

Abstract
High salt (4%NaCl, HS) diet modulates adenosine-induced vascular response through adenosine A2A-receptor (A2AAR). Evidence suggests A2AAR stimulates cyp450-epoxygenases, leading to epoxyeicosatrienoic acids (EETs) generation. The aim of this study was to understand the vascular reactivity to HS and underlying signaling mechanism in the presence or absence of A2AAR. Therefore, we hypothesized that HS enhances adenosine-induced relaxation through EETs in A2AAR+/+, but exaggerates contraction in A2AAR−/−. Organ-bath and Western-blot experiments were conducted in HS and normal salt (NS, 0.18% NaCl)-fed A2AAR+/+ and A2AAR−/− mice aortae. HS produced concentration-dependent relaxation to non-selective adenosine analog, NECA in A2AAR+/+, whereas contraction was observed in A2AAR−/− mice and this was attenuated by A1AR antagonist (DPCPX). CGS-21680 (selective A2AAR-agonist) enhanced relaxation in HS-A2AAR+/+ vs. NS-A2AAR+/+, that was blocked by EETs antagonist (14,15-EEZE). Compared to NS, HS significantly upregulated expression of vasodilators A2AAR and cyp2c29, while vasoconstrictors A1AR and cyp4a in A2AAR+/+ were downregulated. In A2AAR−/− mice, however, HS significantly downregulated the expression of cyp2c29, while A1AR and cyp4a were upregulated compared to A2AAR+/+ mice. Hence, our data suggest that in A2AAR+/+, HS enhances A2AAR-induced relaxation through increased cyp-expoxygenases-derived EETs and decreased A1AR levels, whereas in A2AAR−/−, HS exaggerates contraction through decreased cyp-epoxygenases and increased A1AR levels.
Keywords: HS, NS, A2AAR, cyp4a, cyp2c29, A1AR, relaxation, contraction
Introduction
Adenosine, a breakdown product of ATP, acts as a local modulator with generally cytoprotective function in response to stressful conditions such as ischemia, hypoxia, injury, and inflammation (23, 26). Besides many physiological effects elicited by extracellular adenosine such as inhibition of renin release, platelet aggregation, inflammation, lipolysis and sympathetic neurotransmission (34), its effect on vasculature is crucial for vasoregulation (14) and physiological control of mean arterial pressure (MAP), (1, 19). Adenosine mediates its effects via four adenosine receptors: A1, A2A, A2B, and A3 (10). Activation of A2AAR and A2BAR are known to mediate vasodilation in different vessels (6, 8, 16, 28-29, 32), whereas A1AR and A3AR are believed to be involved in vasoconstriction (12, 14). Adenosine receptors especially A1AR and A2AAR are also involved in the regulation of blood pressure (BP). A2AAR causes renal vasodilation (13) and attenuates tubuloglomerular feedback (5). A2AAR knockout mice as well as rats treated with selective A2AAR antagonist displayed an elevated BP (27), (19). A1AR is involved in water and salt retention (24), mediating tubuloglomerular feedback response (35), constriction of afferent arterioles (38), lowering of heart rate and suppression of renin release (2).
Growing evidence suggests a link between high dietary salt intake and activation of adenosine receptors and A2A receptor in particular (22, 29-30). Data demonstrating upregulation of A2AAR expression in response to salt loading (21, 29), accompanied by enhanced A2AAR mediated vasodilation (6, 21-22, 29-30) further support the role of adenosine A2AAR during high salt (HS) diet. Stimulation of A2AAR is reported to result in an eNOS-independent vasodilation (30) and reduction in MAP (1). A1AR, responsible for vasonstriction, is shown to be downregulated in HS treated animals (21, 29). In addition, elevated levels of adensoine generation have been detected upon the exposure to HS diet in rats (33, 39).
Apart from adenosine, another important vasoregulatory factor associated with HS diet is cyp-epoxygenase enzyme that belongs to cytochrome P450s (cyp450) family (9, 15). Two major subtypes of cyp450 enzymes include cyp-epoxygenases and ω-hydroxylases that play an important role in regulating vascular tone (9, 15, 18). It is well documented that cyp- epoxygenases catalyze the metabolism of arachidonic acid (AA) into epoxyeicosatrienoic acids (EETs), a potent vasodilator and natriuretic agent (3), whereas, ω-hydroxylase metabolizes AA to 20-hydroxyeicosatetraenoic acid (20-HETE), a potent vasoconstrictor (11, 28-29) and natriuretics. HS diet is associated with increase in renal cyp-expoxygenases activity as a protective mechanism against rise in BP (4, 20-21). Inhibition of cyp-epoxygenases activity with clotrimazole is shown to contribute to the surge in MAP and development of salt sensitivity in rats fed HS diet, whereas salt itself was unable to increase blood pressure (25). Unlike EETs, the production of 20-HETE is found to be lower in glomeruli isolated from kidneys of rats fed HS diet than in kidneys of rats fed low salt diet (17). Increased levels of 20-HETE is shown to promote vasoconstriction that contributes to the progression of hypertension (11, 37).
Emerging evidence indicates a close relationship between HS-induced activation of A2AAR and EETs generation through enhanced cyp-epoxygenase activity (7, 30-31). In previous study, we found that non-selective adenosine analaog (NECA) and selective adenosine A2A receptor agonist (CGS-21680) showed enhanced dilation in aortae from mice fed HS compared to mice fed low salt diet and the enhanced dilation in HS diet fed mice was blunted by cyp epoxygenase inhibitor, methylsulfonyl-propargyloxyphenylhexanamide (MSPPOH) (29). Moreover, Liclican et al., (21) reported that inability to upregulate the adenosine A2AAR–EETs pathway in response to salt loading confers salt sensitive hypertension in rats.
Although there is some indication as to the possible adaptive role of A2AAR receptor and cyp epoxygenases mediated vasodilation during HS diet, studies evaluating the ramifications of HS diet in terms of vascular response and biochemical signaling resulting from the A2AAR gene deletion are lacking. On the basis of these prior investigations, it is important to examine the effects of HS diet on vascualr reacitivity in the absence of A2AAR in order to directly evaluate the role of A2AAR and more importantly to understand the underlying mechanism involved in vascular pathophysiological changes. Therefore, using HS and NS fed A2AAR+/+ and A2AAR−/− mice, we aim to determine the role of A2AAR on vascular response and its downstream mechanism.
MATERIALS AND METHODS
The experimental and animal care protocols used in this study were approved by the West Virginia University Institutional Animal Care and Use Committee and carried out according to the principles and guidelines of the Institute of Laboratory Animal Resources Guide for the Care and Use of Laboratory Animals. Ten-weeks-old inbred male and female CD-1 (A2AAR−/− and A2A AR+/+ mice) obtained from Dr. Ledent (Belgium), bred and maintained in our facility at West Virginia University were placed on a whole-grain diet containing either 0.18% NaCl, normal salt (NS) or 4% NaCl, high salt (HS) (TD88311 and TD92100 diets; Teklad, Madison, WI). The initial characterization of A2AAR+/+ and A2AAR−/− mice have been previously described by Ledent et al., (19). In brief, to produce mice on a homogenous genetic background, first-generation heterozygotes were bred for 14 generations to mice on a CD-1 (Charles River Laboratories) outbred background, with selection for the mutant A2AAR gene at each generation by PCR. Fourteenth generation heterozygotes were bred together to generate A2AAR+/+ and A2AAR−/− (1:1, their mate controls) mice (19). All mice were studied 4-5 weeks after assignment to either the NS or HS group.
Isometric tension Muscle bath experiments
HS and NS fed A2AAR+/+ and A2AAR−/− mice were euthanized with pentobarbital sodium (100 mg/kg, i.p.). The aorta was gently removed after thoracotomy, cleaned of fat and connective tissues, and cut transversely into rings of 3-4 mm in length as described previously by us (28-29). Extreme care was taken not to damage the endothelium. The rings were mounted vertically between two stainless steel wire hooks. The two rings were suspended in 10 ml organ baths containing modified Krebs-Henseleit buffer (in 118 mM of NaCl, 4.8 mM of KCl, 1.2 mM of MgSO4, 1.2 mM of KH2PO4, 25 mM of NaHCO3, 11 mM of glucose, and 2.5 mM of CaCl2). The buffer was maintained around pH 7.4 at 37°C. The aortic rings were equlibrated for 60 min with a resting force of 1 g as described previously by us (28-29). At the end of the equilibration period, tissues were contracted with KCl (50 mM) to check the viability. Aortic rings were then constricted with phenylephrine (PE, 10−6 M) and changes in tension were monitored continuously with a fixed range precision force transducer (TSD, 125 C, BIOPAC system). Data were recorded using MP100 WSW, BIOPAC digital acqusition system and analyzed using Acknowledge 3.5.7 software (BIOPAC system). The vascular endothelium was tested to determine whether it was intact, as previously described by our laboratory (28-29) through acetylcholine (ACh, 10−6 M) on precontracted aortic rings with phenylephrine (PE). Preparations were then washed several times with Krebs-Henseleit buffer solution and allowed to equilibrate for 30 min before the experimental protocol began. For all tests, the contraction and relaxation responses are expressed as % decrease or % increase of PE-induced precontraction.
Adenosine agonist-induced vascular response in A2AAR+/+ and A2AAR−/− mice fed HS and NS diet
The responsiveness of pre-contracted aortic rings from A2AAR+/+ and A2AAR−/− mice fed HS and NS diet to non-selective adenosine analog, 5’-N-ethylcarboxamidoadenosine (NECA), the selective A2A receptor agonist, 2-p-(2-carboxyethyl) phenethylamino-5′-N-ethylcarboxamido adenosine hydrochloride (CGS 21680), or selective A1 receptor agonist, 2-chloro-N(6)-cyclopentyladenosine (CCPA) were obtained by cumulative addition of these drugs to the organ bath in 1-log increments to obtain a concentration-response curve (CRC) as previously described (28-29). All concentration response determinations were run in parallel on pairs of rings from either HS (A2AAR+/+ and A2AAR−/−) or NS (A2AAR+/+ and A2AAR−/−).
Effects of A1AR and A2AAR antagonists, and cyp4a inhibitor on NECA and 20-HETE-induced vascular response in A2AAR+/+ and A2AAR−/− mice fed HS and NS diet
Selective A1AR antagonist, 8-cyclopentyl-1,3-dipropylxanthine (DPCPX; 10 μM) or selective A2AAR antagonist (SCH 58261; 10 μM) or selective cyp4a inhibitor, dibromo-dodecenyl-methylsulfimide (DDMS; 10 μM) was added 30 min before contraction of the tissue with PE and was present throughout the experiment. These experiments were performed in parallel on four rings from the same aorta with two serving as control and two treated with either DPCPX or SCH 58261 or DDMS respectively.
Effects of EETs antagonist on CGS 21680-induced vascular response in A2AAR+/+ and A2AAR−/− mice fed HS and NS
Selective EET antagonist, 14,15-epoxyeicosa-5(z)-enoic acid (14,15-EEZE;10 μM) was added 30 min before contraction of the tissue with PE, and was present throughout the experiment. These experiments were performed in parallel on four rings from the same aorta with two serving as control and two treated 14,15-EEZE.
Western-blot analysis
Aortae from HS and NS fed A2AAR+/+ and A2AAR−/− mice were isolated and each sample was homogenized with 130 μL RIPA buffer (Cell Signaling Technology Inc) on wet ice. The samples were transferred to dry ice for 5 min and then thawed on wet ice. After thawing, lysates were sonicated and the samples were vortexed and centrifuged for 5 min at 12,000 rpm at 4°C. Then, the supernatant was stored at −80°C. Protein was measured using Bio-Rad assay based on the Bradford dye procedure with bovine serum albumin (BSA) as a standard. The protein mixture was divided into aliquots and stored at −80°C. At the time of analysis, samples were thawed and 30 μg of total protein per lane was loaded on a slab gel. Proteins were separated by SDS-PAGE using 10% acrylamide gels (1-mm thick). After electrophoresis, the proteins on the gel were transferred to nitrocellulose membrane (Hybond-ECL) by electroelution. Protein transfer was confirmed by employing prestained molecular weight markers (Bio-Rad Laboratories, Hercules, CA). Following blocking with either 5% nonfat dry milk or BSA, the nitrocellulose membranes were incubated with primary antibodies for cyp2c29 (Dr. Zeldin, NIEHS /NIH), cyp4a (Santa Cruz Biotecnology, Santa Cruz, CA), A1AR (Sigma Chemicals) and A2AAR (Alpha Diagnostics). 1:1000 primary antibody concentration was used for all antibodies. β-actin antibody (Santa Cruz Biotechnology, Santa Cruz, CA) was used as an internal control to normalize the target protein expression in each lane. The secondary antibody was a horseradish peroxidase-conjugated anti-rabbit or anti-mouse IgG. The membranes were developed using enhanced chemiluminescence (Amersham BioSciences) and exposed to X-ray film for the appropriate time. The data are presented as the ratio of target protein expression to β-actin.
Chemicals, drugs & antibodies
Phenylephrine hydrochloride and acetylcholine chloride were dissolved in distilled water. NECA, CGS 21680, SCH-58261, CCPA, DPCPX, indomethacin, and L-NAME (Sigma Chemicals, St. Louis, MO) were dissolved in 100% DMSO as 10 mM stock solutions, which were followed by serial dilutions in distilled water. 14,15-EEZE, DDMS and 20-HETE (Dr. Falck) were dissolved in 100% ethanol. Cyp2c9 antibody (Dr. Zeldin, NIEHS /NIH), cyp4a antibody (Santa Cruz Biotecnology), A1AR (Sigma Chemicals, St. Louis, MO) and A2AAR (Alpha Diagnostics) were used for Western-blot experiments.
Statistical Analysis
Statistical data are reported as mean ± SEM. One-way analysis of variance (ANOVA) was used to compare difference among groups, and two ways ANOVA for repeated measure, followed by Tukey post hoc test to compare the vascular responses to antagonist SCH, DPCPX, 14,15-EEZE, DDMS. Differences were considered significant if p<0.05. Further, densitometry of Western-blot analysis (cyp4a, cyp2c29, A1AR and A2AAR) data was expressed as mean ± SEM in arbitrary units. All the statistical analyses were performed using Graph Pad Prism statistical package.
RESULTS
Effects of SCH 58261 on NECA-dependent vascular response in HS and NS diet fed A2AAR+/+ and A2AAR−/− mice
NECA produced enhanced vascular relaxation in HS-fed A2AAR+/+ mice compared to NS-fed A2AAR+/+ mice (p<0.05; Fig. 1a). HS–induced vascular response to NECA was significantly different in A2AAR+/+ vs. A2AAR−/− mice (p<0.05; Fig. 1a). HS diet induced higher relaxation (+17.34 ± 2.50%) to NECA (10−6 M) in A2AAR+/+ mice whereas, HS diet caused contraction (−56.77 ± 3.49%) in A2AAR−/− mice (P<0.05; Fig. 1a). Interestingly no significant difference was observed between aortic responses in HS-fed A2AAR−/− mice and NS-fed A2AAR−/− mice (p>0.05; Fig. 1a). To pharmacologically confirm if HS-mediated relaxation is dependent on A2AAR, we used selective A2A receptor antagonist, SCH 58261 for NECA CRC (Fig.1b). SCH 58261 (1 μM) blocked NECA (10−6 M) dependent relaxation response (+17.34 ± 2.50%) in HS-fed A2AAR+/+ aortae into contraction (−12.20 ± 3.62%, p>0.05; Fig. 1b).
Fig. 1.
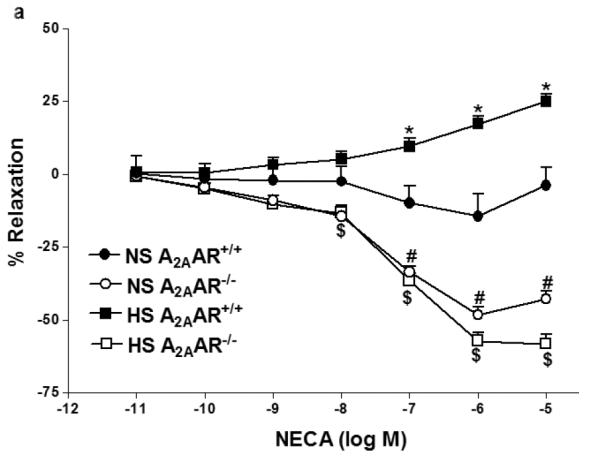
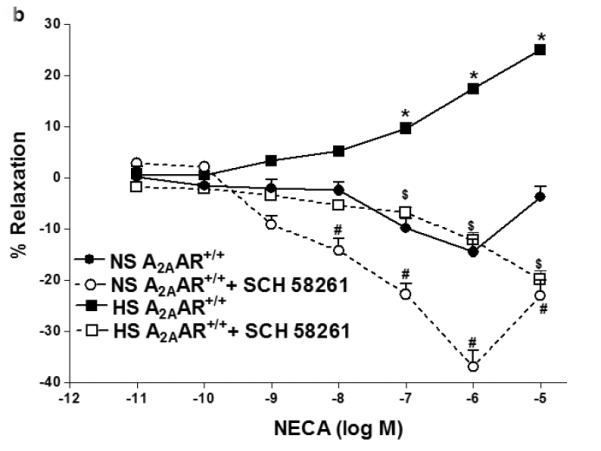
a: NECA-induced vascular responses in aortic rings of A2AAR+/+ and A2AAR−/− mice fed NS and HS diet. Values are means ± SE. *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, $p<0.05 between HS-A2AAR+/+ vs. HS-A2AAR−/− , and #p<0.05 between NS-A2AAR+/+ vs. NS-A2AAR−/−, n = 6. On the y-axis, postive and negative values represent relaxation and contraction, respectively. b: Effects of SCH 58261 (10−6 M) on NECA-induced vascular responses in aortic rings isolated from HS and NS fed mice. Values are means ± SE. *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, $p<0.05 between HS-A2AAR+/+ vs. HS-A2AAR+/+ with SCH 58261, and #p<0.05 between NS-A2AAR+/+ vs. NS-A2AAR+/+ with SCH 58261, n = 6.
Effects of L-NAME, indomethacin and 14,15-EEZE on CGS 21680-dependent vascular response in HS and NS diet fed A2AAR+/+ and A2AAR−/− mice
Selective A2AAR agonist, CGS 21680 demonstrated concentration-dependent relaxation in both HS and NS-fed A2AAR+/+ mice with a significant difference (p<0.05; Fig 2a), but no significant difference was noted between HS and NS diet fed A2AAR−/− mice (p>0.05; Fig. 2a). CGS 21680 (10−6M) produced significantly greater relaxation (+21.4 ± 1.34%) in HS-fed A2AAR+/+ mice than in NS-fed A2AAR+/+ mice (+12.96 ± 1.52; p<0.05; Fig 2a) suggesting an increased A2AAR activity with HS diet.
Fig. 2.
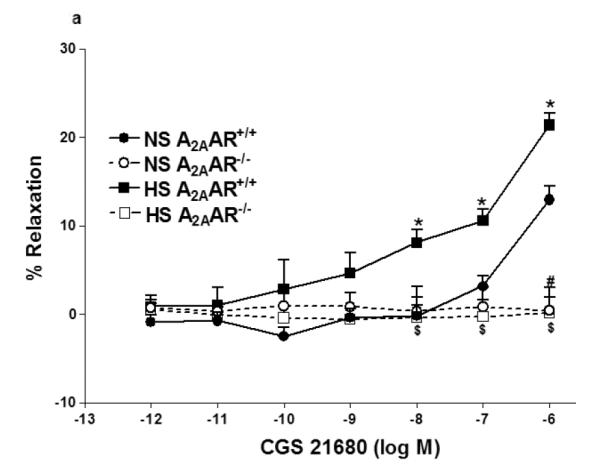
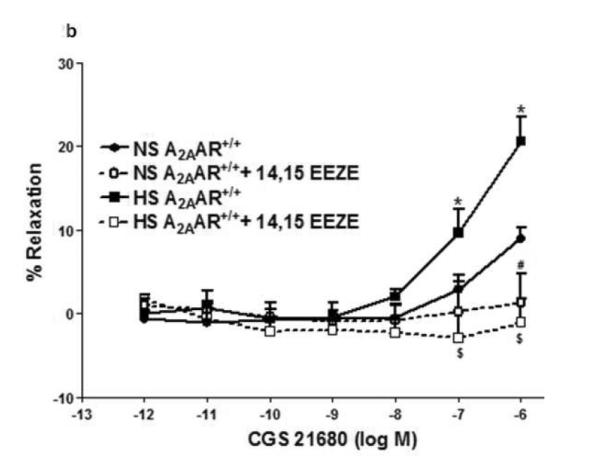
a: CGS 21680-induced vascular responses in aortic rings of A2AAR+/+ and A2AAR−/− mice fed NS and HS diet. Values are means ± SE. *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, $p<0.05 between HS-A2AAR+/+ vs. HS-A2AAR−/−, and #p<0.05 between NS-A2AAR+/+ vs. NS-A2AAR−/−, n = 6. b: Effects of 14,15-EEZE (10−5 M) on CGS 21680-induced vascular response in NS and HS fed A2AAR+/+ aortic rings. Values are mean ± SE. *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, and $p<0.05 between HS-A2AAR+/+ vs. HS-A2AAR+/+ with 14,15-EEZE, n = 6.
Relaxation response to CGS 21680 in both HS and NS-fed A2AAR+/+ mice was neither affected by cycloxygenase inhibitor, indomethacin (1 μM) (p>0.05) nor by e-NOS inhibitor, L-NAME (100 μM) (p>0.05; not shown). However, EETs antagonist, 14,15-EEZE (10 μM) completely abolished CGS 21680-dependent relaxation (from +21.4 ± 1.34% to contraction −1.08 ± 2.95 % at 10−6 M; p<0.05) in HS-fed A2AAR+/+ mice (Fig. 2b). Relaxation to CGS 21680 (10−6 M) in NS-fed A2AAR+/+ mice was also significantly blocked by 14,15-EEZE (10 μM) but to a lower extent compared to HS-fed A2AAR+/+ mice (p<0.05; Fig. 2b). These data suggest that A2AAR mediated relaxation in HS–fed A2AAR+/+ mice is independent of NO and cycloxygenase but dependent on EETs.
Effects of DDMS on NECA-dependent response in HS and NS diet fed A2AAR+/+ and A2AAR−/− mice
In Fig. 3, contraction to NECA (10−6 M) in NS-fed A2AAR+/+ mice was blocked by cyp4a inhibitor DDMS (10 μM), and the response changed from a contraction of −14.42 ± 7.74% to a relaxation of +10.0 ± 3.83% (p<0.05; Fig. 3a). Also, in NS-fed A2AAR−/− mice, DDMS (10 μM) attenuated NECA-induced contraction (from −48.27 ± 2.83% to −22.1 ± 4.37% at 10−6 M; p<0.05; Fig. 3b), suggesting the role of 20-HETE, an arachidonic acid metabolite of cyp4a, in NECA-dependent contraction. Interestingly, NECA-induced contraction in HS-fed A2AAR−/− mice was no different with 10 μM DDMS treatment (without treatment: 57.59 ± 2.84% vs. DDMS treated: 47.03 ± 4.38% at 10−6 M; p>0.05; Fig. 3b). This indicates that 20-HETE does contribute to the contraction in NS-fed A2AAR+/+ and A2AAR−/− mice but not in HS-fed A2AAR−/− mice.
Fig. 3.
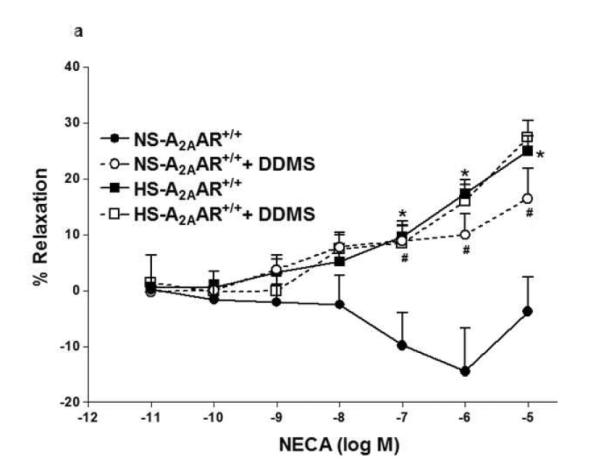
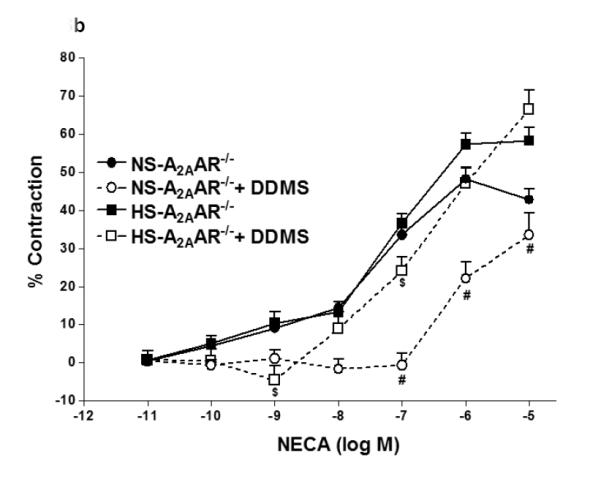
a: Effects of DDMS (10−5 M) on NECA-induced vascular response in HS and NS fed A2A AR+/+ aortic rings. Values are mean ± SE. *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, and #p<0.05 between NS-A2AAR+/+ vs. NS-A2AAR+/+ with DDMS, n = 4-6. b: Effects of DDMS (10−5 M) on NECA-induced vascular response in NS and HS fed A2AAR−/− aortic rings. Values are mean ± SE. #p<0.05 between NS-A2AAR−/− vs. NS-A2AAR−/− with DDMS, and $p<0.05 between HS-A2AAR−/− vs. HS-A2AAR−/− with DDMS, n = 4-6.
Effects of DPCPX on NECA-induced vascular response in HS and NS diet fed A2AAR+/+ and A2AAR−/− mice
Since A1 adenosine receptor is also involved in vasoconstriction (12), we investigated the role of A1AR in NECA-induced contraction in NS-A2AAR+/+ mice (Fig. 4a). Selective A1 antagonist, DPCPX (10 μM) blocked NECA (10−6 M)-dependent contraction (from −14.42 ± 7.74% to relaxation +14.96 ± 3.75%; p<0.05; Fig. 4a) in NS-fed A2AAR+/+ mice. But, DPCPX did not have any significant effect on DPCPX treated HS-fed A2AAR+/+ mice compared to non-treated HS-fed A2AAR+/+ mice (p>0.05; Fig. 4a). Further, DPCPX reversed NECA (10−6 M)-mediated contraction from 51.1 ± 3.01% to +1.02 ± 3.18% in NS-fed A2AAR−/− (p<0.05; Fig. 4b) and 57.29 ± 2.84% to 1.32 ± 5.47% in HS-fed A2AAR−/− (p<0.05; Fig. 4b). No difference was found between 10 μM DPCPX and 0.1 μM DPCPX (data not shown).
Fig. 4.
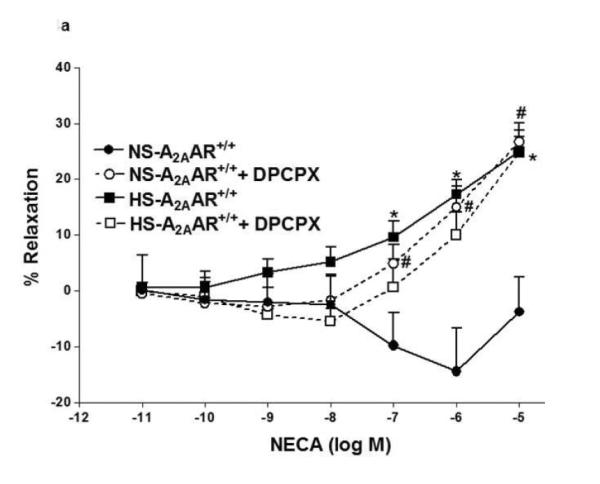
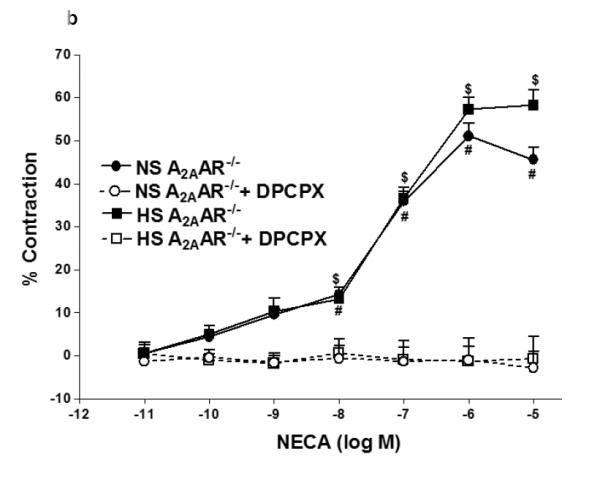
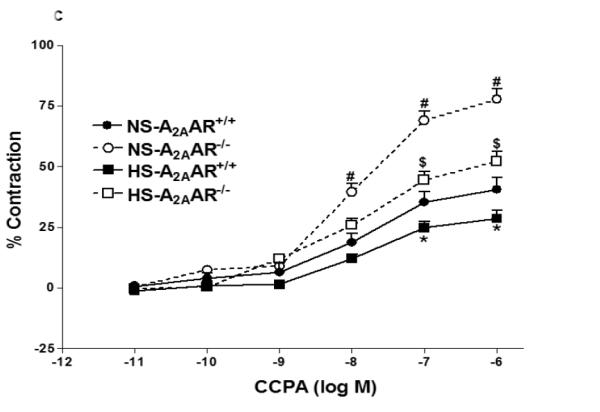
a: Effects of DPCPX (10−5 M) on NECA-induced vascular response in NS and HS fed A2AAR+/+ aortic rings. Values are mean ± SE. #p<0.05 between NS-A2AAR+/+ vs. NS-A2AAR+/+ with DPCPX, and *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, n = 4-6. b: Effects of DPCPX (10−5 M) on NECA-induced vascular response in NS and HS fed A2AAR−/− aortic rings. Values are mean ± SE. #p<0.05 between NS-A2AAR−/− vs. NS-A2AAR−/− with DDMS, and $p<0.05 between HS-A2AAR−/− vs. HS-A2AAR−/− with DDMS, n = 4-6. c: CCPA-induced vascular response in aortic rings isolated from A2A AR+/+ and A2A AR−/− mice fed NS and HS containing diet. Values are means ± SE. *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, and $p<0.05 between HS-A2AAR+/+ vs. HS-A2A AR−/− , and #p<0.05 between NS-A2AAR+/+ vs. NS-A2AAR−/−, n = 4.
CCPA-dependent vascular response in HS and NS diet fed A2AAR+/+ and A2AAR−/− mice
Selective A1 agonist, CCPA yielded higher contraction in A2AAR−/− mice compared to A2AAR+/+ mice regardless of salt content in the diet (Fig. 4c). NS-fed A2AAR−/− and HS-fed A2AAR−/− mice showed robust vascular contraction (77.73 ± 4.26% and 52.08 ± 4.29%) to CCPA (10−6 M) compared to their controls NS-fed A2AAR+/+ and HS-fed A2AAR+/+ (43.05 ± 7.51% and 29.31 ± 4.81%; p<0.05; Fig. 4c), respectively. Interestingly, CCPA (10−6 M) produced significantly lower contraction in HS-fed A2AAR−/− mice (52.08 ± 4.29%) compared to NS-fed A2AAR−/− mice (77.73 ± 4.26%; p<0.05; Fig. 4c).
Effects of DPCPX on 20-HETE-dependent vascular response in HS and NS diet fed A2AAR+/+ and A2AAR−/− mice
We investigated the role of A1AR in 20-HETE-induced contraction in NS/HS-fed A2AAR+/+ and A2AAR−/− mice (Fig. 5a, b). Selective A1 antagonist, DPCPX was unable to block 20-HETE (10−6 M)-induced contraction (Fig. 5a, b) in NS/HS-fed A2AAR+/+ and A2AAR−/− mice, suggesting that 20-HETE is downstream of A1AR.
Fig. 5.
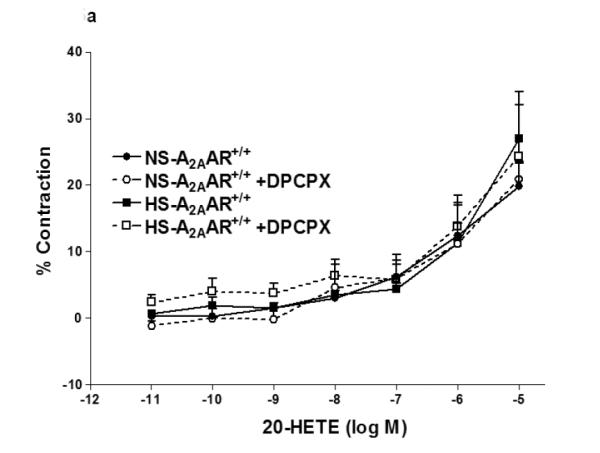
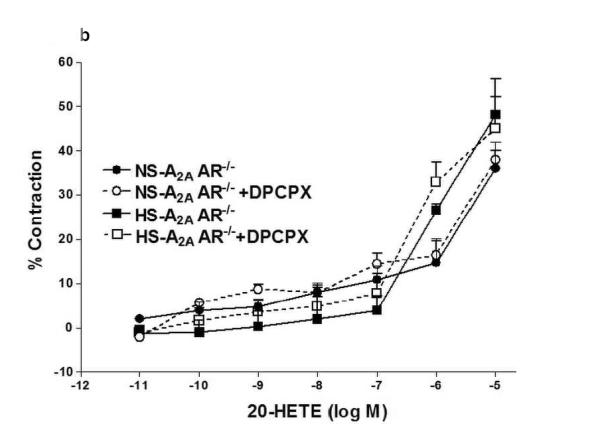
a: Effects of DPCPX (10−7 M) on 20-HETE- induced vascular response in HS and NS fed A2A AR+/+ aortic rings. Values are mean ± SE, n = 3. b: Effects of DPCPX (10−7 M) on 20-HETE-induced vascular response in NS and HS fed A2AAR−/− aortic rings. Values are mean ± SE, n = 3.
A2AAR, cyp2c29, cyp4a and A1AR expression in HS and NS-fed A2AAR+/+ and A2AAR−/− mice
The expression of A2AAR (~45 kDa) protein was upregulated by ~30% in HS-fed A2AAR+/+ mouse aortae (129.7 ± 1.63%) compared with the control NS-fed A2AAR+/+ mouse aortae (100.1 ± 2.16%, p<0.05; Fig. 6).
Fig. 6.
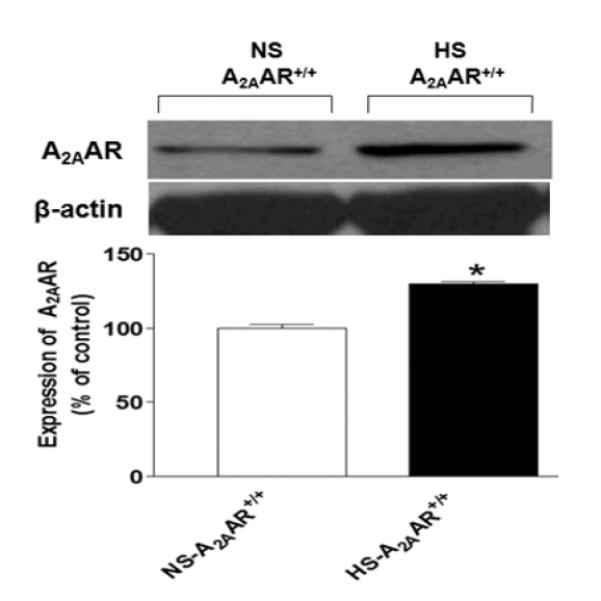
Representative Western-blot and densitometric analysis for A2AAR (~45 kDa) protein in aortae of HS and NS fed A2AAR+/+ mice. Values are mean ± SE, *p<0.05 between NS-A2AAR+/+ vs. HS-A2AAR+/+ mouse aortae, n=4.
Also, cyp2c29 (~55 kDa) protein showed increased expression by ~64 % in HS-fed A2AAR+/+ mouse aortae (164.2 ± 18.50%) compared to the control NS-fed A2AAR+/+ mouse aorta (100 ± 5.55%; p<0.05, Fig. 7). Significantly diminished level of cyp2c29 (~54%) was observed in HS-fed A2AAR−/− mice (59.8 ± 5.20%; Fig. 7) compared to HS-fed A2AAR+/+ mice (164.2 ± 18.50%; p<0.05; Fig. 7).
Fig. 7.
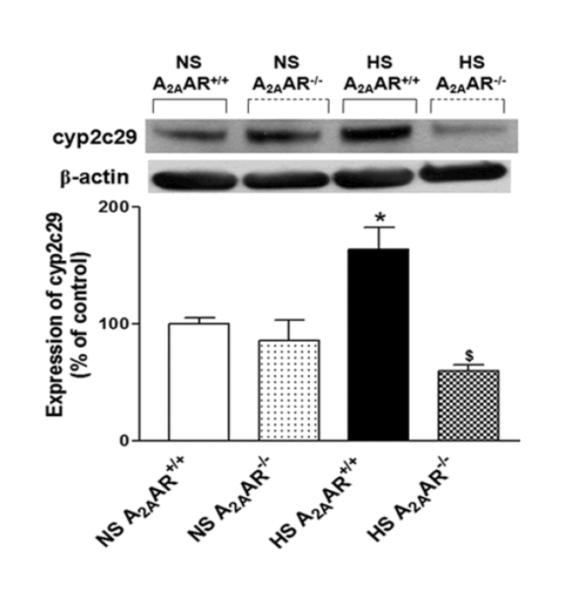
Representative Western-blot and densitometric analysis for cyp2c29 (~50 kDa) protein in aortas of HS and NS fed A2AAR+/+ and A2AAR−/− mice. Values are mean ± SE, *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, and $p<0.05 between HS-A2AAR+/+ vs. HS-A2AAR−/−, n=4.
Substantial elevation in the level of cyp4a (~58 kDa; ~74 %) protein was found in NS-fed A2AAR−/− mice (173.8 ± 12.90%) compared to NS-fed A2AAR+/+ (100.9 ± 1.34%; p<0.05, Fig. 8). On the contrary, cyp4a expression was decreased by ~46 % in HS-fed A2AAR+/+ mice (54.1 ± 7.88%; p<0.05; Fig. 8) compared to the NS-fed A2AAR+/+ mouse aortae (100.9 ± 1.34%). Cyp4a level was ~50% higher in HS-fed A2AAR−/− mouse aortae (103.9 ± 6.53%; p<0.05) compared to HS-fed A2AAR+/+ mice (54.1 ± 7.88%; Fig. 8).
Fig. 8.
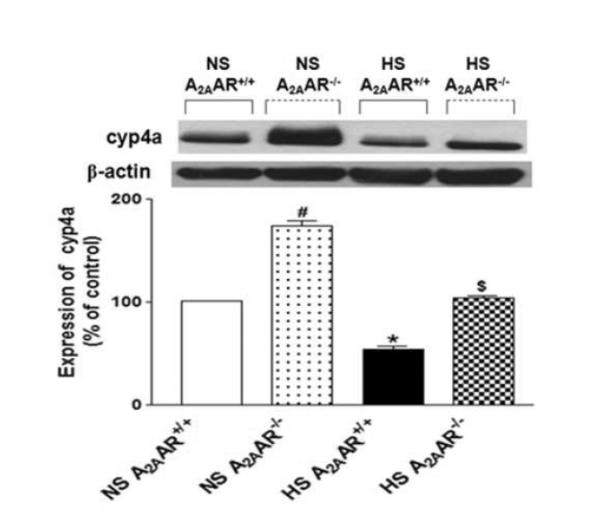
Representative Western-blot and densitometric analysis for cyp4a (~50 kDa) protein in aortas of HS and NS fed A2AAR+/+ and A2AAR−/− mice. Values are mean ± SE, *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, $p<0.05 between HS-A2AAR+/+ vs. HS-A2AAR−/−, and #p<0.05 between NS-A2AAR+/+ vs. NS -A2AAR−/−, n=4.
A1AR (~37 kDa) protein expression observed in both NS-fed A2AAR−/− and HS-fed A2AAR−/− mice (151.60 ± 26.85% and 120.1 ± 11.94%) was ~51.6% and ~45% higher compared to their control counterparts NS and HS-fed A2AAR+/+ mouse aortae (100.01 ± 0.04% and 82.84 ± 9.24%, Fig. 9) respectively. Also, A1AR expression was down-regulated by ~17% in HS-fed A2AAR+/+ mouse (82.84 ± 9.24%) compared to NS-fed A2AAR+/+ mouse aortae (100.0 ± 0.04%, p<0.05; Fig. 9).
Fig. 9.
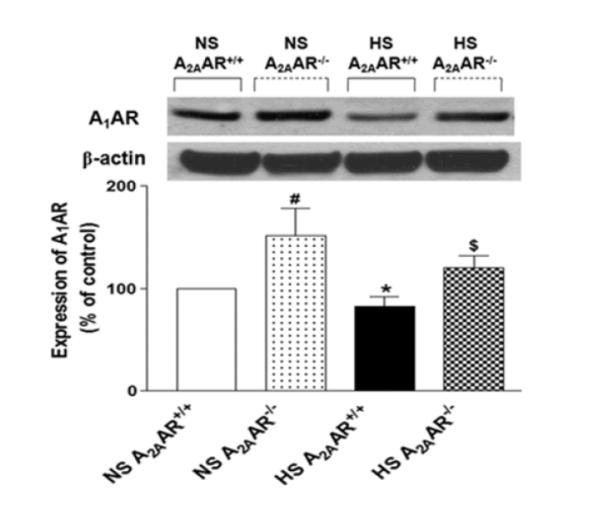
Representative Western-blot and densitometric analysis for A1AR (~37 kDa) protein in aortae of HS and NS fed A2AAR+/+ and A2AAR−/− mice. Values are mean ± SE, *p<0.05 between HS-A2AAR+/+ vs. NS-A2AAR+/+, $p<0.05 between HS-A2AAR+/+ vs. HS-A2AAR−/−, and #p<0.05 between NS-A2AAR+/+ vs. NS-A2AAR−/−, n=4.
Discussion
Our study utilized A2AAR+/+ and A2AAR−/− mice on a HS/NS diet to directly investigate functional and biochemical consequences of HS diet in the presence and absence of A2AAR. The major findings in this study show clear differences in vascular reactivity and signaling cascade between A2AAR+/+ and A2AAR−/− mice on a HS diet. Results from this study confirm that HS diet enhances vascular relaxation through increased A2AAR and cyp-epoxygenases pathway in A2AAR+/+ mice. Moreover, our novel findings suggest that HS diet potentiates contraction through reduced cyp-epoxygenases pathway and increased A1AR in A2AAR−/− mice. One of the interesting findings from our study demonstrates the contribution of 20-HETE in A1AR mediated contraction in NS fed. This suggests the involvement of different signaling mediators downstream of A1AR in vascular contraction observed in NS vs. HS fed A2AAR−/− mice. Hence, we speculate that HS diet in A2AAR−/− mice might induce some pathways downstream of A1AR other than 20-HETE to cause contraction that needs to be explored in future.
Adenosine-induced vascular response in HS/NS-fed A2AAR+/+ and A2AAR−/− mice
As reported earlier by us (28) in mice and in rats pre-glomerular vessels (21-22), in A2AAR+/+ mice, we found enhanced relaxation to NECA (Fig.1a) which was blocked by selective A2A receptor antagonist, SCH 58261 and increased A2AAR protein levels with HS diet compared to NS diet (Fig. 6). However, HS exaggerated contraction to NECA in A2AAR−/− mice (Fig. 1a). Interestingly, we found no significant difference in contraction to NECA between NS-fed A2AAR−/− mice and HS-fed A2AAR−/− mice (Fig. 1a).
Role of A2AAR and cyp-epoxygenases in vascular relaxation
CGS 21680-induced aortic relaxation in HS/NS fed A2AAR+/+ mice were unaffected by cycloxygenase inhibitor, indomethacin and eNOS inhibitor, L-NAME (data not shown), indicating that HS-induced enhanced A2AAR-mediated vascular response is independent of COX and NO. However, EETs antagonist 14,15-EEZE, significantly blocked HS/NS-induced relaxation to CGS 21680. This finding is in agreement with our previous published work (28-29) and others (6-7), and confirms the involvement of EETs in A2AAR-mediated enhanced vasodilatation in mice on HS/NS diet. Supporting the vascular response data, we found that HS diet increased levels of cyp2c29, the enzyme responsible for the production of vasodilatory EETs, compared to NS diet in A2AAR+/+ mice (Fig. 7). On the contrary, we observed substantially reduced expression of cyp2c29 in HS-fed A2AAR−/− mice compared to HS-fed A2AAR+/+ mice aortae, indicating that HS diet fails to up-regulate cy2c29 levels in the absence of A2AAR. Our data suggest that diminished cyp2c29 expression in HS-fed A2AAR−/− mice might account for the magnified contraction noted in these mice, possibly indicating an inability to efficiently adapt to HS in the absence of A2AAR. Dahl salt-sensitive rats are also unable to increase cyp-epoxygenase enzymes in response to HS diet which parallels our findings in HS-fed A2AAR−/− mice (21).
Role of A1AR and cyp4a in vascular contraction
On the other hand, cyp4a enzyme catalyzes the metabolism of AA into 20-HETE, a potent vasoconstrictor. The NECA contraction response was blocked and transformed into relaxation by cyp4a inhibitor DDMS in NS-fed A2AAR+/+ mice (Fig. 3a), while it was significantly lowered in NS-fed A2AAR−/− mice, suggesting the role of 20-HETE in mediating vascular contraction. Compared to NS-fed A2AAR+/+ mice, NS-fed A2AAR−/− mice had substantially increased expression of cyp4a possibly leading to increased generation of 20-HETE. However, DDMS did not affect the contraction observed in HS-fed A2AAR−/− mice, indicating that 20-HETE might not be involved in HS-induced contraction noted in A2AAR−/− mice. We speculate that this discrepancy between the effects of DDMS in NS-fed A2AAR−/− mice and HS-fed A2AAR−/− mice could be due to different signaling mediators involved in these two groups and this finding needs to be further explored. Also, 20-HETE concentration-dependent vascular contraction was not blocked by DPCPX in NS/HS fed A2AAR−/− and A2AAR+/+mice (Fig. 5a and 5b). This indicates that 20-HETE is downstream of A1AR signaling. Western-blot analysis confirmed significantly suppressed levels of cyp4a in HS-fed A2AAR−/− mice compared to NS-fed A2AAR−/− mice possibly resulting in reduced production of 20-HETE with HS diet in A2AAR−/− mice (Fig. 8). This may contribute to enhanced relaxation with HS as a result of decreased 20-HETE production. Our data indicate that levels of vasodilator (cyp-epoxygenase) and vasoconstrictor (ω-hydroxylase) agents appear to counterbalance each other thereby, maintaining vascular tone. Thus an imbalance in these vasoactive agents tends to disrupt this balance and lead to changes in vascular tone.
Similar to our previous finding (29-30), we found HS diet decreased the expression of A1AR protein compared to NS diet fed A2AAR+/+ mice (Fig. 9). Lower levels of vasoconstrictor A1AR could probably aid in increased NECA-induced vascular relaxation in HS diet fed A2AAR+/+ mice. Since the role of A1AR in vascular contraction, and that of A2AAR in vascular relaxation are well established (28, 36), we examined the involvement of A1AR in NECA-induced vascular response. Selective A1AR antagonist DPCPX blocked NECA-dependent contraction in NS-fed A2AAR+/+ mice and changed into a relaxation, suggesting the role of A1AR. Furthermore, NECA-induced contraction in both NS and HS fed A2AAR−/− mice were mitigated by the selective A1 receptor antagonist, DPCPX, suggesting the contribution of A1AR in vascular contraction. In addition, Western-blot data showed higher expression of A1AR in both NS/HS diet fed A2AAR−/− mice compared to respective A2AAR+/+ mice. Nontheless, compared to NS-fed A2AAR−/− mice, HS-fed A2AAR−/− mice displayed reduced A1AR levels. This is an interesting observation which implies that HS diet has a tendency to mitigate the level of A1AR but to a lesser degree in A2AAR−/− mice comapred to A2AAR+/+ mice.
A1AR-mediated vascular response in HS/NS-fed A2AAR+/+ and A2AAR−/− mice
To further examine the A1AR-mediated response in all the four groups, we used selective A1AR agonist CCPA and found that CCPA-dependent response produced significantly greater contraction in both the HS and NS-fed A2AAR−/− mice compared to their respective A2AAR+/+ mice. Corresponding to the A1AR levels, NS-fed A2AAR−/− mice demonstrated highest contraction to CCPA than HS-fed A2AAR−/− mice, suggesting that A1AR-mediated contraction is decreased with HS diet. Also, HS-fed A2AAR+/+ mice had the least contraction to CCPA compared to any other group which corresponds well with the lowest observed level of A1AR in these mice compared to all groups. Overall, these results suggest that besides A2AAR up-regulation, lowering A1AR expression might be another protective mechanism against salt loading.
Conclusion
In conclusion, presence of A2AAR appears to be critical for an adaptive aortic relaxation response to HS intake (4-5 weeks). When the A2AAR is absent, HS diet fails to achieve relaxation response as observed in wild-type counterparts and produces amplified contraction to NECA, possibly through diminished expression and activation of vasodilatory cyp-epoxygenases pathway and enhancing the level and activity of vasoconstrictor factors like A1AR.
Summary
Up-regulation of A2AAR and enhanced vascular relaxation through A2AAR in response to salt loading is most likely an adaptive response as shown in HS fed rat pre-glomerular vessels (21). This adaptive response is lost in the A2AAR null mice fed HS diet confirming a critical role of A2AAR in HS-induced vasodilation. NS and HS diet fed A2AAR−/− mice showed contraction to NECA. Although, the degree of contraction observed in A2AAR−/− mice fed NS diet is similar to A2AAR−/− mice fed HS diet, the mediators responsible for generating the vascular resistance seem to be different. For the most part, cyp4a and A1AR appear to be contributing to the contraction response in NS-fed A2AAR−/− mice. Conversely, it turns out that reduced cyp-epoxygenase expression and activity along with increased A1AR levels might be playing an important role in the contraction response observed in HS-fed A2AAR−/− mice.
Acknowledgements
This work was supported by Bridge Grant funding (WVU), startup funding (WVU) and National Institutes of Health HL-114559 to M. A. Nayeem, HL-094447 and HL 027339 to S. J. Mustafa, GM- 31278 to J. R. Falck, and z01-ES025034 to D. C. Zeldin.
Supported by: Bridge Grant funding (WVU), startup funding (WVU) and National Institutes of Health HL-114559 to M. A. Nayeem; HL-094447 and HL 027339 to S. J. Mustafa; GM- 31278 to J. R. Falck; and z01-ES025034 to D. C. Zeldin.
References
- 1.Andersen H, Jaff MG, Hogh D, Vanhoutte P, Hansen PB. Adenosine elicits an eNOS-independent reduction in arterial blood pressure in conscious mice that involves adenosine A2A receptors. Acta Physiol (Oxf) 2011;203:197–207. doi: 10.1111/j.1748-1716.2010.02218.x. [DOI] [PubMed] [Google Scholar]
- 2.Brown RD, Thoren P, Steege A, Mrowka R, Sallstrom J, Skott O, Fredholm BB, Persson AE. Influence of the adenosine A1 receptor on blood pressure regulation and renin release. Am J Physiol Regul Integr Comp Physiol. 2006;290:R1324–1329. doi: 10.1152/ajpregu.00313.2005. [DOI] [PubMed] [Google Scholar]
- 3.Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circ Res. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- 4.Capdevila JH, Wei S, Yan J, Karara A, Jacobson HR, Falck JR, Guengerich FP, DuBois RN. Cytochrome P-450 arachidonic acid epoxygenase. Regulatory control of the renal epoxygenase by dietary salt loading. J Biol Chem. 1992;267:21720–21726. [PubMed] [Google Scholar]
- 5.Carlstrom M, Wilcox CS, Welch WJ. Adenosine A2A receptor activation attenuates tubuloglomerular feedback responses by stimulation of endothelial nitric oxide synthase. Am J Physiol Renal Physiol. 2011;300:F457–464. doi: 10.1152/ajprenal.00567.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carroll MA, Doumad AB, Li J, Cheng MK, Falck JR, McGiff JC. Adenosine2A receptor vasodilation of rat preglomerular microvessels is mediated by EETs that activate the cAMP/PKA pathway. Am J Physiol Renal Physiol. 2006;291:F155–161. doi: 10.1152/ajprenal.00231.2005. [DOI] [PubMed] [Google Scholar]
- 7.Cheng MK, Doumad AB, Jiang H, Falck JR, McGiff JC, Carroll MA. Epoxyeicosatrienoic acids mediate adenosine-induced vasodilation in rat preglomerular microvessels (PGMV) via A2A receptors. Br J Pharmacol. 2004;141:441–448. doi: 10.1038/sj.bjp.0705640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng MG, Navar LG. Afferent arteriolar vasodilator effect of adenosine predominantly involves adenosine A2B receptor activation. Am J Physiol Renal Physiol. 2010;299:F310–315. doi: 10.1152/ajprenal.00149.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fleming I. Cytochrome p450 and vascular homeostasis. Circ Res. 2001;89:753–762. doi: 10.1161/hh2101.099268. [DOI] [PubMed] [Google Scholar]
- 10.Fredholm BB, AP IJ, Jacobson KA, Klotz KN, Linden J. International Union of Pharmacology. XXV. Nomenclature and classification of adenosine receptors. Pharmacol Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- 11.Frisbee JC, Falck JR, Lombard JH. Contribution of cytochrome P-450 omega-hydroxylase to altered arteriolar reactivity with high-salt diet and hypertension. Am J Physiol Heart Circ Physiol. 2000;278:H1517–1526. doi: 10.1152/ajpheart.2000.278.5.H1517. [DOI] [PubMed] [Google Scholar]
- 12.Hansen PB, Castrop H, Briggs J, Schnermann J. Adenosine induces vasoconstriction through Gi-dependent activation of phospholipase C in isolated perfused afferent arterioles of mice. J Am Soc Nephrol. 2003;14:2457–2465. doi: 10.1097/01.asn.0000086474.80845.25. [DOI] [PubMed] [Google Scholar]
- 13.Hansen PB, Hashimoto S, Oppermann M, Huang Y, Briggs JP, Schnermann J. Vasoconstrictor and vasodilator effects of adenosine in the mouse kidney due to preferential activation of A1 or A2 adenosine receptors. J Pharmacol Exp Ther. 2005;315:1150–1157. doi: 10.1124/jpet.105.091017. [DOI] [PubMed] [Google Scholar]
- 14.Hansen PB, Schnermann J. Vasoconstrictor and vasodilator effects of adenosine in the kidney. Am J Physiol Renal Physiol. 2003;285:F590–599. doi: 10.1152/ajprenal.00051.2003. [DOI] [PubMed] [Google Scholar]
- 15.Harder DR, Campbell WB, Roman RJ. Role of cytochrome P-450 enzymes and metabolites of arachidonic acid in the control of vascular tone. J Vasc Res. 1995;32:79–92. doi: 10.1159/000159080. [DOI] [PubMed] [Google Scholar]
- 16.Hein TW, Belardinelli L, Kuo L. Adenosine A(2A) receptors mediate coronary microvascular dilation to adenosine: role of nitric oxide and ATP-sensitive potassium channels. J Pharmacol Exp Ther. 1999;291:655–664. [PubMed] [Google Scholar]
- 17.Ito O, Roman RJ. Regulation of P-450 4A activity in the glomerulus of the rat. Am J Physiol. 1999;276:R1749–1757. doi: 10.1152/ajpregu.1999.276.6.R1749. [DOI] [PubMed] [Google Scholar]
- 18.Lapuerta L, Chacos N, Falck JR, Jacobson H, Capdevila JH. Renal microsomal cytochrome P-450 and the oxidative metabolism of arachidonic acid. Am J Med Sci. 1988;295:275–279. doi: 10.1097/00000441-198804000-00010. [DOI] [PubMed] [Google Scholar]
- 19.Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2a receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- 20.Liclican EL, Doumad AB, Wang J, Li J, Falck JR, Stier CT, Jr., Carroll MA. Inhibition of the adenosine2A receptor-epoxyeicosatrienoic acid pathway renders Dahl salt-resistant rats hypertensive. Hypertension. 2009;54:1284–1290. doi: 10.1161/HYPERTENSIONAHA.108.123570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liclican EL, McGiff JC, Falck JR, Carroll MA. Failure to upregulate the adenosine2A receptor-epoxyeicosatrienoic acid pathway contributes to the development of hypertension in Dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2008;295:F1696–1704. doi: 10.1152/ajprenal.90502.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liclican EL, McGiff JC, Pedraza PL, Ferreri NR, Falck JR, Carroll MA. Exaggerated response to adenosine in kidneys from high salt-fed rats: role of epoxyeicosatrienoic acids. Am J Physiol Renal Physiol. 2005;289:F386–392. doi: 10.1152/ajprenal.00421.2004. [DOI] [PubMed] [Google Scholar]
- 23.Linden J. Adenosine in tissue protection and tissue regeneration. Mol Pharmacol. 2005;67:1385–1387. doi: 10.1124/mol.105.011783. [DOI] [PubMed] [Google Scholar]
- 24.Macala LJ, Hayslett JP. Basolateral and apical A1 adenosine receptors mediate sodium transport in cultured renal epithelial (A6) cells. Am J Physiol Renal Physiol. 2002;283:F1216–1225. doi: 10.1152/ajprenal.00085.2002. [DOI] [PubMed] [Google Scholar]
- 25.Makita K, Takahashi K, Karara A, Jacobson HR, Falck JR, Capdevila JH. Experimental and/or genetically controlled alterations of the renal microsomal cytochrome P450 epoxygenase induce hypertension in rats fed a high salt diet. J Clin Invest. 1994;94:2414–2420. doi: 10.1172/JCI117608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Miller WL, Thomas RA, Berne RM, Rubio R. Adenosine production in the ischemic kidney. Circ Res. 1978;43:390–397. doi: 10.1161/01.res.43.3.390. [DOI] [PubMed] [Google Scholar]
- 27.Monopoli A, Casati C, Lozza G, Forlani A, Ongini E. Cardiovascular pharmacology of the A2A adenosine receptor antagonist, SCH 58261, in the rat. J Pharmacol Exp Ther. 1998;285:9–15. [PubMed] [Google Scholar]
- 28.Nayeem MA, Poloyac SM, Falck JR, Zeldin DC, Ledent C, Ponnoth DS, Ansari HR, Mustafa SJ. Role of CYP epoxygenases in A2A AR-mediated relaxation using A2A AR-null and wild-type mice. Am J Physiol Heart Circ Physiol. 2008;295:H2068–2078. doi: 10.1152/ajpheart.01333.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nayeem MA, Ponnoth DS, Boegehold MA, Zeldin DC, Falck JR, Mustafa SJ. High-salt diet enhances mouse aortic relaxation through adenosine A2A receptor via CYP epoxygenases. Am J Physiol Regul Integr Comp Physiol. 2009;296:R567–574. doi: 10.1152/ajpregu.90798.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nayeem MA, Zeldin DC, Boegehold MA, Falck JR. Salt modulates vascular response through adenosine A(2A) receptor in eNOS-null mice: role of CYP450 epoxygenase and soluble epoxide hydrolase. Mol Cell Biochem. 2011;350:101–111. doi: 10.1007/s11010-010-0686-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nayeem MA, Zeldin DC, Boegehold MA, Morisseau C, Marowsky A, Ponnoth DS, Roush KP, Falck JR. Modulation by salt intake of the vascular response mediated through adenosine A(2A) receptor: role of CYP epoxygenase and soluble epoxide hydrolase. Am J Physiol Regul Integr Comp Physiol. 2010;299:R325–333. doi: 10.1152/ajpregu.00823.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ponnoth DS, Sanjani MS, Ledent C, Roush K, Krahn T, Mustafa SJ. Absence of adenosine-mediated aortic relaxation in A(2A) adenosine receptor knockout mice. Am J Physiol Heart Circ Physiol. 2009;297:H1655–1660. doi: 10.1152/ajpheart.00192.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Siragy HM, Linden J. Sodium intake markedly alters renal interstitial fluid adenosine. Hypertension. 1996;27:404–407. doi: 10.1161/01.hyp.27.3.404. [DOI] [PubMed] [Google Scholar]
- 34.Spielman WS, Arend LJ. Adenosine receptors and signaling in the kidney. Hypertension. 1991;17:117–130. doi: 10.1161/01.hyp.17.2.117. [DOI] [PubMed] [Google Scholar]
- 35.Sun D, Samuelson LC, Yang T, Huang Y, Paliege A, Saunders T, Briggs J, Schnermann J. Mediation of tubuloglomerular feedback by adenosine: evidence from mice lacking adenosine 1 receptors. Proc Natl Acad Sci U S A. 2001;98:9983–9988. doi: 10.1073/pnas.171317998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tawfik HE, Schnermann J, Oldenburg PJ, Mustafa SJ. Role of A1 adenosine receptors in regulation of vascular tone. Am J Physiol Heart Circ Physiol. 2882005:H1411–1416. doi: 10.1152/ajpheart.00684.2004. [DOI] [PubMed] [Google Scholar]
- 37.Wang J, Schmidt JR, Roman RJ, Anjaiah S, Falck JR, Lombard JH. Modulation of vascular O2 responses by cytochrome 450-4A omega-hydroxylase metabolites in Dahl salt-sensitive rats. Microcirculation. 2009;16:345–354. doi: 10.1080/10739680802698007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weihprecht H, Lorenz JN, Briggs JP, Schnermann J. Vasomotor effects of purinergic agonists in isolated rabbit afferent arterioles. Am J Physiol. 1992;263:F1026–1033. doi: 10.1152/ajprenal.1992.263.6.F1026. [DOI] [PubMed] [Google Scholar]
- 39.Zou AP, Wu F, Li PL, Cowley AW., Jr Effect of chronic salt loading on adenosine metabolism and receptor expression in renal cortex and medulla in rats. Hypertension. 1999;33:511–516. doi: 10.1161/01.hyp.33.1.511. [DOI] [PubMed] [Google Scholar]