of the 24 TCRVb families investigated.
References
- 1.Petty RE, Southwood TR, Manners P, et al. International League of Associations for Rheumatology classification of juvenile idiopathic arthritis: second revision, Edmonton, 2001. J Rheumatol. 2004;31:390–2. [PubMed] [Google Scholar]
- 2.Ravelli A, Martini A. Juvenile idiopathic arthritis. Lancet. 2007;369:767–78. doi: 10.1016/S0140-6736(07)60363-8. [DOI] [PubMed] [Google Scholar]
- 3.Mellins ED, Macaubas C, Grom AA. Pathogenesis of systemic juvenile idiopathic arthritis: some answers, more questions. Nat Rev Rheumatol. 2011;7:416–26. doi: 10.1038/nrrheum.2011.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Packham JC, Hall MA. Long-term follow-up of 246 adults with juvenile idiopathic arthritis: functional outcome. Rheumatology (Oxford) 2002;41:1428–35. doi: 10.1093/rheumatology/41.12.1428. [DOI] [PubMed] [Google Scholar]
- 5.Abinun M, Flood TJ, Cant AJ, et al. Autologous T cell depleted haematopoietic stem cell transplantation in children with severe juvenile idiopathic arthritis in the UK (2000–2007) Mol Immunol. 2009;47:46–51. doi: 10.1016/j.molimm.2008.12.029. [DOI] [PubMed] [Google Scholar]
- 6.de Kleer I, Vastert B, Klein M, et al. Autologous stem cell transplantation for autoimmunity induces immunologic self-tolerance by reprogramming autoreactive T cells and restoring the CD4+ CD25+ immune regulatory network. Blood. 2006;107:1696–702. doi: 10.1182/blood-2005-07-2800. [DOI] [PubMed] [Google Scholar]
- 7.de Kleer IM, Brinkman DM, Ferster A, et al. Autologous stem cell transplantation for refractory juvenile idiopathic arthritis: analysis of clinical effects, mortality, and transplant related morbidity. Ann Rheum Dis. 2004;63:1318–26. doi: 10.1136/ard.2003.017798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wulffraat N, van Royen A, Bierings M, Vossen J, Kuis W. Autologous haemopoietic stem-cell transplantation in four patients with refractory juvenile chronic arthritis. Lancet. 1999;353:550–3. doi: 10.1016/S0140-6736(98)05399-9. [DOI] [PubMed] [Google Scholar]
- 9.Wulffraat NM, Brinkman D, Ferster A, et al. Long-term follow-up of autologous stem cell transplantation for refractory juvenile idiopathic arthritis. Bone Marrow Transplant. 2003;32(Suppl 1):S61–4. doi: 10.1038/sj.bmt.1703946. [DOI] [PubMed] [Google Scholar]
- 10.Brinkman DM, de Kleer IM, ten Cate R, et al. Autologous stem cell transplantation in children with severe progressive systemic or polyarticular juvenile idiopathic arthritis: long-term follow-up of a prospective clinical trial. Arthritis Rheum. 2007;56:2410–21. doi: 10.1002/art.22656. [DOI] [PubMed] [Google Scholar]
- 11.Fassas A, Passweg JR, Anagnostopoulos A, et al. Hematopoietic stem cell transplantation for multiple sclerosis. A retrospective multicenter study. J Neurol. 2002;249:1088–97. doi: 10.1007/s00415-002-0800-7. [DOI] [PubMed] [Google Scholar]
- 12.Burt RK, Georganas C, Schroeder J, et al. Autologous hematopoietic stem cell transplantation in refractory rheumatoid arthritis: sustained response in two of four patients. Arthritis Rheum. 1999;42:2281–5. doi: 10.1002/1529-0131(199911)42:11<2281::AID-ANR4>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 13.van Laar JM, Verburg RJ, Fibbe WE, Breedveld FC. Intensive immunosuppression and autologous stem cell transplantation for patients with severe rheumatoid arthritis: the Leiden experience. J Rheumatol Suppl. 2001;64:25–7. [PubMed] [Google Scholar]
- 14.Song XN, Lv HY, Sun LX, Meng JB, Wang JK, Zhang JQ, Chang YJ. Autologous stem cell transplantation for systemic lupus erythematosus: report of efficacy and safety at 7 years of follow-up in 17 patients. Transplant Proc. 2011;43:1924–7. doi: 10.1016/j.transproceed.2011.03.039. [DOI] [PubMed] [Google Scholar]
- 15.Wedderburn LR, Abinun M, Palmer P, Foster HE. Autologous haematopoietic stem cell transplantation in juvenile idiopathic arthritis. Arch Dis Child. 2003;88:201–5. doi: 10.1136/adc.88.3.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sykes M, Nikolic B. Treatment of severe autoimmune disease by stem-cell transplantation. Nature. 2005;435:620–7. doi: 10.1038/nature03728. [DOI] [PubMed] [Google Scholar]
- 17.Muraro PA, Douek DC, Packer A, et al. Thymic output generates a new and diverse TCR repertoire after autologous stem cell transplantation in multiple sclerosis patients. J Exp Med. 2005;201:805–16. doi: 10.1084/jem.20041679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thiel A, Alexander T, Schmidt CA, et al. Direct assessment of thymic reactivation after autologous stem cell transplantation. Acta Haematol. 2008;119:22–7. doi: 10.1159/000117824. [DOI] [PubMed] [Google Scholar]
- 19.Darlington PJ, Touil T, Doucet JS, et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol. 2013;73:341–54. doi: 10.1002/ana.23784. [DOI] [PubMed] [Google Scholar]
- 20.Bomberger C, Singh-Jairam M, Rodey G, Guerriero A, Yeager AM, Fleming WH, Holland HK, Waller EK. Lymphoid reconstitution after autologous PBSC transplantation with FACS-sorted CD34+ hematopoietic progenitors. Blood. 1998;91:2588–600. [PubMed] [Google Scholar]
- 21.Wedderburn LR, Jeffery R, White H, Patel A, Varsani H, Linch D, Murray K, Woo P. Autologous stem cell transplantation for paediatric-onset polyarteritis nodosa: changes in autoimmune phenotype in the context of reduced diversity of the T- and B-cell repertoires, and evidence for reversion from the CD45RO+ to RA+ phenotype. Rheumatology (Oxford) 2001;40:1299–307. doi: 10.1093/rheumatology/40.11.1299. [DOI] [PubMed] [Google Scholar]
- 22.Koehne G, Zeller W, Stockschlaeder M, Zander AR. Phenotype of lymphocyte subsets after autologous peripheral blood stem cell transplantation. Bone Marrow Transplant. 1997;19:149–56. doi: 10.1038/sj.bmt.1700624. [DOI] [PubMed] [Google Scholar]
- 23.Pannetier C, Cochet M, Darche S, Casrouge A, Zoller M, Kourilsky P. The sizes of the CDR3 hypervariable regions of the murine T-cell receptor β chains vary as a function of the recombined germ-line segments. Proc Natl Acad Sci U S A. 1993;90:4319–23. doi: 10.1073/pnas.90.9.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorski J, Yassai M, Zhu X, Kissela B, Kissella B, Keever C, Flomenberg N. Circulating T cell repertoire complexity in normal individuals and bone marrow recipients analyzed by CDR3 size spectratyping. Correlation with immune status. J Immunol. 1994;152:5109–19. [PubMed] [Google Scholar]
- 25.de Kleer IM, Kamphuis SM, Rijkers GT, et al. The spontaneous remission of juvenile idiopathic arthritis is characterized by CD30+ T cells directed to human heat-shock protein 60 capable of producing the regulatory cytokine interleukin-10. Arthritis Rheum. 2003;48:2001–10. doi: 10.1002/art.11174. [DOI] [PubMed] [Google Scholar]
- 26.Prakken AB, van Eden W, Rijkers GT, Kuis W, Toebes EA, de Graeff-Meeder ER, van der Zee R, Zegers BJ. Autoreactivity to human heat-shock protein 60 predicts disease remission in oligoarticular juvenile rheumatoid arthritis. Arthritis Rheum. 1996;39:1826–32. doi: 10.1002/art.1780391108. [DOI] [PubMed] [Google Scholar]
- 27.van Eden W, van der Zee R, Prakken B. Heat-shock proteins induce T-cell regulation of chronic inflammation. Nat Rev Immunol. 2005;5:318–30. doi: 10.1038/nri1593. [DOI] [PubMed] [Google Scholar]
- 28.Mackall CL, Fleisher TA, Brown MR, et al. Distinctions between CD8+ and CD4+ T-cell regenerative pathways result in prolonged T-cell subset imbalance after intensive chemotherapy. Blood. 1997;89:3700–7. [PubMed] [Google Scholar]
- 29.Roord ST, de Jager W, Boon L, Wulffraat N, Martens A, Prakken B, van Wijk F. Autologous bone marrow transplantation in autoimmune arthritis restores immune homeostasis through CD4+ CD25+ Foxp3+ regulatory T cells. Blood. 2008;111:5233–41. doi: 10.1182/blood-2007-12-128488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Primer sequences.
Table S2. Absolute cell counts for patient 1.
Figure S1. T-cell receptor β variable region (TCRVb) expression before and after autologous stem cell transplantation (ASCT). Frequency of expression of 24 TCRVb families was assessed using flow cytometry before (top left), and 1 month (top centre), 19 months (top right), 23·5 months (bottom left) and 37·5 months (bottom centre) after transplant in the CD8+ T-cell subset in patient 2. Gated on live CD8+ T cells and then each TCRVb population. Pie charts show relative proportion of Frequency of total cells positive for each TCRVb shown (in greyscale), with other (in white) representing frequency of CD8+ T cells not accounted for by any of the 24 TCRVb families investigated.