

Abstract
Deficiencies of lysosomal β-D-galactosidase can result in GM1 gangliosidosis, a severe neurodegenerative disease characterized by massive neuronal storage of GM1 ganglioside in the brain. Currently there are no available therapies that can even slow the progression of this disease. Enzyme enhancement therapy utilizes small molecules that can often cross the blood brain barrier, but are also often competitive inhibitors of their target enzyme. It is a promising new approach for treating diseases, often caused by missense mutations, associated with dramatically reduced levels of functionally folded enzyme. Despite a number of positive reports based on assays performed with patient cells, skepticism persists that an inhibitor-based treatment can increase mutant enzyme activity in vivo. To date no appropriate animal model, i.e., one that recapitulates a responsive human genotype and clinical phenotype, has been reported that could be used to validate enzyme enhancement therapy. In this report, we identify a novel enzyme enhancement-agent, N-nonyl-deoxygalactonojirimycin, that enhances the mutant β-galactosidase activity in the lysosomes of a number of patient cell lines containing a variety of missense mutations. We then demonstrate that treatment of cells from a previously described, naturally occurring feline model (that biochemically, clinically and molecularly closely mimics GM1 gangliosidosis in humans) with this molecule, results in a robust enhancement of their mutant lysosomal β-galactosidase activity. These data indicate that the feline model could be used to validate this therapeutic approach and determine the relationship between the disease stage at which this therapy is initiated and the maximum clinical benefits obtainable.
Keywords: Lysosomal storage disease, β-D-galactosidase, Morquio disease type B, Endoplasmic reticulum quality control, Large animal model, Small molecule therapy
1. Introduction
Deficiencies of lysosomal β-D-galactosidase (β-Gal,2 EC 3.2.1.23) caused by mutations in the gene GLB1 (3p12.33), can result in two very different clinical phenotypes that were originally thought to reflect two different lysosomal storage diseases (LSDs). The first, GM1 gangliosidosis (GM1, OMIM 230500) is characterized by massive neuronal storage of GM1 ganglioside in the brain and occurs in infantile (type 1), juvenile (type 2) and adult chronic (type 3) forms. Four mis-sense mutations are frequently associated with GM1, R482H in type 1 Italian patients, R208C in type 1 American patients and R201C or I51T in type 2 or type 3 Japanese patients, respectively. The second, Morquio disease type B (OMIM 253010), which is primarily associated with a W273L missense mutation in Caucasian patients, presents with generalized skeletal dysplasias resulting from the storage of oligosaccharides derived from keratan sulfate, and little neurological involvement, i.e. these patients do not store GM1 ganglioside [1]. Both diseases typically afflict infants or young children and currently only symptomatic relief and supportive therapy can be offered to them.
In most LSDs, a clinical phenotype does not develop unless genetic mutations lead to at least an 80% reduction in normal levels of the affected enzyme activity. Thus, there is a surprisingly low “critical threshold” of activity required to prevent substrate storage and GM1 [2]. At present, the main approach used to treat selected forms of LSDs is enzyme replacement therapy (ERT). ERT was initially developed and remains the most effective method for treating type 1 (non-neurological) Gaucher Disease [3]. However, ERT is limited by the fact that the recombinant enzyme is not distributed homogeneously throughout the body; e.g. it does not cross the blood brain barrier and in the case of type 1 Gaucher Disease, does not effectively alleviate bone crises. Additionally, its very high cost (≥ $150,000/patient/year) limits its availability to many patients [4].
Two small molecule-based therapies have been proposed to address the limitations of ERT. The first is substrate reduction therapy (SRT) that attempts to limit the storage of non-degraded substrate by using small molecules to inhibit its synthesis in vivo. This approach has shown some promise in treating Gaucher Disease, but is not as effective as ERT [5,6]. Neither ERT nor SRT has been attempted for GM1.
The second small molecule approach is enzyme enhancement therapy (EET) [7,8], which is still under investigation, but has shown some promising preclinical results in at least four enzyme deficiencies [3,9] with several Phase I and Phase II clinical trials being completed (e.g. [10]). EET utilizes small molecules called pharmacological chaperones (PCs) and is based on the theory that an exogenous low molecular weight competitive inhibitor, used at sub-inhibitory concentrations, can stabilize and thus enhance the folding of its target enzyme in the endoplasmic reticulum (ER). Proper folding and in some cases oligomerization, are required for the passage of proteins by the ER’s quality control system, avoiding its associated degradation system, and transport to their site of action, e.g. the lysosome, resulting in a net increase in catalytic activity. It is believed that once the PC-enzyme complex reaches the lysosome, the stored substrates; e.g., GM1 ganglio-side, β-galactose-containing oligosaccharides and glycoconjugates, and keratan sulfate in the case of β-Gal deficiencies; will displace the PC and continue to stabilize the enzyme [3]. However, the ideal PC would bind tightest at the neutral pH of the ER and weakest or not at all at the acidic pH of the lysosome [11], e.g. Ambroxol for Gaucher Disease [12]. Like SRT, EET has the potential to treat the CNS, but is limited to a subgroup of “responsive” mutations. All the responsive mutations described to date appear to produce small but detectable levels of residual mutant enzyme activity [13,14], e.g. GM1 caused by a missense mutation [15]. The attractiveness of this strategy resides in its applicability to a wide range of both inherited and acquired pathologic conditions associated with protein misfolding; e.g. Adult Tay-Sachs [7] and ischemic diseases [16], respectively. On the negative side, each target protein requires a different molecule and not all mutations, not even all missense mutations, will be responsive.
While animal models, such as knockout (K/O) mice, can be used to test ERT or SRT, suitable models in which EET can be validated are lacking. This is because K/O mice do not synthesize any mutant protein whose stability and intracellular transport could be enhanced by treatment with a PC. Furthermore, mouse models frequently do not mimic the human disease; e.g., the K/O mouse model of Tay-Sachs disease develops a very mild phenotype due to a metabolic bypass pathway not present in humans [17,18]. For diseases affecting the CNS such as GM1, mouse models are even more problematic because of their smaller brain size (~1000-fold smaller than human) and different organization.
Transgenic expression of a missense mutation on a K/O mouse background is a new approach that has had some success, but often produces an animal with a very mild or no clinical phenotype, possibly because overexpression of a mutant enzyme with a missense mutation can result in significantly higher levels of residual activity than is found endogenously in patient cells [19,20]. Suzuki and colleagues produced such a mildly affected mouse model of chronic GM1 by the introduction of a human transgene containing a p.R201C point mutation [21]. Even their original K/O mice, despite GM1 ganglioside accumulation in the brain, show no overt clinical phenotype until 4–5 months of age [22]. They have reported administering N-octyl-4-epi-β-valienamine (NOEV) orally for up to 16 weeks to these transgenic mice [23]. After 8 weeks of NOEV treatment, total GM1 ganglioside levels in the brain were reduced by ~3 fold and remained at that level despite another 8 week treatment. They also reported that β-Gal activity increased in the brain during treatment, reaching up to 30 to 40% of wild type levels. No toxicity/adverse effects were reported. Finally, another study of these mice treated with NOEV using three motor assessment tests reported a small, but significant decrease in the rate at which their motor skills deteriorate over a 12–14 month period versus untreated controls [24]. The group also used NOEV to treat GM1 patient fibroblasts with various genotypes and from this survey estimated that EET could be effective in 20–40% of patients [25].
Larger animal models that more closely correspond to the human condition are needed to evaluate the efficacy of new EET-agents. Naturally occurring GM1 has been recorded in cats, dogs, sheep and cattle [1]. GM1 Shiba dogs cannot be used for testing EET-agents, because they are homozygous for a deletion mutation, c.Δ C1647 in the coding region of the protein, which results in no β-Gal (protein or enzyme activity) being made [26,27]. We have recently reported that a naturally occurring feline model of GM1 is caused by a missense mutation [28], p.R483P, which aligns with human p.R482. A similar substitution, p.R482H, has been associated with type 1 GM1 in humans [29], making the results from this model directly applicable to a defined human patient population. In the recently published crystal structure of human β-Gal [30], p. Arg482 (a totally buried residue) appears to play an important role in the interdomain interactions within the functional enzyme through the formation of an ion pair with E131 (in the TIM barrel domain), as well as hydrogen bonds with H412, D491 and F492 (within the β-domain 1). The feline β-Gal deficiency disease, initially described in 1971, is a nearly identical replica of human type 2, juvenile GM1 both clinically and biochemically [31], as well as at the molecular level [28].
Since the GM1 feline (described above) fits most of the criteria as a model that could be used in the future to test the efficacy of EET, we investigated whether fibroblasts from these animals would also respond to treatment with a PC. To accomplish this goal we first characterized a previously untested competitive inhibitor of lysosomal β-Gal, N-nonyl-deoxygalactonojirimycin (NN-DGJ), as a PC for our collection of human GM1 fibroblasts containing a variety of missense mutations.
NN-DGJ is a long-alkyl-chain iminosugar derivative, part of a large class of compounds also described as “sugar mimetics”, the polyhydroxylated alkaloids. Many of them are naturally occurring in plants and microorganism species and have generated sustained interest over time, as some act as general inhibitors of glycosidases while others are very potent and highly selective for only one type of glycosidase. Their potential therapeutic applications range from anti-cancer agents (swainsonine), immuno-stimulants, anti-diabetic agents (Miglitol), antiviral agents and inhibitors of glycosphingolipid synthesis [32]. This class of compounds is of particular interest to LSDs as some have been shown to be effective for SRT; e.g. N-butyl-deoxygalactonojirimycin (NB-DGJ) for Gaucher Disease, which is more specific and thus has fewer side effects [33] than the only currently approved SRT-agent NB-DNJ (Zavesca or Miglustat) [34], and others acting as PCs for EET, e.g., DGJ for Fabry disease caused by deficiencies in α-galactosidase (α-Gal) [35].
We demonstrate that the NN-DGJ derivative of DGJ is the most potent and specific inhibitor of human β-Gal, and that it acts as a PC for a number of GM1 fibroblast lines bearing a number of missense mutations. Finally we show that this small molecule can also produce a robust enhancement of mutant lysosomal β-Gal activity in feline GM1 fibroblasts, confirming the suitability of this animal model for the future evaluation of EET.
2. Material and methods
2.1. Chemical reagents
NN-DGJ and NB-DGJ were purchased from Toronto Research Chemical Inc. (Canada) and DGJ, HCl salt (DGJ) from Sigma-Aldrich Ltd. (Canada). All compounds were dissolved in DMSO to obtain stock solutions. The following fluorogenic substrates also from Sigma-Aldrich Ltd., 4-methylumbelliferyl-β-D-galactopyranoside (MU-βGal), 4-methylumbelliferyl-N-acetylglucosaminide (MUG) and 4-methylumbelliferyl-α-D-galactopyranoside (MU-αGal) were used to assay the lysosomal enzymes β-galactosidase (β-Gal), total hexosaminidase (Hex) and α-galactosidase (α-Gal), respectively. Taurodeoxycholic acid (TC), sodium salt was purchased from Calbiochem (CA, USA) and DMSO from EMD Chemicals Inc. (Germany). Other chemicals used were analytical grade reagents from general laboratory suppliers. A Concanavalin A fraction of enriched glycoprotein including wild type lysosomal enzymes, e.g., β-Gal and α-Gal [36,37] was used for the in vitro characterization of NN-DGJ.
2.2. Cell lines, tissue culture and small molecule treatment
Primary skin fibroblast cultures established from various GM1 (infantile, juvenile or adult clinical phenotype) or Morquio B disease patients with different mutations in their GLB1 gene were obtained from our own collection of cells (HSC tissue culture services), through collaboration or purchased from Coriell Cell Repositories (NJ, USA). Some characteristics of the cell lines i.e. mutations, used in this study are indicated in Table 1 along with their response to NN-DGJ. Immortalized fibroblasts cell lines from normal (SV3) and GM1 cats (GM1 SV3) were generated in the D. Martin laboratory [28] by calcium phosphate transfection of primary fibroblasts with the plasmid pSV3-DHFR (American Type Culture Collection) containing the large T antigen of Simian Virus 40.
Table 1.
Characteristics of the human and feline GM1 Gangliosidosis/Morquio disease type B fibroblasts.
| Fibroblasts | Clinical features at presentation | Phenotypea | β-Gal. RAb NR/Rc | Mutations |
|---|---|---|---|---|
| Hu 4032d | Hepatosplenomegaly (HSM), cherry red spots (CRS), hypotrophic cardiomyopathy (HCM) | Infantile (<18 month) | <1% NR | p.R351X/p.R351X |
| Hu 4992 | CRS, HSM, developmental delay, hypotonia, kyphoscoliosis | Infantile (6 month) | 1% R | p.R148S/p.D332N (S532G)e |
| Hu 8981 | HSM, no skeletal or cardiac involvement | Juvenile (34 month) | 6% NR | p.R148S/p.R482H |
| Hu 14771 | Behavioral problems, developmental regression, seizures, wheelchair bound by 17 year old | Juvenile (9.5 year) | 3% R | p.R201H/IVS14-2A>G |
| Hu 3633 | Extrapyramidal signs, no HSM/cardiac involvement | Adult (adulthood) | 6% R | p.R201H/p.W509C |
| Hu 4080 | From Suzuki et al., [1] | Morquio B (11 year) | 5% NR | p.W273L/p.R482H |
| Hu 10213 | Short stature, scoliosis, corneal clouding, joint restriction | Morquio B (8 year) | 6% NR | p.G438E/p.G438E |
| Fe GM1 SV3 | From Martin et al., [28] | Juvenile (3 to 4 month) | 9% R | p.R483P/p.R483Pf |
Age at presentation.
β-Galactosidase residual activity expressed as % of wild type.
NR for non-responding and R for responding to NN-DGJ see Table 2.
Coriell cell repository GM 056052A, used as negative control for PC treatment.
Mutation characterized as polymorphism [15].
Align with R482 in human.
All cells were grown in α-minimal essential medium (α-MEM) from Wisent Inc. (Canada) in the presence of 1% antibiotics (penicillin and streptomycin, Gibco BRL, Canada) and supplemented with Fetal Bovine Serum (FBS, Wisent Inc., Canada) at 10% for human cells or 1% for cat cells, and incubated at 37 °C in a humidified atmosphere with 5% CO2.
Treatment of human and cat cell lines (normal or mutant) with candidate drugs were performed in 24 or 96 well plates (Falcon BD, Canada), or Petri dishes (10 cm). Using tissue culture containers of different sizes allows us to either work simultaneously with multiple drug concentrations (dose response curves) or to obtain the larger cell numbers needed for some analyses (western blot). The cell lines grown to 60–80% confluency were rinsed with phosphate buffered saline (PBS) and growth medium, containing the test-drug at a defined concentration or solvent only (mock treated), was added. The cells were returned to the incubator for the specific period of time dictated by the experiment, rinsed with cold PBS, then processed as required for each analysis (lysis, staining, etc.). The range of concentrations used, the drugs tested and the lengths of incubation in the presence of the small molecules are indicated in the text or in the legends of the figures.
2.3. Enzyme assays and protein determination
A similar protocol was used for the determination of either β-Gal or α-Gal activity using an enriched glycoprotein fraction from human placenta (for IC50s, Ki, Km and heat stability measurements), or β-Gal and Hex activity in cell lysates (see below for the experimental conditions). Preparation of cell lysates from cells grown and treated in tissue culture plates (10 cm diameter) was as follows: the cell suspensions centrifuged at low speed after washing and scraping in cold PBS, were resuspended in appropriate buffer and lysed by repeated freezing-thawing followed by a second centrifugation (benchtop at ~13,000 g) for 30 min at 4 °C to separate cellular debris. The resulting supernatant or cell lysate was transferred to a new tube and used for enzyme assays/western blot analysis, or kept at −20 °C for later use. When cells were grown/treated in 24 well plates, the cell lysate preparation was performed “in situ”. Each well was rinsed 3 times with cold PBS, then 150 μL of lysis buffer was added and the plate placed at 4 °C for ~1 h. Lysis buffer for enzyme activity assays consisted of McIlvaine citrate (0.1 M) — phosphate (0.2 M) buffer (CP) at pH 4.3 containing TC 0.4% (w/v) and Triton X100 0.4% (v/v).
β-Gal, Hex and α-Gal enzyme activities were determined in all cases using 96 well plates (in triplicate or quadruplicate) by hydrolysis of their respective fluorescent substrate. Stock solutions of the fluorogenic substrate were prepared in CP buffer at 0.56 mM for MU-βGal, 3.2 mM for MUG and 6 mM for MU-αGal at pH 4.3, 4.1 and 4.5, respectively for the three enzyme activities measured. For the enriched glycoprotein fraction “enzyme mix” (see below) was prepared and for cell lysates, an aliquot (2–10 μL) was used following the experimental protocol previously reported [38] to measure the fluorescence generated by liberation of 4-methyl umbelliferone (MU). In both cases, the dilution performed or the volume of cell lysate assayed and the time of incubation were adjusted to obtain meaningful fluorescence readings relative to the level of enzyme present (normal activity or mutant enzyme with as low as 1% of WT).
Protein determinations were performed either with the Biorad assay (Biorad, USA) or the Pierce BCA (bicinchoninic acid) Protein Assays (Thermo Scientific) when Triton X100 was present. Quantification of duplicate assays was obtained from bovine serum albumin (BSA) standard curves according to manufacturer’s protocol.
2.4. Enzyme kinetics: Km, IC50 and Ki
A series of ten dilutions of the MU-βGal stock substrate ranging from 0.011 to 0.504 mM was prepared in CP at pH 4.3 and distributed in triplicate in a 96 well plate in a constant volume (45 μL). An aliquot (5 μL) of an enzyme mix containing enriched human placental β-Gal in CP pH 4.3 plus BSA 0.25% (w/v) was added to each well. Substrate blanks (3 wells) with no enzyme added were also included. The enzymatic reaction was incubated for 1 h at 37 °C, stopped by addition of 2-amino-2-methyl-1-propanol (pH 10,5) and the fluorescence generated quantified as relative fluorescence units (RFUs) [38]. The RFUs for each MU-βGal concentration were calculated relative to the RFUs measured in the absence of substrate. The resulting data expressed as mean of the triplicate assays plus or minus one standard deviation were analyzed by non-linear regression curve-fitting using Prism 5 (Graph Pad Software, Inc.) to calculate the Km of β-Gal.
A serial dilution of a 17 mM NN-DGJ stock solution in DMSO was made and mixed with the enzyme (as above). Each assay performed in 96 well plates (N=3 or 4), consisted of 24 μL of the enzyme mix to which was sequentially added 1 μL of each of the NN-DGJ dilutions followed by a fixed volume of the MU-βGal substrate (25 μL at 0.56 mM). The enzymatic reactions were incubated, stopped and the fluorescence quantified as above. Blank substrate and assay with no NN-DGJ (“0” concentration) were also included. The enzyme activity for each of the 7 NN-DGJ concentrations tested (4.7 nM to 3.4 μM) was calculated relative to the enzyme activity measured in the absence of inhibitor. These sets of experiments were performed at pH 5. The data were expressed as mean of the triplicate/quadruplicate assays plus or minus one standard deviation and a graph generated using a non linear fit to a sigmoidal dose response curve of the logarithmic concentration of NN-DGJ as a function of the enzyme activity with no NN-DGJ using Prism 5 (Graph Pad Software, Inc.), allowing for the IC50 or 50% inhibitory concentration of NN-DGJ for human placental β-Gal to be calculated. Similar protocols were used to generate dose response curves for NN-DGJ and α-Gal, as well as DGJ (stock at 100.2 mM) and NB-DGJ (stock at 22.80 mM) for both β-Gal and α-Gal. The range of the serial dilutions was modified until meaningful data for each compound tested were obtained with each of the two enzymes. Corresponding IC50s were calculated as above when satisfactory inhibition was detected.
Apparent Km of human placental β-Gal for MU-βGal in the presence of various concentrations of NN-DGJ was next determined. A set of experiments was conducted in the same way as those for determining the Km of β-Gal for MU-βGal (see above), with supplemental addition of NN-DGJ to the enzyme mix to obtain a range of inhibitor concentrations from 10 to 200 nM. Each set of data corresponding to one concentration of NN-DGJ (6 in total) was used to generate a curve (as for Km above) allowing the calculation of the apparent or observed Km of β-Gal for MU-βGal substrate in the presence of each inhibitor concentration. Using a non-linear regression fit the apparent Km (with their standard deviation) and their inhibitor concentration were tested for competitive inhibition using Prism 5 (Graph Pad Software, Inc.).
2.5. Testing for pH influence on NN-DGJ inhibitor properties
Dose response curves at three more pHs (4.3, 5.5 and 6.5) using a serial dilution of the 17 mM DMSO stock of NN-DGJ in water and the enzyme mix (as for IC50 above) were repeated, but the range of NN-DGJ concentration was extended (11 concentrations) to 0.58 nM and 4 μM. Data, expressed as a mean of triplicate assays plus or minus one standard deviation, were used to generate sigmoidal dose–response curves (data not shown) and processed as above to obtain the IC50 value of NN-DGJ for β-Gal at each pH.
2.6. Heat inactivation assays
The effect of NN-DGJ on the stability of β-Gal was determined by attenuation of heat denaturation experiments with the enriched human placental β-Gal fraction (60 or 120 nM) or cell lysate from GM1 feline fibroblasts (100 or 200 nM) prepared as described for the enzyme assays. All the steps were carried out on ice prior to the final enzyme assay. Enzyme mixes were prepared at pH 7 and contained either NN-DGJ or DMSO to obtain the same concentration than in the NN-DGJ mix, and 15 μL transferred to pre-labeled sets of PCR tubes (4 sets of 3 tubes per experiment). One set of PCR tubes containing the enzyme/NN-DGJ mix and another containing the enzyme/DMSO mix were kept on ice (zero time points) when the other sets were simultaneously transferred to a PCR machine pre-heated at 48 °C. The heat denaturation step and the calculation of the T50 of β-Gal in the absence or presence of NN-DGJ were done as previously reported [38].
2.7. Western blot analysis and densitometry quantification
Western blot analyses of reduced and denatured total proteins (40 μg) from the different human GM1 fibroblasts lysates were carried out as previously described [39]. Primary antibodies were a rabbit polyclonal IgG anti-human β-Gal (Callahan [15] proprietary antibody), and a rabbit polyclonal IgG1 anti-GAPDH (Invitrogen, Canada). Membranes were stripped between β-Gal and GAPDH labeling. Secondary antibody was donkey anti-rabbit HRP conjugated from Jackson Laboratory (Maine, USA). Two specific immunoreactive bands can be visualized for β-Gal corresponding to the precursor and the mature protein displaying an apparent molecular mass of ~84 and ~64 kDa respectively. The intermediate band is non-specific. The GAPDH band corresponds to the expected ~39 kDa.
Quantification of the immunoreactive bands visualized on the radiographic films was performed after scanning and processing of the images through Image J (rsbweb.nih.gov/ij/). For each fibroblast line the densitometry value of each band corresponding to the precursor or mature β-Gal was first divided by the densitometry value for GAPDH band as a normalization step. In total cell homogenates from wild-type fibroblasts over 95% of β-Gal was present as the mature form. Then for each GM1 fibroblast line the normalized value of the precursor band after treatment by NN-DGJ was compared to the value without treatment allowing the estimation of potential variation of each of the two protein forms produced by the PC treatment.
2.8. Indirect immunofluorescence staining and confocal microscopy image analyses
Indirect immunofluorescence and confocal microscopy imaging for human or feline fibroblasts in the presence or absence of NN-DGJ were performed as previously reported [28].
3. Results and discussion
3.1. In vitro characterization of NN-DGJ as a competitive inhibitor of human lysosomal β-Gal
Most PCs reported to date have also been co-factors or inhibitors of their target enzyme. Since DGJ, as well as its N-butyl derivative, NB-DGJ, have been shown to be inhibitors of β-Gal with IC50s ~25 μM [40], we decided to test and compare another commercially available derivative of DGJ, N-nonyl-DGJ (NN-DGJ), as an inhibitor of β-Gal. These three derivatives were first tested in vitro using a glycoprotein-enriched Concanavalin A fraction of a human placental extract high in lysosomal enzymes such as α-Gal and β-Gal [36,37]. The β-Gal activity in this gly-coprotein fraction displayed a Km of 0.53±0.04 mM for the artificial 4MU-βGal substrate, a value comparable to the previously reported value of 0.43 mM determined using the purified enzyme [15]. Since both β-Gal and α-Gal recognize similar substrates, we determined the IC50s of the three DGJ derivatives for both enzymes at their respective pH optima (Fig. 1). Interestingly, while DGJ was a low μM inhibitor of α-Gal (~20-fold stronger than for β-Gal), NB-DGJ was a poor inhibitor, as previously reported [35,41], and no significant inhibitory effect on the enzyme was detected in the presence of up to 340 μM NN-DGJ. On the other hand NN-DGJ was found to be an ~30-fold better inhibitor of β-Gal than NB-DGJ. Thus, the IC50 values indicate that the addition of an N-alkyl side chain dramatically increases the specificity of the DGJ-derivative for β-Gal and that the strength of inhibition is further increased when the N-alkyl-chain is lengthened from 4 to 9 hydrocarbons.
Fig. 1.

IC50 curves of DGJ and two N-alkyl derivatives for human α-Gal and β-Gal. A: The IC50 values of DGJ, NB-DGJ and NN-DGJ (±SD) were calculated by non linear regression analysis from the data points in the graphs shown in B and C for β-Gal and α-Gal enzymes, at their pH optima. *DGJ is in phase III clinical trial as a PC for the treatment of Fabry disease; **The inhibition measured was too weak to allow for the calculation of an IC50. B: Serial dilutions of DGJ (open circle), NB-DGJ (open square) and NN-DGJ (black triangle) were incubated in presence of fixed amounts of an enriched fraction of β-Gal. The relative enzyme activity determined in the presence of each of the inhibitor concentrations tested (N=4) was calculated relative to the enzyme activity measured in the absence of that inhibitor (N=4) and plotted on the Y axis, versus the [inhibitor] (mM) plotted on a logarithmic scale on the X axis. Data are expressed as mean+SD. C: Serial dilutions of DGJ, NB-DGJ and NN-DGJ were incubated in presence of fixed amounts of an enriched fraction of α-Gal. The same experimental protocol and data analysis as described in B was followed, except α-Gal enzyme activity was measured.
Since NN-DGJ represents the DGJ derivative with the lowest IC50 and the greatest specificity for human β-Gal, we next determined its effect on the apparent Vmax and Km of the enzyme using the MU-βGal substrate. Increasing concentrations of the inhibitor did not significantly alter the apparent Vmax, but increased the apparent Km, confirming NN-DGJ acts as a competitive inhibitor. Non-linear regression analysis of the experimental data produced a Ki of 185±12 nM.
3.2. NN-DGJ is a better inhibitor at neutral as compared to acidic pH and stabilizes β-Gal against thermal denaturation
In order to promote the release of a PC once the mutant enzyme has been folded and transported out of the ER, a desirable characteristic is that the PC binds tighter at the neutral pH of the ER than at the acidic pH of the lysosome [7]. Thus IC50s of NN-DGJ were determined at different pHs. The IC50s decreased from 120±23 nM at pH 4.3 (the pH optimum of the enzyme), to 60±12 nM at pH 5, and 40± 6 nM at pH 5.5 or 6.5.
An effective PC stabilizes its target protein during folding in the ER. This property can be indirectly evaluated by examining the ability of the PC to protect the enzyme against heat inactivation. In the absence of NN-DGJ the T50 (time at which 50% of the initial enzyme activity is lost) of WT β-Gal activity at pH 7 and 48 °C was 4.5±1.5 min. In contrast, the T50 value increased by ~3- (12.7±2.5 min) or 4- fold (18.5±1.5 min) in the presence of 60 or 120 nM of NN-DGJ, respectively. Like most lysosomal enzymes β-Gal is more stable at acidic pH with a T50 in the absence of NN-DGJ at pH 5 and 48 °C of 18.5±2.5 min. Taken together with the stronger binding affinity of NN-DGJ to β-Gal at a more neutral pH (above), these data indicate that NN-DGJ displays an in vitro behavior consistent with acting as a PC for human β-Gal in vivo.
3.3. Ex vivo (in cellulo) characterization of NN-DGJ as a PC using a human GM1 fibroblast line
We initially used primary human fibroblasts (4992) derived from a patient with infantile GM1 bearing two missense mutations, p.R148S/p.D332N [15] to test the ability of NN-DGJ to enhance the residual activity levels of mutant lysosomal β-Gal. Frequently the infantile forms of LSDs are caused by null mutations and are not amenable to EET. However this cell line was known to have low, but detectable levels of residual β-Gal enzyme activity (<1%) and low levels of β-Gal protein visualized as cross-reacting material by western blot analysis (CRM positive) [15], suggesting that it might respond to PC-treatment. GM1 fibroblasts were grown for 5 days with various amounts of NN-DGJ (0.17 to 17 μM) added to the growth media and a dose response curve generated. The activity of β-Gal in treated versus mock-treated (DMSO) cells was increased by >2 fold at concentration starting at ~1 μM of NN-DGJ, reaching ~3 fold between 1.7 and 2.6 μM (Fig. 2). Total Hex activity, also determined in the cell lysates, remained essentially unchanged with NN-DGJ treatment, indicating that over this concentration range it produced no toxic effects on the cultured cells nor did it result in a general enhancement of multiple lysosomal enzyme activities. These data also demonstrate that NN-DGJ can penetrate the plasma membrane, but that concentrations ~15 fold greater than the IC50 are necessary in the growth media to produce significant PC activity. As the concentration of 1.2 μM was the lowest that also produced a robust “fold enhancement” it was used thereafter for testing other human GM1 fibroblasts (see below).
Fig. 2.
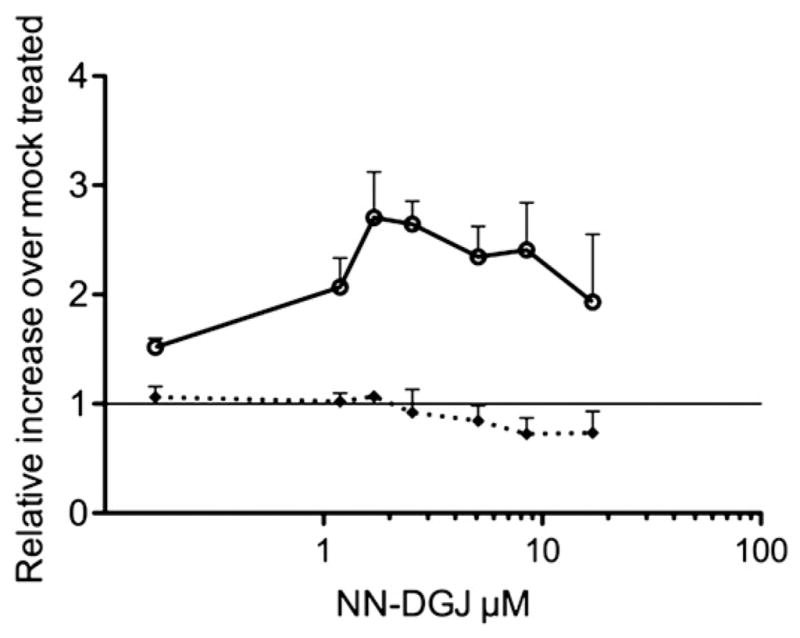
Dose response of human infantile GM1 fibroblasts to NN-DGJ: Primary skin fibroblasts (4992, Table 1) were grown in presence of increasing concentrations of NN-DGJ for 5 days. Enzyme activities for β-Gal (open circle) and total hexosaminidase (black diamond) were measured (N=3) in cell lysates. The enzyme activities in the treated cells are expressed as fold increase relative to mock treated cell (Y=1 represents no enhancement) and plotted versus the [NN-DGJ] (μM) on a logarithmic scale. Data are expressed as mean+SD.
To visualize the effect of NN-DGJ on intracellular transport of mutant β-Gal to lysosomes, treated and untreated GM1 fibroblasts (4992) were next examined by indirect immunofluorescence staining followed by confocal microscopy imaging and compared to normal (wild type, WT) fibroblasts (Fig. 3). In WT fibroblasts, β-Gal (in green) displayed a punctate pattern predominantly co-localizing with Lamp-1 (in red) a marker of the lysosomal compartment (left “Merge” panel), with limited co-localization with PDI (in red), a marker for the ER compartment (right “Merge” panel). In contrast in DMSO treated GM1 fibroblasts, β-Gal had a very diffused pattern of staining, co-localizing predominantly with PDI (GM1+DMSO panels). Addition of NN-DGJ (1.2 μM for 3 days) to the fibroblasts growth media produced an increased fluorescence staining (green) for β-Gal, which was more punctate and predominantly co-localized with Lamp-1 (GM1+NN-DGJ panels). Taken together, the 2–3 fold increase in β-Gal enzyme activity in conjunction with the increased localization of β-Gal protein to lysosomes observed in human GM1 fibroblasts treated with >1 μM of NN-DGJ for 5 days, strongly support the conclusions that NN-DGJ acts as a PC for this form of mutant β-Gal.
Fig. 3.
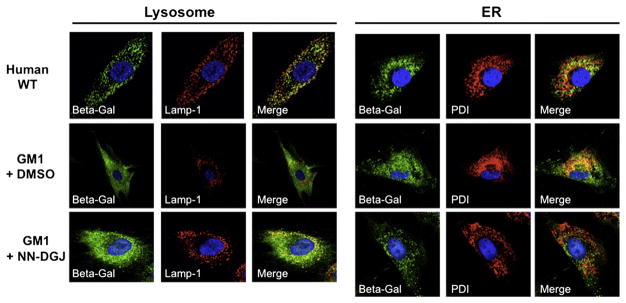
NN-DGJ treatment restores the intra-cellular localization of human β-Gal: Human GM1 fibroblasts (4992, Table 1) were grown in the absence or presence of 1.2 μM of NN-DGJ and indirect immunofluorescence staining followed by confocal image analyses performed. Wild type fibroblasts were processed in parallel as controls for the staining pattern. All cells were probed with a rabbit polyclonal IgG raised against the β-Gal precursor protein (green). Antibodies for sub-cellular localization (red) included lysosomal associated membrane protein-1 (Lamp-1, left panel) and protein disulfide isomerase (PDI, right panel) to identify lysosomes and the ER, respectively. Nuclei were stained with DAPI. Merged images are shown to help with visualization of the co-localization of β-Gal with Lamp-1 or PDI (yellow-orange). WT, wild type human fibroblasts; GM1+DMSO, 4992 GM1 fibroblasts grown in the absence of NN-DGJ, but in the presence of DMSO used as solvent; GM1+NN-DGJ, 4992 GM1 fibroblasts grown in the presence of 1.2 μM of NN-DGJ for 3 days. The cell shown in each pannel is representive of at least 10 cells visualized.
3.4. NN-DGJ also acts as a PC for other mutations affecting human β-Galactosidase
To test the efficacy of NN-DGJ on enhancing the residual activity of other β-Gal mutations, six additional human GM1 patient-derived fibroblast lines were treated for 5 days with 1.2 μM of NN-DGJ. In total, the fibroblasts were derived from patients diagnosed with infantile (n=2), juvenile (n=2) and adult (n=1) forms of GM1, as well as Morquio B disease (n=2). These fibroblasts have been collected over the years from patients seen at The Hospital for Sick Children, purchased or acquired through collaboration (as indicated). Phenotypes, clinical features, residual levels of β-Gal enzyme activity and genotypes are indicated in Table 1. The fold-increases of β-Gal enzyme activities after NN-DGJ treatment for the responsive fibroblasts are given in Table 2.
Table 2.
Longer PC treatment can increase the level of response in some mutant cells.
| GM1 fibroblasts | 4032a | 4992 | 14771 | 3633 | 4080 | Feline GM1 SV3 |
|---|---|---|---|---|---|---|
| +NN-DGJ 1 dose over 5 days | 1.4±0.45 | 4.1±0.63 | 4.9±1.05 | 7.3±1.05 | 0.9±0.20 | 6.0b±0.20 |
| +NN-DGJ 3 doses over 15 days | 1.2±0.05 | 4±0.60 | 7.8±1.10 | 13.8±0.25 | 1.26±0.35 | NAc |
Fibroblast 4032 is used as negative control for pharmacological chaperoning.
Over 3 days only.
Not applicable, GM1-SV3 fibroblasts are immortalized, very fast growing cells that become over confluent after 5 days and began lifting off.
As expected fibroblasts (4032) derived from a patient with an infantile phenotype resulting from an homozygous non-sense mutation (p.R351X/p.R351X, CRM negative) did not show any enhancement of β-Gal activity after NN-DGJ treatment and were used as a negative control for chaperone therapy. Fibroblasts from the juvenile (8981, p.R148S/p.R482H) GM1 and the two Morquio B patients (4080, p.W273L/p.R482H and 10213, p.G438E/p.G438E) were similarly non-responsive. In contrast, fibroblasts 14771 (p.R201H/IVS14-2A>G) had an ~5 fold increase in β-Gal activity, which would raise the residual activity to ~15% of normal, suggesting that for this patient, EET would be beneficial. Lastly, an impressive 7-fold increase was seen for the fibroblasts derived from an adult GM1 patient (p.R201H/p.W509C), suggesting that both mutations are likely responsive. The positions of these mutations were then mapped onto the recent X-ray crystallographically-derived 3D structure of the β-Gal monomer (Fig. 4) reported by Ohto et al. [30].
Fig. 4.

Localization of the missense mutations analyzed in this report on the crystal structure of the human β-Gal monomer: The β-Gal monomeric structure is shown in ribbon representation with the bound NN-DGJ (in green) inserted inside the active site pocket, and the mutated amino acid residues are represented as spheres within the diagram. The different domains of the β-Gal are indicated (TIM barrel domain, in pale pink; β domain 1, in purple; β domain 2, in orange and TIM- β1 loop, in blue). Our data identified p.R201H (dark green, on the surface of the protein) as a mutation that strongly responds to NN-DGJ (14771 and 3633, Table 1 and 2), while p.W273L (4080, Table 1, involved in ligand recognition), p.G438E (10213, Table 1, on the surface of the protein), p.R482H (4080 and 8981, Table 1, core residue in the interdomain), and p.R148S (8981, Table 1, core residue in the intradomain) are nonresponsive mutations (in red). Taken together these data also indicate that p.W509C (3633, Table 1, core residue in the intradomain) and p.D332N (4992, Table 1, on the surface of the protein) are responsive mutations (lighter green). (For interpretation of the references to color in this figure legend, the reader is referred to the web version of the article.)
To confirm that the increases in β-Gal activity levels (Table 2) were the result of increased β-Gal protein levels (Fig. 5A), lysates from NN-DGJ treated and untreated fibroblasts were analyzed by Western blotting. The immunoreactive bands were scanned for quantification by densitometry, and the values normalized using the loading control GAPDH. The values obtained for the 84 kDa precursor (ER) or the 64 kDa mature (lysosomal) forms of the protein with and without NN-DGJ were plotted on a stacked bar graph (Fig. 5B). The increases in the intensity of the mature β-Gal roughly correlated with the increases in enzyme activity measurements in the various GM1 cells treated with NN-DGJ.
Fig. 5.

NN-DGJ treatment can increase β-Gal protein levels in GM1 fibroblasts expressing different β-Gal missense mutations: A: Western blot analysis of 40 μg total protein from either untreated wild type fibroblasts (WT) lysate or lysates from three GM1 fibroblast lines with different mutations in their GLB1 genes [14771, 4992, and 3633 (4032 negative control) see Table 1] grown for 5 days in the absence (−) or presence of 1.2 μM NN-DGJ (+). GAPDH was used as loading control. The proprietary antibody against human β-Gal precursor protein (JW Callahan) recognizes the β-Gal precursor (84 kDa, predominantly ER) and the mature (64 kDa, active lysosomal) forms of the enzyme. A non-specific band can be seen at ~75 kDa (x). B: Densitometry quantification of the CRM bands was performed using image J and the amount of either precursor or mature β-Gal protein was estimated after normalization to the corresponding GAPDH band intensity. Data are displayed as stacked bars. The lower open bars correspond to the relative densities of the precursor form, while the upper shaded bars correspond to the densities of the mature form of β-Gal.
It is interesting to compare the effects of PC-treatment on the precursor (likely an inactive ER form) and mature (the active lysosomal form) of β-Gal bearing different mutations. Whereas the most responsive line (adult 3633) shows a 50% decrease in the precursor form coupled with an ~10-fold increase in mature form, the second most responsive line (juvenile 14771) produces an increase of ~2-fold in the precursor and a 3-fold in the mature forms after NN-DGJ treatment. Since these lines share a p.R201H allele (Table 1), these data suggest that the quality control in the ER is capable of turning over the product of the single missense mutation in the juvenile line; e.g., p.R201H, a surface residue (Fig. 4), which may therefore be inhibiting protein folding; but has some difficulty in turning-over the second missense mutation in the adult line; e.g., p.W509C, a core intradomain residue (Fig. 4), which therefore may be promoting protein misfolding. If these suppositions are correct, then treatment with NN-DGJ is able to both inhibit protein misfolding and enhance protein folding of mutant forms of β-Gal.
3.5. NN-DGJ treatment of patient fibroblasts shows an additive effect after administration of multiple dosing over 15 days
Since EET would involve chronic delivery of drug to the patients, we investigated if prolonged (15 days with 3 doses) vs. short (5 days with one dose) treatment with NN-DGJ (1.2 μM in fresh media every 5 days) would result in further increases in β-Gal activity for the 5 fibroblast lines tested in Table 1. The data presented in Table 2 show that GM1 fibroblasts 4032 and Morquio B 4080, which did not show any response to NN-DGJ after 5 days, were still nonresponsive after the extended treatment. Similarly, fibroblasts 4992 showed the same fold-enhancement (4-fold increase) to both short and prolonged NN-DGJ treatment. By contrast, in the two most responsive fibroblast lines, 14771 (juvenile) and 3633 (adult), the levels of β-Gal enzyme activities were increased a further ~2-fold after the extended treatment (relative to the mock treated cells).
To date more than 130 mutations associated with GM1 have been described. Out of the 7 GM1 missense mutants we report here, four have never been tested in patient cells for their ability to respond to EET. They are D332N (type 1) and W509C (type 3), which we now report as being responsive, and R148S (type 1/2) and R482H (common in Italian type 1 patients), which we find to be nonresponsive. In agreement with others, we found that R201C/H (common in Japanese type 2 patients) is responsive, while W273L (common in Caucasian Morquio B patients) is nonresponsive [25,40,42]. Of the 11 mutations associated with Morquio B mapped to the new crystal structure of β-Gal by Ohto et al. [30], only two were found to be involved in ligand binding, W273L and Y83C/H. Another Morquio B mutation that we examined, G438E, mapped to the surface of the protein. We found that this mutation, like W273L, was not responsive to NN-DGJ treatment. However, G438E has previously been reported to be mildly responsive to another derivative of DGJ [43]. Additionally one type 2, Y333H, and one type 3, Y270D, mutation were also considered to affect ligand binding and 11 of 39 mutations associated with type 1 affected surface residues. Thus, as concluded by Ohto et al., even with the new information provided by the crystal structure it is still not possible to explain the detrimental phenotypes resulting from most of the β-Gal missense mutations. It follows that, outside of those residues that are directly involved in either substrate binding or catalysis, there are also insufficient data available to predict which missense mutations will respond to EET.
3.6. The GM1 feline model of juvenile human GM1 is responsive to NN-DGJ
Feline GM1 fibroblasts were grown in the presence of increasing concentration of NN-DGJ for 3 days in parallel with mock treated cells. The NN-DGJ treated cat cells exhibited an increase in β-Gal residual activity of ~6 fold between 0.7 and 2.1 μM of NN-DGJ, relative to non-treated cells (Fig. 6). In contrast, total Hex activity in the same samples remained unaffected. The 6-fold increase of β-Gal residual enzyme activity with the unchanged values of Hex demonstrates that NN-DGJ acts as a specific PC for the cat enzyme and is non-toxic towards the feline cells.
Fig. 6.
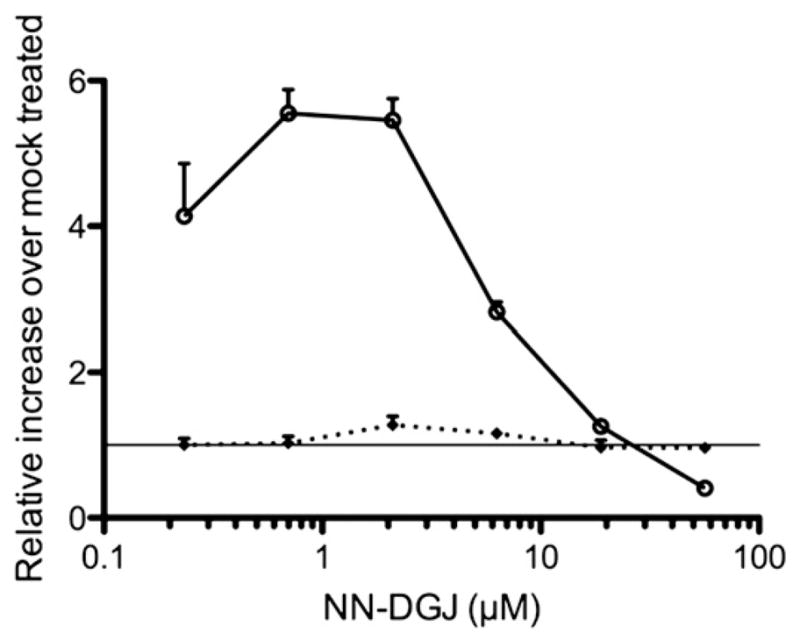
Dose response of feline juvenile GM1 fibroblasts to NN-DGJ: Fibroblasts derived from GM1 cats were grown in the presence of increasing concentration of NN-DGJ for 3 days. Enzyme activities for β-Gal (circle) and total hexosaminidase (diamond) were measured in cell lysates. The enzyme activities in the treated cells (N=3) are expressed as fold increase relative to mock treated cell (Y=1 represents no enhancement) and plotted versus the [NN-DGJ] (μM) on a logarithmic scale. Data are expressed as mean+SD.
3.7. NN-DGJ protects the mutant feline β-Gal against heat denaturation and partially corrects its lysosomal targeting deficiency
Cell lysates containing feline p.R483H mutant β-Gal (~6% residual WT activity) were mixed with NN-DGJ or DMSO and incubated at 48 °C for up to 30 min, the remaining enzyme activity was compared to untreated controls. In the absence of NN-DGJ the T50 of the enzyme was 6 min. In the presence of 100 nM of NN-DGJ the T50 increased to 13 min and in the presence of 200 nM it increased to 19 min.
To examine lysosomal targeting of cat β-Gal, treated and untreated GM1 feline fibroblasts (GM1) were examined by indirect immunofluorescence staining followed by confocal imaging and compared to normal (wild type, WT) feline fibroblasts (Fig. 7). In “Feline WT” fibroblasts, β-Gal (in green) displayed a pattern comparable to the one observed for WT human fibroblasts (Fig. 3), i.e. a punctate pattern preferentially co-localizing with Lamp-2 (left “Merge” panel). The staining intensity for β-Gal in the untreated GM1 fibroblasts (GM1+DMSO panels) is reduced and with fractions that co-localize with either Lamp-2 or PDI. As observed for the human GM1 fibroblasts, the addition of NN-DGJ (1.2 μM for 3 days) to the cat GM1 fibroblast growth media (GM1+ NN-DGJ panels) resulted in a strong increase in fluorescence staining for β-Gal (in green), consistent with the ~6 fold enhancement previously observed in the enzyme activity, which now predominantly co-localizes with Lamp-2.
Fig. 7.
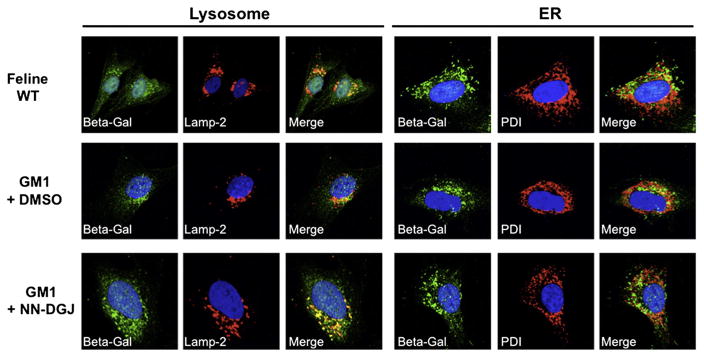
NN-DGJ treatment restores the intra-cellular localization of feline β-Gal: Feline GM1 fibroblasts were grown in the absence or presence of 1.2 μM of NN-DGJ and indirect immunofluorescence staining followed by confocal images analyses were performed. Wild type cat fibroblasts were processed in parallel as controls for the staining pattern. All cells were probed with a rabbit polyclonal IgG raised against the β-Gal precursor protein (green). Antibodies for sub-cellular localization (red) included lysosomal associated membrane protein-2 (Lamp-2, left panel) and protein disulfide isomerase (PDI, right panel) to identify lysosomes and the ER, respectively. Nuclei were stained with DAPI. Merged images are shown to help with visualization of the co-localization of β-Gal with Lamp-2 or PDI (yellow-orange). WT, wild type feline N SV3 fibroblasts; GM1+NN-DGJ, feline GM1-SV3 fibroblasts (p.R483P/p.R483P) grown in the presence of 1.2 μM of NN-DGJ for 3 days; GM1-SV3+DMSO, GM1 feline fibroblasts were grown in the absence of NN-DGJ but in the presence of DMSO used as solvent. The cell(s) shown in each pannel is representive of at least 10 cells visualized.
The hypothesis that treatment with an inhibitor can lead to increased residual enzyme activity is often viewed as counter intuitive. Thus, despite the many promising results obtained using patient cells, EET remains a controversial therapeutic strategy for LSDs. The major roadblock in validating this approach remains the lack of a suitable animal model. This is particularly true for the neurodegenerative LSDs like GM1, as ganglioside accumulation occurs during embryonic and early neonatal periods in the infantile and juvenile onset forms, indicating that early intervention may be necessary to effectively treat the disease [44]. At present it is not known to what extent any preexisting neurological damage can be reversed, even if it were possible to correct the enzyme deficiency. Stopping the progression of the disease may be the best possible outcome achievable for any therapy. Identification of an appropriate animal model would allow investigators to explore the effects on the clinical outcome of the initiation of treatment before GM1 storage becomes extensive or deleterious, i.e. administration at birth or even to the pregnant female, or at different times during the evolution of the disease to determine when or if reversal of the clinical phenotype can be obtained.
The feline GM1 model represents the first large animal model to be described in the literature, that is suitable to test the validity of EET in vivo. The feline mutation, p.R483P, is very similar to mutations observed in the aligned Arg482 residue in some GM1 patients, e.g., p.R482H or p.R482C. However in humans such mutations are usually associated with an infantile phenotype [45], whereas affected felines present with a phenotype more consistent with juvenile GM1 in humans [31]. Additionally the fibroblasts from patient 8981 (p.R148S/p.R482H) or 4080 (p.W273L/p.R482H) fail to show any β-Gal enhancement after treatment with NN-DGJ, despite the ~6-fold increase we document for the GM1 feline cells. Coupled with the more severe phenotype expressed in humans, these observations suggest that the substitutions at p.R482 in humans produces a more highly destabilized protein than does the substitution at the aligned residue in cats. Despite these differences, feline GM1 cells express 6% residual β-Gal activity, which can be increased to ~40% by NN-DGJ treatment. Achieving a similar level of enzyme enhancement in vivo, with NN-DGJ or any other candidate PC molecule, should result in an improved clinical outcome in these animals if the PC can be displaced by the stored substrate and/or the acidic pH, once the PC/β-Gal complex enters the lysosome.
4. Conclusion
The increases in mutant β-Gal enzyme activity, and in its staining intensity and co-localization with Lamp-1 or -2 in the immunofluorescence images of NN-DGJ treated human or feline GM1 cells, are strong indications that NN-DGJ is acting as a PC in cell lines from either species. Additionally, since the feline model biochemically, molecularly and clinically parallels the human juvenile form of GM1, it represents the first large animal model that can be exploited to validate and optimize the EET approach for treating lysosomal storage disorders associated with protein misfolding.
Acknowledgments
This work was supported by Canadian Institutes for Health Research (CIHR) grants (Team grant CTP-82944, and Operating grant MOP-119487) to DJM, and a US Lysosomal Storage Disease Research Consortium grant to DRM. The authors thank M. Woodside and P. Paroutis (Hospital for Sick Children Imaging Facility) for their technical assistance with confocal microscopy. The Lamp-1 and -2 antibodies used in this report were obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by the University of Iowa, Department of Biological Sciences, Iowa City, IA 52242.
Footnotes
Non-standard abbreviations used: β-D-galactosidase, β-Gal; lysosomal storage diseases, LSDs, GM1 gangliosidosis, GM1; enzyme replacement therapy, ERT; substrate reduction therapy, SRT; N-butyl-deoxynojirimycin, NB-DNJ; enzyme enhancement therapy, EET; pharmacological chaperone, PC; N-octyl-4-epi-β-valienamine, NOEV; N-nonyl-deoxygalactonojirimycin, NN-DGJ; N-butyl-deoxygalactonojirimycin, NB-DGJ; 1-deoxygalactonojirimycin, DGJ; α-D-galactosidase, α-Gal; 4-methylumbelliferyl-β-D-galactopyranoside, MU-βGal; 4-methylumbelliferyl-N-acetylglucosaminide, MUG; 4-methylumbelliferyl-α-D-galactopyranoside, MU-αGal; taurodeoxycholic acid, TC; immortalized fibroblasts from normal, N SV3, or GM1 gangliosidosis cats, GM1 SV3; McIlvaine 0.1 M citrate — 0.2 M phosphate buffer, CP; 4-methylumbelliferone, MU; wild-type, WT; inhibitor concentration at which 50% of the original enzyme activity remains, IC50; time at which 50% of the original enzyme activity remains (at a given temperature), T50; protein disulfide isomerase, PDI; lysosomal associated membrane protein, Lamp.
Conflict of interests
The authors declare no con3ict of interest.
References
- 1.Suzuki Y, Nanba E, Matsuda J, Higaki K, Oshima A. β-Galactosidase deficiency (β-galactosidase): GM1 gangliosidosis and Morquio B disease. In: Valle D, Beaudet AL, Volgelstein B, Kinzler KW, Antonarakis SF, Ballabio A, editors. The On-Line Metabolic Basis of Inherited Disease. McGraw-Hill; New York: 2008. pp. 1–101. http://www.ommbid.com/OMMBID/a/c.html/lysosomal_disorders/galactosidase_deficiency_galactosidosis_gm101_gangliosidosis_morquio_disease. [Google Scholar]
- 2.Conzelmann E, Sandhoff K. Partial enzyme deficiencies: residual activities and the development of neurological disorders. Dev Neurosci. 1983;6:58–71. doi: 10.1159/000112332. [DOI] [PubMed] [Google Scholar]
- 3.Desnick RJ. Enzyme replacement and enhancement therapies for lysosomal diseases. J Inherit Metab Dis. 2004;27:385–410. doi: 10.1023/B:BOLI.0000031101.12838.c6. [DOI] [PubMed] [Google Scholar]
- 4.Cabrera-Salazar MA, Novelli E, Barranger JA. Gene therapy for the lysosomal storage disorders. Curr Opin Mol Ther. 2002;4:349–358. [PubMed] [Google Scholar]
- 5.Pastores GM, Barnett NL. Substrate reduction therapy: miglustat as a remedy for symptomatic patients with Gaucher Disease type 1. Expert Opin Investig Drugs. 2003;12:273–281. doi: 10.1517/13543784.12.2.273. [DOI] [PubMed] [Google Scholar]
- 6.Belmatoug N, Burlina A, Giraldo P, Hendriksz CJ, Kuter DJ, Mengel E, Pastores GM. Gastrointestinal disturbances and their management in miglustat-treated patients. J Inherit Metab Dis. 2011;34:991–1001. doi: 10.1007/s10545-011-9368-7. [DOI] [PubMed] [Google Scholar]
- 7.Tropak MB, Mahuran D. Lending a helping hand, screening chemical libraries for compounds that enhance beta-hexosaminidase A activity in GM2 gangliosidosis cells. FEBS J. 2007;274:4951–4961. doi: 10.1111/j.1742-4658.2007.06040.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sawkar AR, Cheng WC, Beutler E, Wong CH, Balch WE, Kelly JW. Chemical chaperones increase the cellular activity of N370S beta -glucosidase: a therapeutic strategy for Gaucher Disease. Proc Natl Acad Sci U S A. 2002;99:15428–15433. doi: 10.1073/pnas.192582899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vellodi A. Lysosomal storage disorders. Br J Haematol. 2005;128:413–431. doi: 10.1111/j.1365-2141.2004.05293.x. [DOI] [PubMed] [Google Scholar]
- 10.Clarke JT, Mahuran DJ, Sathe S, Kolodny EH, Rigat BA, Raiman JA, Tropak MB. An open-label Phase I/II clinical trial of pyrimethamine for the treatment of patients affected with chronic GM2 gangliosidosis (Tay-Sachs or Sandhoff variants) Mol Genet Metab. 2011;102:6–12. doi: 10.1016/j.ymgme.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lieberman RL, D’Aquino JA, Ringe D, Petsko GA. Effects of pH and iminosugar pharmacological chaperones on lysosomal glycosidase structure and stability. Biochemistry. 2009;48:4816–4827. doi: 10.1021/bi9002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maegawa GH, Tropak MB, Buttner JD, Rigat BA, Fuller M, Pandit D, Tang L, Kornhaber GJ, Hamuro Y, Clarke JT, Mahuran DJ. Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher Disease. J Biol Chem. 2009;284:23502–23516. doi: 10.1074/jbc.M109.012393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Okumiya T, Ishii S, Kase R, Kamei S, Sakuraba H, Suzuki Y. Alpha-galactosidase gene mutations in Fabry disease: heterogeneous expressions of mutant enzyme proteins. Hum Genet. 1995;95:557–561. doi: 10.1007/BF00223869. [DOI] [PubMed] [Google Scholar]
- 14.Ishii S, Kase R, Okumiya T, Sakuraba H, Suzuki Y. Aggregation of the inactive form of human alpha-galactosidase in the endoplasmic reticulum. Biochem Biophys Res Commun. 1996;220:812–815. doi: 10.1006/bbrc.1996.0486. [DOI] [PubMed] [Google Scholar]
- 15.Zhang S, Bagshaw R, Hilson W, Oho Y, Hinek A, Clarke JT, Callahan JW. Characterization of beta-galactosidase mutations Asp332–>Asn and Arg148–>Ser, and a polymorphism, Ser532–>Gly, in a case of GM1 gangliosidosis. Biochem J. 2000;348(Pt 3):621–632. [PMC free article] [PubMed] [Google Scholar]
- 16.Perlmutter DH. Chemical chaperones: a pharmacological strategy for disorders of protein folding and trafficking. Pediatr Res. 2002;52:832–836. doi: 10.1203/00006450-200212000-00004. [DOI] [PubMed] [Google Scholar]
- 17.Phaneuf D, Wakamatsu N, Huang JQ, Borowski A, Peterson AC, Fortunato SR, Ritter G, Igdoura SA, Morales CR, Benoit G, Akerman BR, Leclerc D, Hanai N, Marth JD, Trasler JM, Gravel RA. Dramatically different phenotypes in mouse models of human Tay-Sachs and Sandhoff diseases. Hum Mol Genet. 1996;5:1–14. doi: 10.1093/hmg/5.1.1. [DOI] [PubMed] [Google Scholar]
- 18.Sango K, Yamanaka S, Hoffmann A, Okuda Y, Grinberg A, Westphal H, McDonald MP, Crawley JN, Sandhoff K, Suzuki K, Proia RL. Mouse models of Tay-Sachs and Sandhoff diseases differ in neurologic phenotype and ganglioside metabolism. Nat Genet. 1995;11:170–176. doi: 10.1038/ng1095-170. [DOI] [PubMed] [Google Scholar]
- 19.Brown CA, Mahuran DJ. Beta-hexosaminidase isozymes from cells co-transfected with alpha and beta cDNA constructs: analysis of alpha subunit missense mutation associated with the adult form of Tay-Sachs disease. Am J Hum Genet. 1993;53:497–508. [PMC free article] [PubMed] [Google Scholar]
- 20.Wu BM, Tomatsu S, Fukuda S, Sukegawa K, Orii T, Sly WS. Overexpression rescues the mutant phenotype of L176F mutation causing beta-glucuronidase deficiency mucopolysaccharidosis in two Mennonite siblings. J Biol Chem. 1994;269:23681–23688. [PubMed] [Google Scholar]
- 21.Matsuda J, Suzuki O, Oshima A, Yamamoto Y, Noguchi A, Takimoto K, Itoh M, Matsuzaki Y, Yasuda Y, Ogawa S, Sakata Y, Nanba E, Higaki K, Ogawa Y, Tominaga L, Ohno K, Iwasaki H, Watanabe H, Brady RO, Suzuki Y. Chemical chaperone therapy for brain pathology in G(M1)-gangliosidosis. Proc Natl Acad Sci U S A. 2003;100:15912–15917. doi: 10.1073/pnas.2536657100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hahn CN, del Pilar Martin M, Schroder M, Vanier MT, Hara Y, Suzuki K, Suzuki K, d’Azzo A. Generalized CNS disease and massive GM1 ganglioside accumulation in mice defective in lysosomal acid beta-galactosidase. Hum Mol Genet. 1997;6:205–211. doi: 10.1093/hmg/6.2.205. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki Y, Ichinomiya S, Kurosawa M, Ohkubo M, Watanabe H, Iwasaki H, Matsuda J, Noguchi Y, Takimoto K, Itoh M, Tabe M, Iida M, Kubo T, Ogawa S, Nanba E, Higaki K, Ohno K, Brady RO. Chemical chaperone therapy: clinical effect in murine G(M1)-gangliosidosis. Ann Neurol. 2007;62:671–675. doi: 10.1002/ana.21284. [DOI] [PubMed] [Google Scholar]
- 24.Suzuki Y, Ichinomiya S, Kurosawa M, Matsuda J, Ogawa S, Iida M, Kubo T, Tabe M, Itoh M, Higaki K, Nanba E, Ohno K. Therapeutic chaperone effect of N-Octyl 4-Epi-beta-valienamine on murine G(M1)-gangliosidosis. Mol Genet Metab. 2012;106:92–98. doi: 10.1016/j.ymgme.2012.02.012. [DOI] [PubMed] [Google Scholar]
- 25.Iwasaki H, Watanabe H, Iida M, Ogawa S, Tabe M, Higaki K, Nanba E, Suzuki Y. Fibroblast screening for chaperone therapy in beta-galactosidosis. Brain Dev. 2006;28:482–486. doi: 10.1016/j.braindev.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Yamato O, Endoh D, Kobayashi A, Masuoka Y, Yonemura M, Hatakeyama A, Satoh H, Tajima M, Yamasaki M, Maede Y. A novel mutation in the gene for canine acid beta-galactosidase that causes GM1 gangliosidosis in Shiba dogs. J Inherit Metab Dis. 2002;25:525–526. doi: 10.1023/a:1021280007739. [DOI] [PubMed] [Google Scholar]
- 27.Yamato O, Masuoka Y, Yonemura M, Hatakeyama A, Satoh H, Kobayashi A, Nakayama M, Asano T, Shoda T, Yamasaki M, Ochiai K, Umemura T, Maede Y. Clinical and clinico-pathologic characteristics of Shiba dogs with a deficiency of lysosomal acid beta-galactosidase: a canine model of human GM1 gangliosidosis. J Vet Med Sci. 2003;65:213–217. doi: 10.1292/jvms.65.213. [DOI] [PubMed] [Google Scholar]
- 28.Martin DR, Rigat BA, Foureman P, Varadarajan GS, Hwang M, Krum BK, Smith BF, Callahan JW, Mahuran DJ, Baker HJ. Molecular consequences of the pathogenic mutation in feline GM1 gangliosidosis. Mol Genet Metab. 2008;94:212–221. doi: 10.1016/j.ymgme.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Georgiou T, Stylianidou G, Anastasiadou V, Caciotti A, Campos Y, Zammarchi E, Morrone A, D’Azzo A, Drousiotou A. The Arg482His mutation in the beta-galactosidase gene is responsible for a high frequency of GM1 gangliosidosis carriers in a Cypriot village. Genet Test. 2005;9:126–132. doi: 10.1089/gte.2005.9.126. [DOI] [PubMed] [Google Scholar]
- 30.Ohto U, Usui K, Ochi T, Yuki K, Satow Y, Shimizu T. Crystal structure of human beta-galactosidase: the structural basis of GM1 gangliosidosis and Morquio B diseases. J Biol Chem. 2012;287:1801–1812. doi: 10.1074/jbc.M111.293795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Baker HJ, Jr, Lindsey JR, McKhann GM, Farrell DF. Neuronal GM1 gangliosidosis in a Siamese cat with beta-galactosidase deficiency. Science. 1971;174:838–839. doi: 10.1126/science.174.4011.838. [DOI] [PubMed] [Google Scholar]
- 32.Butters TD, Mellor HR, Narita K, Dwek RA, Platt FM. Small-molecule therapeutics for the treatment of glycolipid lysosomal storage disorders. Philos Trans R Soc Lond B. 2003;358:927–945. doi: 10.1098/rstb.2003.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Andersson U, Butters TD, Dwek RA, Platt FM. N-butyldeoxygalactonojirimycin: a more selective inhibitor of glycosphingolipid biosynthesis than N-butyldeoxynojirimycin, in vitro and in vivo. Biochem Pharmacol. 2000;59:821–829. doi: 10.1016/s0006-2952(99)00384-6. [DOI] [PubMed] [Google Scholar]
- 34.Platt FM, Neises GR, Dwek RA, Butters TD. N-butyldeoxynojirimycin is a novel inhibitor of glycolipid biosynthesis. J Biol Chem. 1994;269:8362–8365. [PubMed] [Google Scholar]
- 35.Asano N, Ishii S, Kizu H, Ikeda K, Yasuda K, Kato A, Martin OR, Fan JQ. In vitro inhibition and intracellular enhancement of lysosomal alpha-galactosidase A activity in Fabry lymphoblasts by 1-deoxygalactonojirimycin and its derivatives. Eur J Biochem. 2000;267:4179–4186. doi: 10.1046/j.1432-1327.2000.01457.x. [DOI] [PubMed] [Google Scholar]
- 36.Mahuran D, Lowden JA. The subunit and polypeptide structure of hexosaminidases from human placenta. Can J Biochem. 1980;58:287–294. doi: 10.1139/o80-038. [DOI] [PubMed] [Google Scholar]
- 37.Hubbes M, D’Agrosa RM, Callahan JW. Human placental beta-galactosidase: characterization of the dimer and complex forms of the enzyme. Biochem J. 1992;285:827–831. doi: 10.1042/bj2850827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rigat B, Mahuran D. Diltiazem, a L-type Ca(2+) channel blocker, also acts as a pharmacological chaperone in Gaucher patient cells. Mol Genet Metab. 2009;96:225–232. doi: 10.1016/j.ymgme.2008.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan X, Tkachyova I, Sinha A, Rigat B, Mahuran D. Characterization of the biosynthesis, processing and kinetic mechanism of action of the enzyme deficient in mucopolysaccharidosis IIIC. PLoS One. 2011;6:e24951. doi: 10.1371/journal.pone.0024951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tominaga L, Ogawa Y, Taniguchi M, Ohno K, Matsuda J, Oshima A, Suzuki Y, Nanba E. Galactonojirimycin derivatives restore mutant human beta-galactosidase activities expressed in fibroblasts from enzyme-deficient knockout mouse. Brain Dev. 2001;23:284–287. doi: 10.1016/s0387-7604(01)00216-9. [DOI] [PubMed] [Google Scholar]
- 41.Sugawara K, Tajima Y, Kawashima I, Tsukimura T, Saito S, Ohno K, Iwamoto K, Kobayashi T, Itoh K, Sakuraba H. Molecular interaction of imino sugars with human alpha-galactosidase: insight into the mechanism of complex formation and pharmacological chaperone action in Fabry disease. Mol Genet Metab. 2009;96:233–238. doi: 10.1016/j.ymgme.2008.12.017. [DOI] [PubMed] [Google Scholar]
- 42.Fantur KM, Wrodnigg TM, Stutz AE, Pabst BM, Paschke E. Fluorous iminoalditols act as effective pharmacological chaperones against gene products from GLB1 alleles causing G(M1)-gangliosidosis and Morquio B disease. J Inherit Metab Dis. 2012;35:495–503. doi: 10.1007/s10545-011-9409-2. [DOI] [PubMed] [Google Scholar]
- 43.Fantur K, Hofer D, Schitter G, Steiner AJ, Pabst BM, Wrodnigg TM, Stutz AE, Paschke E. DLHex-DGJ, a novel derivative of 1-deoxygalactonojirimycin with pharmacological chaperone activity in human G(M1)-gangliosidosis fibroblasts. Mol Genet Metab. 2010;100:262–268. doi: 10.1016/j.ymgme.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 44.Takaura N, Yagi T, Maeda M, Nanba E, Oshima A, Suzuki Y, Yamano T, Tanaka A. Attenuation of ganglioside GM1 accumulation in the brain of GM1 gangliosidosis mice by neonatal intravenous gene transfer. Gene Ther. 2003;10:1487–1493. doi: 10.1038/sj.gt.3302033. [DOI] [PubMed] [Google Scholar]
- 45.Mosna G, Fattore S, Tubiello G, Brocca S, Trubia M, Gianazza E, Gatti R, Danesino C, Minelli A, Piantanida M. A homozygous missense arginine to histidine substitution at position 482 of the beta-galactosidase in an Italian infantile GM1 gangliosidosis patient. Hum Genet. 1992;90:247–250. doi: 10.1007/BF00220071. [DOI] [PubMed] [Google Scholar]