
Table 1. Data-collection and refinement statistics for Rv2971.
Values in parentheses are indicative of the highest resolution shell.
| Data collection | |
| Diffraction source | MX2, Australian Synchrotron |
| Detector | ADSC Quantum 315r CCD |
| Space group | P3221 |
| Unit-cell parameters (Å, °) | a = b = 86.27, c = 86.29, α = 90.0 β = 90.0 γ = 120.0 |
| Wavelength (Å) | 0.95370 |
| Resolution range (Å) | 38.58–1.60 (1.66–1.60) |
| Total No. of reflections | 525473 (76789) |
| No. of unique reflections | 49373 (4857) |
| Completeness (%) | 100.0 (100.0) |
| Mean I/σ(I)† | 12.72 (2.35) |
| R merge ‡ (%) | 11.4 (91.7) |
| R p.i.m. ‡ (%) | 3.7 (29.1) |
| Multiplicity | 10.6 (10.8) |
| Structural refinement | |
| Resolution range (Å) | 38.59–1.60 (1.63–1.60) |
| R work § (%) | 13.60 (22.67) |
| R free § (%) | 16.90 (27.59) |
| R.m.s.d.,¶ bond lengths (Å) | 0.006 |
| R.m.s.d.,¶ bond angles (°) | 1.05 |
| Ramachandran plot | |
| Favoured (%) | 98.20 |
| Allowed (%) | 1.80 |
| Outliers (%) | 0.00 |
| No. of modelled non-H atoms | |
| Macromolecules | 2128 |
| Ligands | 15 |
| Water | 311 |
| Protein residues | 276 |
| Wilson B factor (Å2) | 18.51 |
| Average B factors (Å2) | |
| Protein atoms | 20.40 |
| Ligand molecules | 20.40 |
| Water molecules | 33.70 |
| PDB code | 4otk |
†
I is the integrated intensity and σ(I) is the estimated standard deviation of that intensity.
‡
R
merge = 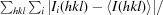
 where Ii(hkl) is the scaled intensity of the ith measurement and 〈I(hkl)〉 is the mean intensity for that reflection. R
p.i.m. = R
merge divided by the multiplicity.
where Ii(hkl) is the scaled intensity of the ith measurement and 〈I(hkl)〉 is the mean intensity for that reflection. R
p.i.m. = R
merge divided by the multiplicity.
§
R
work = 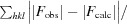
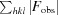 for all data excluding the 5% that comprise the R
free used for cross-validation.
for all data excluding the 5% that comprise the R
free used for cross-validation.
¶
Root-mean-square deviation from ideal values (Engh & Huber, 1991 ▶).