

Abstract
SAG (Sensitive to Apoptosis Gene), also known as RBX2 or ROC2, is a RING protein required for the activity of Cullin-RING ligase (CRL). Our recent study showed that Sag total knockout caused embryonic lethality at E11.5–12.5 days with associated defects in vasculogenesis. Whether Sag is required for de novo vasculogenesis in embryos and angiogenesis in tumors is totally unknown. Here, we report that Sag endothelial deletion also causes embryonic lethality at E15.5 with poor vasculogenesis. Sag deletion in primary endothelial cells or knockdown in MS-1 endothelial cells inhibits migration, proliferation and tube formation with p27 accumulation being responsible for the suppression of migration and proliferation. Furthermore, Sag deletion significantly inhibits angiogenesis in an in vivo Matrigel plug assay, and tumor angiogenesis and tumorigenesis in a B16F10 melanoma model. Finally, MLN4924, an investigational small molecule inhibitor of NEDD8-activating enzyme (NAE) that inhibits CRL, suppresses in vitro migration, proliferation, and tube formation, as well as in vivo angiogenesis and tumorigenesis. Taken together, our study, using both genetic and pharmaceutical approaches, demonstrates that Sag is essential for embryonic vasculogenesis and tumor angiogenesis, and provides the proof-of-concept evidence that targeting Sag E3 ubiquitin ligase may have clinical value for anti-angiogenesis therapy of human cancer.
Keywords: Angiogenesis, anti-angiogenesis therapy, MLN4924, Sag cKO mouse model, SAG-CRL E3 ligase, vasculogenesis
Introduction
CRL (Cullin-RING ligase) is the multi-complex E3 ubiquitin ligase, consisting of four components: an adaptor protein (e.g. SKP1), one of eight cullin family members (e.g. Cul-1), a substrate recognizing receptor (e.g. F-box protein Skp2), and one of two RING family proteins: RBX1/ROC1 and RBX2/ROC2, also known as SAG (Sensitive to Apoptosis Gene). 1–3 While the receptor protein determines the substrate specificity, the cullin-RING components constitute the core ubiquitin ligase activity. 3, 4 Activity of CRL also requires cullin neddylation, which disrupts inhibitory binding by CAND1 and confers on the enzyme an active conformation. 5–8 Various combinations of different family members of CRL components constitute CRL as the largest class of E3 ubiquitin ligases that, by targeting a variety of substrate proteins, control many important biological processes, including cell cycle progression, signal transduction, transcription, DNA replication, and tumorigenesis. 1, 2, 9, 10
SAG, an evolutionarily conserved small RING-containing protein with 113 amino acids, was originally cloned in our laboratory as a redox inducible antioxidant protein and later characterized as the second member of the RBX/ROC RING component of CRL E3 ubiquitin ligases. 11–13 In response to various stimuli, such as ROS, mitogen and hypoxia, SAG is induced at the transcriptional level by transcription factors AP-1 14 and HIF-1, 15 respectively. Induced SAG then recruits other components of CRL E3s to promote the ubiquitination and degradation of various substrates, including p27, 16 c-Jun, 14 pro-caspase-3, 17 IκBα, 18, 19 HIF-1α, 15 NOXA, 20 and Nf-1 21 in a cell context, temporal, and spatial dependent manner. Functionally, we and others have previously shown that ectopic SAG expression protects cells from apoptosis induced by redox, 11, 22 hypoxia 23 and various apoptosis-inducing agents [for review, see 24], and promotes the S-phase entry and cell proliferation under serum starved conditions. 25 Likewise, SAG knockdown by anti-sense or siRNA transfection inhibits tumor cell growth, 26 and enhances apoptosis induced by etoposide and TRAIL. 17 SAG knockdown or knockout also enhances cellular sensitivity to radiation. 18, 20 Most recently, we showed that total Sag knockout in the mouse causes embryonic lethality at E11.5–12.5 which is associated with overall growth retardation, massive apoptosis, and diminished vasculogenesis. 21 However, it has not been determined whether defective vasculogenesis upon Sag disruption plays a causal role in embryonic lethality, nor has the potential role of Sag in tumor angiogenesis been determined.
In this study, we addressed these important issues by the use of a Sag conditional KO mouse model in which selective Sag deletion in endothelial cells was driven by Tie2-Cre. We report here that Sag endothelial deletion also causes embryonic lethality but at a later stage of E15.5, again with defective vasculogenesis and proliferation, indicating its causal role in vasculogenesis and embryonic viability. We also report that Sag is required for in vitro endothelial cell migration and tube formation, and in vivo tumor angiogenesis using a B16F10 melanoma/Sagfl/fl model. Mechanistic and rescuing studies indicated that p27 plays, at least in part, a key role. Finally, we found that inhibition of CRL activity via cullin deneddyation by MLN4924, a small molecule inhibitor of NEDD8-activating enzyme, causes accumulation of p27 and suppresses in vitro migration and tube formation and in vivo tumor angiogenesis. Taken together, our study provides the first in vivo demonstration that 1) Sag is required for embryonic vasculogenesis and tumor angiogenesis and 2) small molecule inhibitors of cullin neddylation (such as MLN4924) may have potential for future development as a novel class of anti-angiogenesis agents.
RESULTS
Endothelial targeted Sag deletion causes embryonic lethality with reduced vasculogenesis and proliferation
Our recent study revealed that Sag total knockout via a gene trap approach caused embryonic lethality at E11.5–12.5 days, which is associated with poor vasculogenesis in both yolk sac and embryos. 21 To define the primary role of Sag in endothelial cells, we first generated a Sag conditional knockout mouse model in which Cre/LoxP mediated excision removes the exon 1 of the Sag gene, leading to a frame-shift mutation to produce a small peptide of 34 amino acids from exon 2 and part of exon 3. This peptide does not contain the functional RING domain and is a part of the non-functional Sag splicing variant (hSAG-MU1) 22 (manuscript in revision elsewhere). To achieve targeted Sag deletion in endothelial cells, we crossed Sagfl/fl mice with Tie2-Cre mice. Intercrossing of resolving Tie2-Cre;Sagfl/+ males with Sagfl/fl females would give rise to the Tie2-Cre;Sagfl/fl mice with Sag endothelial deletion, if it were not embryonic lethal. Of 72 newborn pulps genotyped, none of them carried the Tie2-Cre+;Sagfl/fl background (Table 1), indicating that Sag endothelial deletion also causes embryonic lethality. We next defined at which stage of development the Tie2-Cre+;Sagfl/fl; embryos die by dissecting uteri of pregnant mice at E13.5 and E15.5. Genotyping of E13.5 embryos showed a 1:1 ratio of Tie2-Cre+;Sagfl/fl vs. Tie2-Cre−;Sagfl/fl; background, whereas the ratio becomes 2:1 when E15.5 embryos were dissected (Table 1). We, therefore, concluded that Sag endothelial deletion causes embryonic lethality around day E15.5.
Table 1.
Genotypes of offspring of Tie2-Cre;Sag fl/+ x Sag fl/fl crossing
| Sagfl/+ | Sagfl/fl | ||||
|---|---|---|---|---|---|
|
| |||||
| Age | Tie2-Cre(−) | Tie2-Cre(+) | Tie2-Cre(−) | Tie2-Cre(+) | Total |
| Newborn | 33 | 25 | 14 | 0 | 72 |
| E15.5 | 29 | 32 | 15 | 8 | 84 |
| E13.5 | 18 | 16 | 10 | 10 | 54 |
| Expected ratio | 25% | 25% | 25% | 25% | 100% |
Dissection of Tie2-Cre+;Sagfl/fl embryos at E13.5 or E15.5 revealed that Sag endothelial deletion caused slight growth retardation, with smaller embryos as compared to the heterozygous littermates (Fig. 1A&B). On the other hand, Sag endothelial deletion remarkably inhibited vasculogenesis. The Tie2-Cre+;Sagfl/fl embryos are pale (Fig. 1A) with bleeding in various areas of the body (Fig. 1A, right panel), likely resulting from leaking of red blood cells from the defective vessels. The CD31 whole-mount staining of the E13.5 embryos revealed a significant reduction in the blood vessel density (Fig. 1B). Consistently, immunofluorescence analysis of Tie2-Cre+;Sagfl/fl; embryonic sections showed a remarkable reduction in endothelial cells (by CD 31, Fig. 1C&D), overall reduced proliferation (by Ki67, Fig. 1C&E), but not apoptosis (by cleaved caspase-3, Fig. S1A). Thus, Sag appears to play a primary role in endothelial proliferation and differentiation, and Sag deletion is a direct cause of poor embryonic vasculogenesis.
Figure 1. Sag endothelial deletion disrupts vascular development in mouse embryos.
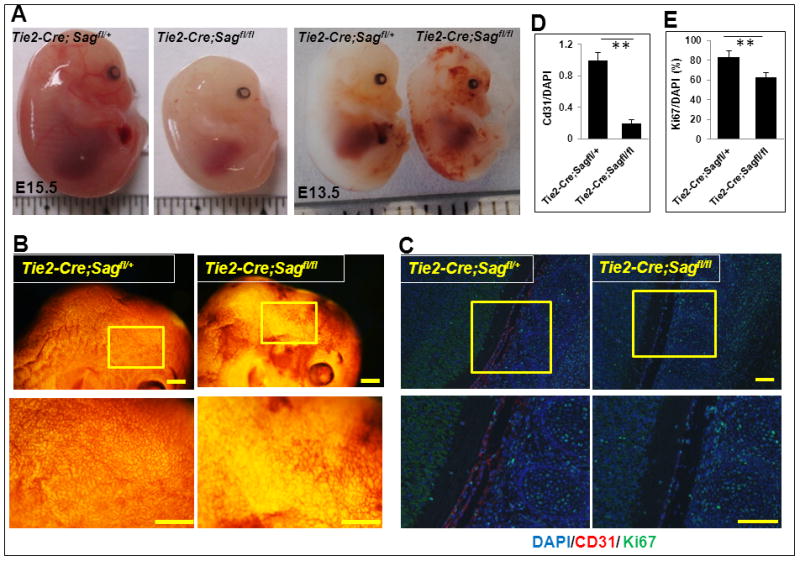
(A) Appearance of mouse embryos at E15.5 (left) and E13.5 (right). (B) Appearance of the brain regions of mouse embryos at E13.5 after CD31 whole-mount staining. Scale bar represents 3 mm. (C) Sagittal sections of control and mutant embryos were stained with antibodies against CD31, Ki67, and DAPI at the spinal cord areas. Scale bar represents 100 μm. (D and E) Quantification of CD31 positive cells (D) and Ki67 positive cells (E). The data were normalized as the ratio of CD31+/DAPI+ cells and represented as fold change with CD31+/DAPI+ value setting at 1 in Tie2-Cre;Sagfl/+ embryos (mean ± SEM, n=3) (D). The number of cells expressing Ki67 was normalized to the total number of DAPI positive cells as a percentage (mean ± SEM, n=3) (E).
Sag endothelial deletion inhibits in vitro migration, tube formation and proliferation
Given that both total KO and conditional KO of Sag showed endothelial and vascular defects, we next generated primary endothelial cells (ECs) from the lung and heart tissues of Sagfl/fl mice and assessed the potential role of Sag in endothelial migration, tube formation and proliferation in vitro. We first confirmed that the floxed Sag allele can be excised by Ad-Cre infection. Indeed, infection of ECs with Ad-Cre caused the excision of floxed Sag allele to give rise to a 257 bp PCR fragment, as compared to a ~1.6 kb PCR fragment in Ad-GFP-infected control cells which retained the floxed Sag allele (Fig 2A, top). We then confirmed that deletion of the floxed Sag allele completely eliminates the Sag protein expression (2A, bottom). Consistent with in vivo data, Sag deletion in endothelial cells significantly inhibits in vitro migration (Fig. 2B), tube formation (Fig. 2C) and proliferation (Fig. 2D), but has little, if any, effect on apoptosis (Fig. S1B). We further examined potential accumulation of known Sag substrates associated with growth suppression (p21 and p27) and apoptosis (Bim and Noxa) 24. Consistently, Sag deletion caused accumulation of p21 and p27, moderate increase in Bim, but not Noxa (Fig. 2E). Finally, we determined potential effect of Sag deletion on expression of VEGF and VEGFR-2 and found a minimal effect, if any (Fig. S2).
Figure 2. Sag endothelial deletion reduces migration, tube formation and proliferation.
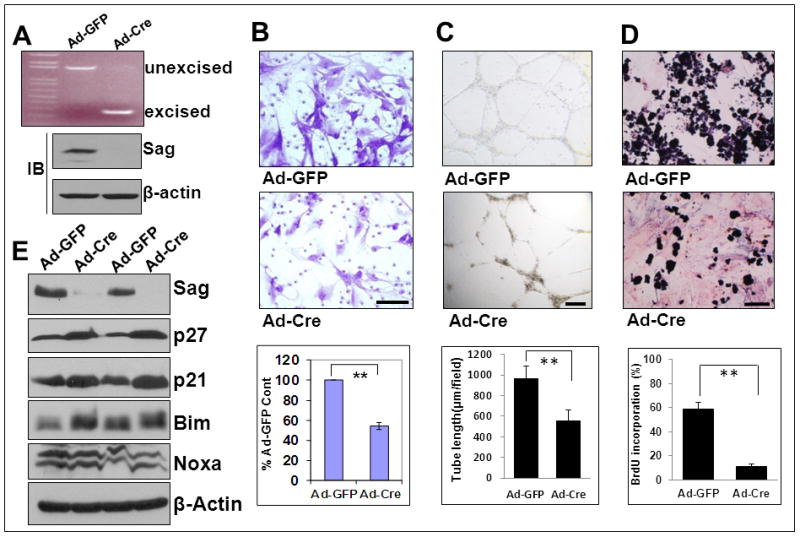
Primary ECs were isolated from lung and heart tissues of 4 week-old Sagfl/fl mice. Cells at passage 2 were infected with Ad-Cre or Ad-GFP (6×10E7 pfu) in 5 ml medium for 72 hrs. One portion of cells was subjected to PCR (A, top), a second portion for IB to detect Sag (A, bottom), and a third portion was used for a Boyden chamber migration assay (B), tube formation assay (C) and BrdU-based proliferation assay (D). Shown is mean ± SEM from three individual mice (B, C & D). Scale bar represents 50 μm. Two independent pairs of primary cultures of ECs were subjected to IB to detect accumulation of the indicated Sag-CRL E3 substrates (E).
Sag knockdown in MS-1 endothelial cells also inhibits in vitro migration, tube formation and proliferation
We next extended our observation to immortalized mouse endothelial MS-1 cells and found that lentivirus-based Sag knockdown caused accumulation of p21 and p27, moderate increase in Bim, but not Noxa (Fig. 3A), and suppression of migration, tube formation and proliferation (Fig. 3B–D). Suppression of endothelial migration was also observed in human HUVEC endothelial cells upon lentivirus-mediated SAG knockdown (Fig. S3A–C).
Figure 3. Sag knockdown reduces migration, tube formation and proliferation in MS1 cells.
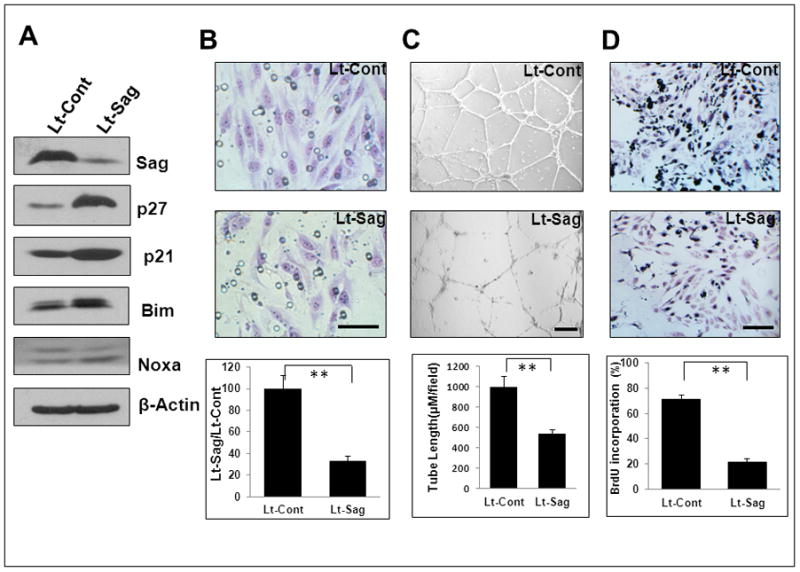
Mouse endothelial MS-1 cells were infected with Lt-Sag and Lt-Cont for 72 hrs. One portion was subjected to IB (A), and the other portion was used for a Boyden chamber migration assay (B), tube formation assay (C) and BrdU-based proliferation assay (D). Shown is mean ± SEM from three independent experiments. Scale bar represents 50 μm.
p27 knockdown partially rescues the suppression of migration and proliferation, but not tube formation, induced by Sag deletion
We next investigated potential mechanisms by which Sag deletion alters endothelial properties. We focused our study on p27, since 1) p27 is a Sag substrate 16 which is accumulated upon Sag deletion in endothelial cells (Fig. 2E and 3A), and 2) p27 has been previously implicated in neoangiogenesis, invasion and vascular remodeling. 27, 28 MS-1 cells were infected with lentivirus targeting Sag, along with the scrambled control, followed by p27 knockdown via siRNA (Fig. 4A). p27 knockdown partially rescued Sag knockdown-induced suppression of endothelial migration (Fig. 4B) and proliferation (Fig. 4C), but not tube formation (Fig. 4D), suggesting that p27-induced growth suppression affects endothelial migration, but not tube formation.
Figure 4. p27 deletion partially rescues Sag deletion-induced defects in migration and proliferation, but not tube formation.
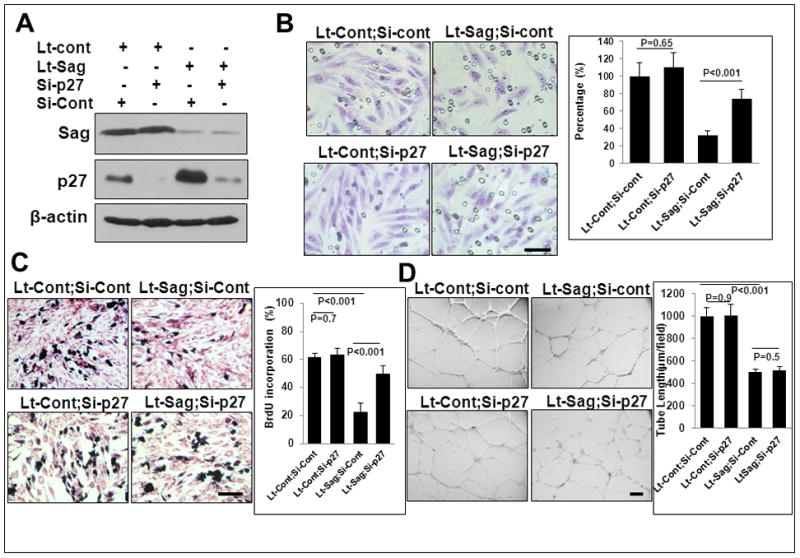
MS1 cells were infected with Lt-Sag and Lt-Cont and co-transfected with siRNA oligonucleotides targeting p27, along with scrambled control siRNA for 72 hrs. One portion was subjected to IB (A), and the other portion was used for assays of migration (B), proliferation (C) and tube formation (D). Shown is mean ± SEM from three independent experiments. Scale bar represents 50 μm.
Sag EC deletion inhibited in vivo angiogenesis and tumorigenesis
We next used two in vivo models to determine the effect of Sag endothelial deletion on angiogenesis. In an in vivo matrigel plug assay, we injected the matrigel mixed with Ad-Cre or Ad-GFP control, plus or minus VEGF, into Sagfl/fl mice (left flank for Ad-Cre and right flank for Ad-GFP). We euthanized mice seven days later and harvested the entire matrigel plug. Expectedly, VEGF induced remarkable angiogenesis when matrigel was mixed with the Ad-GFP control, as evidenced by the bloody matrigel plugs (Fig. 5A, top) and the presence of CD31-positive vessels (Fig. 5A, bottom). In contrast, VEGF-induced angiogenesis was nearly completely inhibited if Ad-Cre was present, which deleted Sag in host blood vessels during angiogenesis into matrigel plugs (Fig. 5A).
Figure 5. Sag EC deletion inhibits in vivo angiogenesis and tumorigenesis.
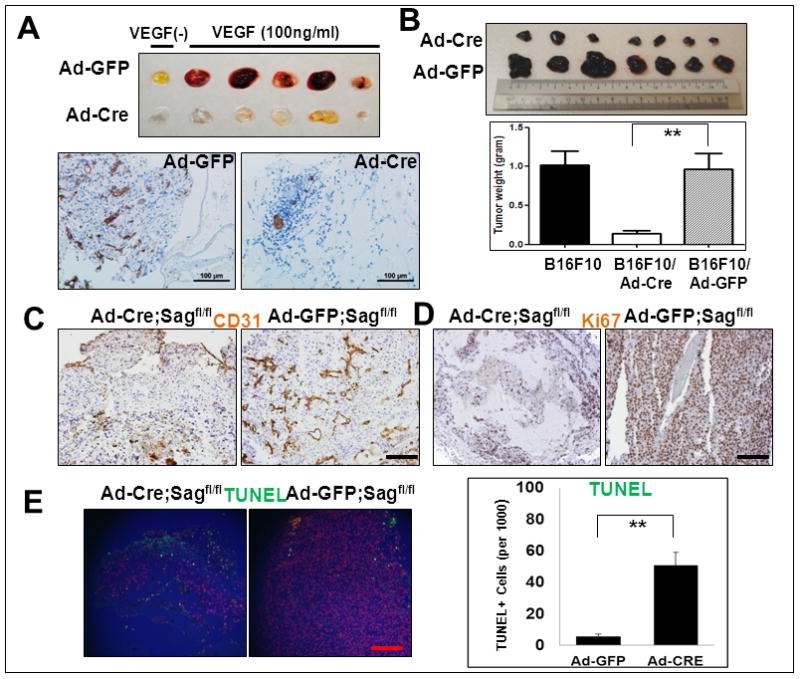
Sagfl/fl mice were injected with matrigel mixed with Ad-Cre (on the left flank) or Ad-GFP control (on the right flank) in the presence or absence of VEGF. After 7 days, mice were euthanized and the matrigel plugs were harvested and photographed (A, top panel), then fixed, sectioned and stained with CD31 antibody (A, bottom panels). Sagfl/fl mice were injected with B16F10 mouse melanoma cells (5×105) mixed with Matrigel and Ad-Cre or Ad-GFP as indicated (right before injection without pre-virus infections). Twelve days later, tumors were harvested and photographed (B, left panel). Shown is mean ± SEM from 7 mice in each group, except B16F10 only control group (n=3) (B, right panel). The tumor tissues were fixed, sectioned and stained for CD31 (for blood vessels, C), Ki67 (for proliferation, D) and TUNEL assay (for apoptosis, E). Scale bar represents 100 μm.
We further determined the effect of Sag EC deletion on in vivo angiogenesis and tumorigenesis of B16F10 melanoma cells, a well-established in vivo tumor angiogenesis model 29. B16F10 cells were mixed with Matrigel containing Ad-Cre or Ad-GFP control virus and injected into Sagfl/fl mice on the left or right flank, respectively. As an additional control, B16F10 cells mixed with Matrigel without any virus were also injected. Tumors were harvested 12 days post-injection, weighed and sectioned for staining with various antibodies. While Ad-GFP had no effect on tumor growth, as compared to the control, Ad-Cre infection caused remarkable reductions in tumor mass (Fig 5B), blood vessel density (by CD31 staining, Fig. 5C), and rate of cell proliferation (by Ki67 staining, Fig. 5D), as well as extensive apoptosis (by TUNEL, Fig. 5E). We also excluded possible toxicity associated with Ad-Cre virus infection by repeating the experiment in Sagfl/+ mice and found that in vivo tumor growth was comparable between Ad-Cre and Ad-GFP control (Fig. S4). Thus, Sag endothelial deletion inhibits the development of host blood vessels into implanted tumors (inhibition of angiogenesis), leading to growth retardation and apoptosis. We further determined angiogenesis capacity in skin papillomas induced by DMBA/TPA 19 and found a significantly increased vessel density in tumors derived from SAG transgenic mice, as compared to those from non-transgenic control mice (Fig. S5), which likely contributes to increased tumor size seen in SAG transgenic mice. 19 Thus, both loss-of-function and gain-of-function studies support the notion that Sag plays a key role in and is essential for tumor angiogenesis.
MLN4924 suppresses in vitro migration, tube formation and proliferation
It is well-established that activation of CRL ligases requires cullin neddylation. 5–8 MLN4924, a potent inhibitor of NEDD8-activating enzyme (NAE), was recently found as a novel class of anticancer agents that inhibits CRL activity via cullin deneddylation and causes accumulation of tumor suppressive substrates to induce apoptosis. 30 We treated MS-1 endothelial cells with MLN4924 as a small molecule approach to inactivate CRL (including Sag-associated ligases) via cullin deneddylation (Fig. 6D, top panel), and found that MLN4924 significantly inhibited their migration (Fig. 6A), tube formation (Fig. 6B), and proliferation (Fig. 6C), but not apoptosis (Fig. S6). Consistent with Sag knockdown, MLN4924 treatment also caused a dose dependent accumulation of p21, p27, and Bim, but not Noxa (Fig. 6D).
Figure 6. CRL inhibitor MLN4924 suppresses migration, tube formation and proliferation in vitro.
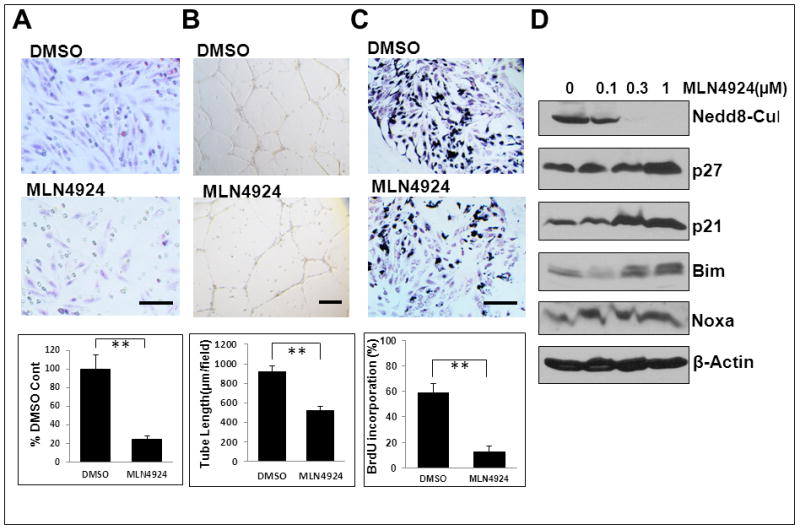
Mouse endothelial MS-1 cells were treated with MLN4924 for 24 hrs. One portion was used for assays of migration (A), tube formation (B) and BrdU incorporation (C). Shown is mean ± SEM from three independent experiments. Scale bar represents 50 μm. The other portion was used for IB with indicated Abs (D).
p27 knockdown partially rescues MLN4924-induced suppression of migration and proliferation, but not tube formation
We then determined whether p27 accumulation also plays a crucial role in the observed suppressive effects of MLN4924. MS-1 cells were transfected with siRNA targeting p27, along with the scrambled control to silence p27 (Fig. 7A). Consistent with Sag knockdown study (Fig. 4), p27 knockdown also partially rescued MLN4924-induced suppression of endothelial migration (Fig. 7B) and proliferation (Fig. 7C), but not tube formation (Fig. 7D).
Figure 7. p27 knockdown partially rescues MLN4924-induced migration and proliferation, but not tube formation.
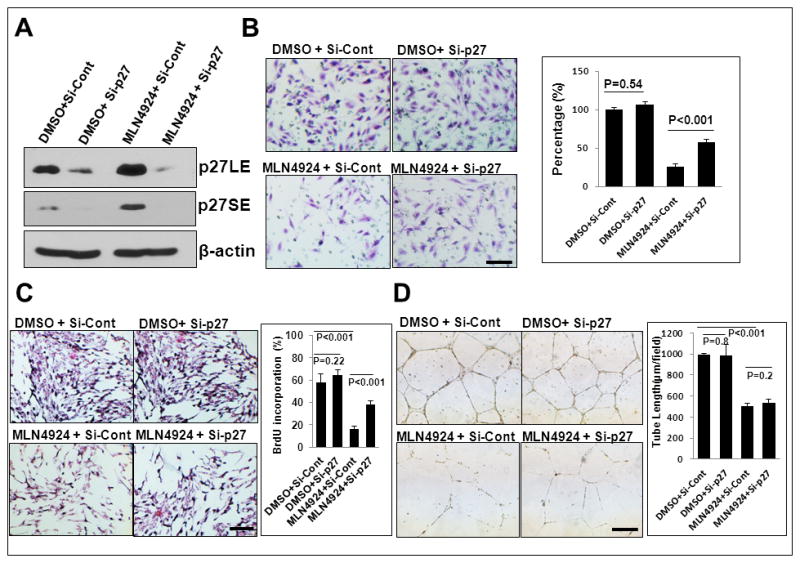
MS1 cells were first transfected with siRNA targeting p27 and si-Cont for 72 hrs, followed by MLN4924 treatment for 24 hrs. One portion was subjected to IB (A), and the other portion was used for assays of migration (B), proliferation (C) and tube formation (D). Shown is mean ± SEM from three independent experiments. Scale bar represents 50 μm.
MLN4924 inhibits tumor angiogenesis and growth in an in vivo B16F10 model
Finally, we determined the effect of MLN4924 on in vivo tumor angiogenesis, again using the B16F10 model. Sagfl/fl mice were treated with MLN4924 subcutaneously at a non-toxic dose regimen (60 mg/kg, once a day, 5 days per week) 30, 31 for 12 days beginning at 24 hrs post B16F10 inoculation. Tumors were harvested at day 13, weighed and snap-frozen for Western blotting or sectioned for staining with various antibodies. We first confirmed that MLN4924 indeed reached the tumor site by showing a significant inhibition of cullin neddylation in two independent tumor tissues (Fig. 7A). Similar to Sag deletion, MLN4924 treatment caused significant reductions in tumor mass (Fig. 8B), vessel density (Fig. 8C), and proliferation (Fig. 8D), as well as extensive apoptosis (Fig. 8E). MLN4924 treatment also caused a dose dependent accumulation of p21, p27, and Bim, but not Noxa in B16F10 cells (Fig. 8F), indicating a mechanism similar to that of Sag knockdown.
Figure 8. CRL inhibitor MLN4924 suppresses tumor angiogenesis in vivo.
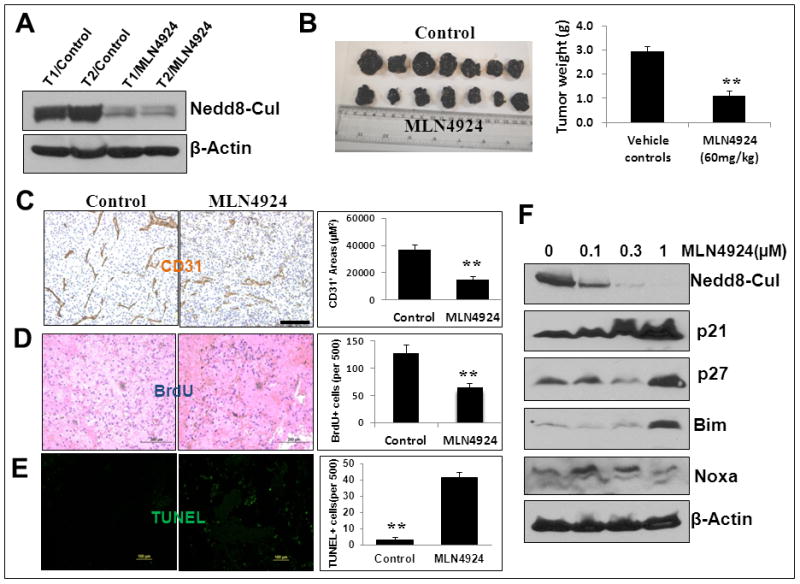
Sagfl/fl mice were injected with B16F10 mouse melanoma cells (5×105) mixed with Matrigel. Starting on the next day MLN4924 was administrated (60 mg/kg, s.c.) once a day, 5 days a week for 12 days. Tumors were then harvested and weighed. Two pairs of tumor tissues (T) were subjected to IB with indicated antibodies (A). Tumor weight is shown as mean ± SEM from seven mice in each group (B). The tumor tissues were fixed, sectioned and stained for CD31 (for blood vessels, C), BrdU (for cell proliferation, D), and TUNEL (for apoptosis, E). **: p<0.01. Scale bar represents 100 μm. (F) B16F10 cells were treated with MLN4924 at different concentrations for 24 hrs and subjected to IB using indicated Abs.
DISCUSSION
Our recent study using a genetrap-based Sag total KO mouse model revealed that Sag is essential for embryonic development, since Sag inactivation causes embryonic lethality associated with poor vasculogenesis, robust apoptosis, and defects in neural development. 21 However, it is unclear whether defective vasculogenesis is causally related to the embryonic death. Here we showed that targeted Sag endothelial deletion driven by Tie2-Cre also causes embryonic lethality (Fig. 1), indicating a causal relationship, although it occurs at the later time-point during embryogenesis. It is noteworthy that the lethality happens in the wild type background of Rbx1, a Sag family gene, again indicating non-redundant function of two family members during embryogenesis. 21, 32 Consistently, the follow-up in vitro assays using both primary and immortalized endothelial cells revealed that Sag deletion or knockdown inhibits migration, tube formation and proliferation (Fig. 2&3), but has a minimal effect, if any, on apoptosis (Fig. S1).
We next focused our mechanistic studies on Sag substrates known to regulate proliferation and apoptosis and found that upon Sag deletion, p27 is remarkably accumulated, along with a moderate increase in p21 and a minimal increase in Bim, but not Noxa. It has been previously reported that in addition to inhibiting cell cycle progression as an inhibitor of cyclin dependent kinases, p27 also inhibits cell migration, tumor cell invasion and metastasis via binding to and inhibiting the function of microtubule destabilizing protein stathmin, 28, 33 although its role in this aspect is still controversial in some cellular contexts. 34, 35 We therefore examined whether p27 accumulation indeed plays a crucial role in the observed phenotypic changes by simultaneous knockdown of Sag and p27 in MS-1 endothelial cells. Our results clearly demonstrate that p27 is responsible, at least in part, for the observed suppression of proliferation and migration induced by Sag knockdown (Fig. 4). Interestingly, p27 knockdown is unable to rescue the suppression of endothelial tube formation (Fig. 4), indicating that mechanistically it is independent of p27. Our results suggest that p27 accumulation upon Sag ablation affects endothelial cell proliferation and migration, but has a minimal effect on tube formation. Nevertheless, the underlying mechanism that drives the suppression of tube formation upon Sag deletion warrants a future investigation.
Having established an essential role of Sag in embryonic vasculogenesis, we next investigated potential roles of Sag in tumor angiogenesis. It is well-established that tumorigenesis and angiogenesis are intertwined processes that promote each other. During tumorigenesis, when tumor mass reaches a certain size, tumor cells secrete pro-angiogenic factors to promote proliferation and migration of host endothelial cells to the tumor mass to initiate neovascularization, leading to development of blood vessels. 36 Angiogenesis, in turn, promotes rapid tumor growth and metastasis. 37–39 Indeed, using a matrigel plug assay, we showed that VEGF-induced angiogenesis is near completely inhibited by Sag deletion (Fig. 5). More importantly, using the B16F10 mouse melanoma model, we showed that Sag deletion during tumor neovascularization significantly inhibits angiogenesis in the tumor mass, leading to reduced vascular density, and consequent suppression of proliferation and induction of apoptosis. This eventually causes remarkably reduced tumor size (Fig. 5). Thus, Sag could be an attractive anti-angiogenesis and anti-tumorigenesis target.
We further extended the observations made by our genetic approach via Sag deletion or knockdown to a small molecule approach using MLN4924, an investigational inhibitor of CRL E3 ligases via cullin deneddylation. 30 Consistently, we found that MLN4924 significantly suppresses migration, tube formation and proliferation of MS-1 endothelial cells, and at the same time induces accumulation of p21 and p27 (Fig. 6). We further confirmed a critical role played by p27 in suppression of migration and proliferation by a simultaneous p27 knockdown experiment (Fig. 7).
Finally, we explored the anti-angiogenesis and anti-tumorigenesis activity of MLN4924 again using a B16F10 melanoma cell model. Our results clearly demonstrate that MLN4924 reaches the tumor site and suppresses tumorigenesis by inhibiting angiogenesis and proliferation, and also induces apoptosis (Fig. 8). It is anticipated that compared to permanent Sag deletion, the anti-angiogenesis effect of MLN4924 is less, since the compound was only given once a day for 12 days and the drug effect is temporary. Mechanistically, similar to Sag knockdown, MLN4924 treatment also caused accumulation of p21, p27 and Bim, but not Noxa. It should be noted that MLN4924 is a NAE inhibitor and would probably inhibit other cellular neddylation reactions, 40, 41 although cullins are the only known physiological substrates. 40, 42 Nevertheless, given the fact that MLN4924 phenocopies Sag endothelial deletion biologically and mechanistically, the major effect of MLN4924 against tumor angiogenesis is likely through the inhibition of CRL E3s. Furthermore, MLN4924 is the first-in-class and only NAE inhibitor currently in phase I clinical trials for anticancer therapy. 43–45 Our study using both genetic and small molecule approaches provides proof-of-concept for future development of inhibitors of cullin neddylation (such as MLN4924) as a novel class of anti-angiogenesis agents through inactivation of CRL E3 ligases.
MATERIALS AND METHODS
Mouse studies
The Sagfl/fl conditional KO mouse model was generated with exon 1 flanked with loxP sites (manuscript submitted elsewhere). Tie2-Cre transgenic mice were a gift from Dr. Jun-Lin Guan. 46 All procedures were approved by the University of Michigan Committee on Use and Care of Animals. Animal care was provided in accordance with the principles and procedures outlined in the National Research Council Guide for the Care and Use of Laboratory Animals.
PCR-based genotyping
Genomic DNA was isolated from mouse tail tips and yolk sac and was genotyped using the primer set of PSag-KO-F: 5′-TTCTGGCCAGGTGTGGTGATATC-3′, and PSag-KO-G: 5′-CTTAGCCTTGGTTGTGTAGAC-3′ to detect floxed allele (140 bp) and wild type allele (105 bp). The primer set for detecting the removal of the Sag targeting fragment is PSag-KO-Seq-B: 5′-GTAACTCCAGACAATGCTCGCT-3′ and PSag-KO-Seq-R: 5′-TGAGTTCCAGGACAGCCAGGG-3′ with sag deletion (275 bp) or without Sag deletion (1.6 kb). The primer set for detecting the Tie2-Cre mice is Tie2-F1: 5′-CAGAACCTGAAGATGTTCGCG-3 and Tie2-R2: 5′-CACCGTCAGTACGTGAGATA-3′ to detect a 400 bp fragment.
Whole-mount immunostaining on embryos
Whole-mount CD31 immunostaining was performed on E13.5 embryos, as described 21. Briefly, embryos were fixed in 10% formalin, dehydrated by methanol, quenched by H2O2, and blocked in 4% BSA for 2 hrs. The samples were stained by incubating with anti-CD31 (rat monoclonal MEC13.3, BD Biosciences) at 4°C overnight, followed by peroxidase conjugated secondary antibodies. The embryos were developed in DAB solution (Vector), and photographed on a dissecting microscope (modelS6D; Leica) with a progressive 3CCD camera (Sony).
Immunofluorescence
Paraffin sections were deparaffinized, rehydrated, and analyzed by immunofluorescence, as described. 47 Briefly, the sections were incubated with primary antibodies in blocking solution overnight. The antibodies used are as follows: Ki67 (BD Biosciences), cleaved caspase3 (Cell signaling) and CD31 (Dako). The secondary antibodies were conjugates of Alexa Fluor 488 or Alexa Fluor 555 (Invitrogen). DAPI (Invitrogen) was used as nuclear counter-staining. The stained sections were examined under a fluorescence microscope (Olympus).
Immunohistochemistry staining
Embryos or tissues were fixed in 10% formalin and embedded in paraffin 19. Five-μm-thick sections were cut for H&E staining and examined under a microscope. Immunohistochemistry was performed using the ABC Vectastain kit (Vector Laboratories) with antibodies against Ki67 (BD Biosciences), cleaved caspase3 (Cell Signaling Technology) and CD31 (Dako). The sections were developed with DAB and counterstained with haematoxylin.
Culture of ECs and adenovirus infection
Primary endothelial cells (ECs) from hearts and lungs of Sagfl/fl mice were isolated using magnetic bead (Dynabead M-450; Invitrogen) purification with rat anti–mouse PECAM-1 (BD), as described. 46, 48 In brief, lung and heart tissues were harvested, minced, and then digested with 0.2% of type I collagenase (Worthington Biomedical) at 37°C for 1–2 hrs. The digested tissues were mechanically dissociated using vigorous flushing through a metal cannula, passed through a 70-μm filter (BD Biosciences), and then centrifuged at 400 xg for 5 min at 4°C. The cells were resuspended in cold Dulbecco’s PBS and then incubated with magnetic beads (M-450; sheep anti–rat IgG Dynabeads; Invitrogen) coated with rat anti–murine CD31 Ab (PECAM-1; clone MEC 13.3; BD Biosciences) at 4°C. The beads were washed several times in PBS, resuspended in growth medium [DMEM medium supplemented 100 μg/ml EC mitogen (Biomedical Technologies), endothelial cell growth factor (100x) (Sigma) and VEGF (30 ng/ml, PeproTech). L-glutamine, nonessential amino acids, and Na pyruvate at standard concentrations] and cultured in 100-mm tissue culture dishes pre-coated with 0.1% gelatin (Sigma-Aldrich) at 37°C in a 5% CO2 incubator.
The recombinant adenoviruses encoding Cre recombinase or lacZ control were purchased from Gene Transfer Vector Core (University of Iowa, Iowa City, IA) and used to infect the primary ECs. For most experiments, 108 plaque-forming units were used for a 10-cm dish. To increase efficiency, a second infection was performed after 9–12 hrs. No detectable cellular toxicity was observed.
siRNA knockdown
The lenti-virus-based siRNA knockdown of Sag (Lt-SAG), along with scrambled siRNA control (Lt-Cont) was performed as described 14. For double silencing, cells were infected with Lt-Sag or Lt-Cont for 48 to 72 hrs in 60-mm dishes. Cells were then split into 3 60-mm dishes and transiently transfected with si-Cont or si-p27 using Lipofectamine 2000. Forty-eight hrs later, cells were harvested for proliferation, migration and tube formation assay. The sequence for these siRNA oligonucleotides are as follows: Si-Cont: 5′-AUUGUAUGCGA UCGCAGAC-3′; and Si-p27 (pooled from Santa Cruz Biotechnology).
Immunoblotting analysis
Cells with or without MLN4924 treatment were harvested, lysed in a Triton X-100 lysis buffer and subjected to immunoblotting analysis. 49 SAG monoclonal antibody was raised against the RING domain (AA44-113). 20 Other antibodies were purchased commercially as follows: p21 (BD Transduction Labs), p27 (BD PharMingen), NOXA and VEGF (Millipore), Bim, Parp and Cleaved-Caspase3 (Cell Signaling), VEGFR2 (Santa Cruz Biotechology), and β-Actin (Sigma, MO).
Migration assay
ECs infected with Ad-LacZ or Ad-Cre were subjected to Boyden chamber migration assay (BD Biosciences) according to the manufacturer’s instructions. Briefly, the top chambers were seeded with 3×104 ECs in 250 μL serum free medium, and the bottom chambers were filled with 500 μL of complete Medium 200PRF (Life Technologies), supplemented with low serum growth supplement (LSGS) (Invitrogen) and VEGF (30 ng/ml, PeproTech). Cells were allowed to migrate for 12 hours. Stationary cells on the top surface of the membrane were scraped with a cotton swab, and the migrated cells were fixed with methanol for 30 min. After being washed three times, the cells were stained with 1% crystal violet. Images were taken using an inverted microscope (Olympus).
Tube formation assay
ECs infected with Ad-LacZ or Ad-Cre were plated on a thin layer of Matrigel (BD Biosciences) at 104 cells/well of a 96-well plate and cultured in Medium 200PRF (Life Technologies), supplemented with LSGS (Invitrogen) and VEGF (30 ng/ml, PeproTech), and incubated for 8 to 16 hrs to allow the formation of a tubular structure. The tube length was measured and calculated by Nikon NIS-Elements BR Version 4.0 software.
TUNEL assay
ECs infected with Ad-LacZ or Ad-Cre were assessed for apoptosis by TUNEL assay using the In Situ Cell Death Detection Kit (Roche), according to the manufacturer’s recommendations.
BrdU incorporation assay
ECs after 48-hrs infection with Ad-LacZ or Ad-Cre were serum starved for 18 hrs to arrest cells at the G0 phase. BrdU (100μg/ml) was then added into the culture medium. A BrdU incorporation assay was performed as described previously 32. Cells were fixed in 4% PFA-PBS, and BrdU positive cells detected with a 5-Bromo-2′-deoxy-uridine labeling and detection Kit II (Roche).
In vivo Matrigel plug assay
A total of 0.5 mL Matrigel (BD Biosciences) mixed with Ad-Cre (2.5 × 108 PFU) or Ad-GFP (2.5 × 108 PFU), 20 units of heparin (Sigma) in the absence or presence of VEGF (100ng/mL) (PeproTech), was subcutaneously injected into 6 week-old Sagfl/fl mice. After 7 days, the intact Matrigel plugs were collected and photographed. Plugs were embedded and dissected, the sections were stained with CD31 (Dako).
In vivo anti-angiogenesis assay
B16F10 melanoma cells (5×105) were mixed with Matrigel containing Ad-Cre (5×107 pfu) or Ad-GFP (2.5×108 pfu) control virus and inoculated subcutaneously in both flanks of mice with a genotype of Sagfl/fl or Sagfl/+ at 6–8 weeks of age (one side for Ad-Cre, another side for Ad-GFP). After 12 days, mice were euthanized, and tumors were removed and weighed. A portion of each tumor was fixed, sectioned and stained with antibodies against CD31, Ki67 and TUNEL.
MLN4924 antitumor in vivo assay
B16F10 melanoma cells (5×105) were mixed with matrigels and inoculated subcutaneously in both flanks of mice with genotype of Sagfl/fl at age of 6–8 weeks. The next day MLN4924 (60 mg/kg, s.c.) was administrated once a day, 5 days a week for 12 days. Mice in the control group received 10% 2-hydroxypropyl-β-cyclodextrin (HPBCD) as the vehicle control. 30, 31 Mice were euthanized at the end of the last dosage; their tumors were harvested and weighed. A portion of each tumor was fixed, sectioned and stained with antibodies against CD31, BrdU and TUNEL.
Statistical analysis
The paired Student’s t test was used for statistical analysis, using SAS software for two paired samples.
Supplementary Material
Acknowledgments
We thank Millennium Pharmaceuticals, Inc. for providing MLN4924. This work is supported by the NCI grants (CA118762, CA156744, CA170995, and CA171277) to YS.
Footnotes
Conflicts of Interest
The authors declare no conflict of interest.
Dedication
This manuscript is dedicated to the fond memory of Victor Fung, Ph.D., a former Program Officer at NCI and former Scientific Review Officer of the Cancer Etiology study section of CSR, NIH, for his wisdom, compassion, integrity, his love of sciences and the arts, his incredible culinary skills, and above all, his contributions to the career development of so many investigators during his own distinguished career.
References
- 1.Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399–434. doi: 10.1146/annurev.biochem.78.101807.093809. [DOI] [PubMed] [Google Scholar]
- 2.Nakayama KI, Nakayama K. Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer. 2006;6:369–381. doi: 10.1038/nrc1881. [DOI] [PubMed] [Google Scholar]
- 3.Zhao Y, Sun Y. Cullin-RING Ligases as Attractive Anti-cancer Targets. Curr Pharm Des. 2013;19:3215–3225. doi: 10.2174/13816128113199990300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wu K, Fuchs SY, Chen A, Tan P, Gomez C, Ronai Z, et al. The SCF(HOS/beta-TRCP)-ROC1 E3 ubiquitin ligase utilizes two distinct domains within CUL1 for substrate targeting and ubiquitin ligation. Mol Cell Biol. 2000;20:1382–1393. doi: 10.1128/mcb.20.4.1382-1393.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Duda DM, Borg LA, Scott DC, Hunt HW, Hammel M, Schulman BA. Structural insights into NEDD8 activation of cullin-RING ligases: conformational control of conjugation. Cell. 2008;134:995–1006. doi: 10.1016/j.cell.2008.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Goldenberg SJ, Cascio TC, Shumway SD, Garbutt KC, Liu J, Xiong Y, et al. Structure of the Cand1-Cul1-Roc1 complex reveals regulatory mechanisms for the assembly of the multisubunit cullin-dependent ubiquitin ligases. Cell. 2004;119:517–528. doi: 10.1016/j.cell.2004.10.019. [DOI] [PubMed] [Google Scholar]
- 7.Kamura T, Conrad MN, Yan Q, Conaway RC, Conaway JW. The Rbx1 subunit of SCF and VHL E3 ubiquitin ligase activates Rub1 modification of cullins Cdc53 and Cul2. Genes Dev. 1999;13:2928–2933. doi: 10.1101/gad.13.22.2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamoah K, Oashi T, Sarikas A, Gazdoiu S, Osman R, Pan ZQ. Autoinhibitory regulation of SCF-mediated ubiquitination by human cullin 1’s C-terminal tail. Proc Natl Acad Sci U S A. 2008;105:12230–12235. doi: 10.1073/pnas.0806155105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willems AR, Schwab M, Tyers M. A hitchhiker’s guide to the cullin ubiquitin ligases: SCF and its kin. Biochim Biophys Acta. 2004;1695:133–170. doi: 10.1016/j.bbamcr.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 10.Jia L, Sun Y. SCF E3 ubiquitin ligases as anticancer targets. Curr Cancer Drug Targets. 2011;11:347–356. doi: 10.2174/156800911794519734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Duan H, Wang Y, Aviram M, Swaroop M, Loo JA, Bian J, et al. SAG, a novel zinc RING finger protein that protects cells from apoptosis induced by redox agents. Mol Cell Biol. 1999;19:3145–3155. doi: 10.1128/mcb.19.4.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Y, Tan M, Duan H, Swaroop M. SAG/ROC/Rbx/Hrt, a zinc RING finger gene family: molecular cloning, biochemical properties, and biological functions. Antioxid Redox Signal. 2001;3:635–650. doi: 10.1089/15230860152542989. [DOI] [PubMed] [Google Scholar]
- 13.Swaroop M, Bian J, Aviram M, Duan H, Bisgaier CL, Loo JA, et al. Expression, purification, and biochemical characterization of SAG, a RING finger redox sensitive protein. Free Radicals Biol Med. 1999;27:193–202. doi: 10.1016/s0891-5849(99)00078-7. [DOI] [PubMed] [Google Scholar]
- 14.Gu Q, Tan M, Sun Y. SAG/ROC2/Rbx2 is a novel activator protein-1 target that promotes c-Jun degradation and inhibits 12-O-tetradecanoylphorbol-13-acetate-induced neoplastic transformation. Cancer Res. 2007;67:3616–3625. doi: 10.1158/0008-5472.CAN-06-4020. [DOI] [PubMed] [Google Scholar]
- 15.Tan M, Gu Q, He H, Pamarthy D, Semenza GL, Sun Y. SAG/ROC2/RBX2 is a HIF-1 target gene that promotes HIF-1alpha ubiquitination and degradation. Oncogene. 2008;27:1404–1411. doi: 10.1038/sj.onc.1210780. [DOI] [PubMed] [Google Scholar]
- 16.He H, Gu Q, Zheng M, Normolle D, Sun Y. SAG/ROC2/RBX2 E3 ligase promotes UVB-induced skin hyperplasia, but not skin tumors, by simultaneously targeting c-Jun/AP-1 and p27. Carcinogenesis. 2008;29:858–865. doi: 10.1093/carcin/bgn021. [DOI] [PubMed] [Google Scholar]
- 17.Tan M, Gallegos JR, Gu Q, Huang Y, Li J, Jin Y, et al. SAG/ROC-SCFbeta-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia. 2006;8:1042–1054. doi: 10.1593/neo.06568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tan M, Zhu Y, Kovacev J, Zhao Y, Pan ZQ, Spitz DR, et al. Disruption of Sag/Rbx2/Roc2 induces radiosensitization by increasing ROS levels and blocking NF-kB activation in mouse embryonic stem cells. Free Radic Biol Med. 2010;49:976–983. doi: 10.1016/j.freeradbiomed.2010.05.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gu Q, Bowden GT, Normolle D, Sun Y. SAG/ROC2 E3 ligase regulates skin carcinogenesis by stage-dependent targeting of c-Jun/AP1 and IkappaB-alpha/NF-kappaB. J Cell Biol. 2007;178:1009–1023. doi: 10.1083/jcb.200612067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia L, Yang J, Hao X, Zheng M, He H, Xiong X, et al. Validation of SAG/RBX2/ROC2 E3 Ubiquitin Ligase as an Anticancer and Radiosensitizing Target. Clin Cancer Res. 2010;16:814–824. doi: 10.1158/1078-0432.CCR-09-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tan M, Zhao Y, Kim SJ, Liu M, Jia L, Saunders TL, et al. SAG/RBX2/ROC2 E3 Ubiquitin Ligase Is Essential for Vascular and Neural Development by Targeting NF1 for Degradation. Dev Cell. 2011;21:1062–1076. doi: 10.1016/j.devcel.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y. Alteration of SAG mRNA in human cancer cell lines: Requirement for the RING finger domain for apoptosis protection. Carcinogenesis. 1999;20:1899–1903. doi: 10.1093/carcin/20.10.1899. [DOI] [PubMed] [Google Scholar]
- 23.Yang GY, Pang L, Ge HL, Tan M, Ye W, Liu XH, et al. Attenuation of ischemia-induced mouse brain injury by SAG, a redox- inducible antioxidant protein. J Cereb Blood Flow Metab. 2001;21:722–733. doi: 10.1097/00004647-200106000-00010. [DOI] [PubMed] [Google Scholar]
- 24.Sun Y, Li H. Functional characterization of SAG/RBX2/ROC2/RNF7, an antioxidant protein and an E3 ubiquitin ligase. Protein Cell. 2013;4:103–116. doi: 10.1007/s13238-012-2105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Duan H, Tsvetkov LM, Liu Y, Song Y, Swaroop M, Wen R, et al. Promotion of S-phase entry and cell growth under serum starvation by SAG/ROC2/Rbx2/Hrt2, an E3 ubiquitin ligase component: association with inhibition of p27 accumulation. Mol Carcinog. 2001;30:37–46. doi: 10.1002/1098-2744(200101)30:1<37::aid-mc1011>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 26.Huang Y, Duan H, Sun Y. Elevated expression of SAG/ROC2/Rbx2/Hrt2 in human colon carcinomas: SAG does not induce neoplastic transformation, but its antisense transfection inhibits tumor cell growth. Mol Carcinog. 2001;30:62–70. [PubMed] [Google Scholar]
- 27.Diez-Juan A, Castro C, Edo MD, Andres V. Role of the growth suppressor p27Kip1 during vascular remodeling. Curr Vasc Pharmacol. 2003;1:99–106. doi: 10.2174/1570161033386709. [DOI] [PubMed] [Google Scholar]
- 28.Schiappacassi M, Lovat F, Canzonieri V, Belletti B, Berton S, Di Stefano D, et al. p27Kip1 expression inhibits glioblastoma growth, invasion, and tumor-induced neoangiogenesis. Mol Cancer Ther. 2008;7:1164–1175. doi: 10.1158/1535-7163.MCT-07-2154. [DOI] [PubMed] [Google Scholar]
- 29.Park AY, Shen TL, Chien S, Guan JL. Role of Focal Adhesion Kinase Ser-732 Phosphorylation in Centrosome Function during Mitosis. J Biol Chem. 2009;284:9418–9425. doi: 10.1074/jbc.M809040200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458:732–736. doi: 10.1038/nature07884. [DOI] [PubMed] [Google Scholar]
- 31.Wei D, Li H, Yu J, Sebolt JT, Zhao L, Lawrence TS, et al. Radiosensitization of human pancreatic cancer cells by MLN4924, an investigational NEDD8-activating enzyme inhibitor. Cancer Res. 2012;72:282–293. doi: 10.1158/0008-5472.CAN-11-2866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan M, Davis SW, Saunders TL, Zhu Y, Sun Y. RBX1/ROC1 disruption results in early embryonic lethality due to proliferation failure, partially rescued by simultaneous loss of p27. Proc Natl Acad Sci U S A. 2009;106:6203–6208. doi: 10.1073/pnas.0812425106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baldassarre G, Belletti B, Nicoloso MS, Schiappacassi M, Vecchione A, Spessotto P, et al. p27(Kip1)-stathmin interaction influences sarcoma cell migration and invasion. Cancer Cell. 2005;7:51–63. doi: 10.1016/j.ccr.2004.11.025. [DOI] [PubMed] [Google Scholar]
- 34.Wander SA, Zhao D, Slingerland JM. p27: a barometer of signaling deregulation and potential predictor of response to targeted therapies. Clin Cancer Res. 2011;17:12–18. doi: 10.1158/1078-0432.CCR-10-0752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang XQ, Lui EL, Cai Q, Ching WY, Liu KS, Poon RT, et al. p27Kip1 promotes migration of metastatic hepatocellular carcinoma cells. Tumour Biol. 2008;29:217–223. doi: 10.1159/000152939. [DOI] [PubMed] [Google Scholar]
- 36.Folkman J. Tumor angiogenesis. Adv Cancer Res. 1985;43:175–203. doi: 10.1016/s0065-230x(08)60946-x. [DOI] [PubMed] [Google Scholar]
- 37.Grunstein J, Roberts WG, Mathieu-Costello O, Hanahan D, Johnson RS. Tumor-derived expression of vascular endothelial growth factor is a critical factor in tumor expansion and vascular function. Cancer Res. 1999;59:1592–1598. [PubMed] [Google Scholar]
- 38.Carmeliet P. Angiogenesis in health and disease. Nat Med. 2003;9:653–660. doi: 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]
- 39.Folkman J. Angiogenesis: an organizing principle for drug discovery? Nat Rev Drug Discov. 2007;6:273–286. doi: 10.1038/nrd2115. [DOI] [PubMed] [Google Scholar]
- 40.Deshaies RJ, Emberley ED, Saha A. Control of cullin-RING ubiquitin ligase activity by Nedd8. In: Groettrup M, editor. Conjugation and Deconjugation of Ubiquitin Family Modifiers: Subcellular Biochemistry. 2010. pp. 41–56. [DOI] [PubMed] [Google Scholar]
- 41.Xirodimas DP. Novel substrates and functions for the ubiquitin-like molecule NEDD8. Biochem Soc Trans. 2008;36:802–806. doi: 10.1042/BST0360802. [DOI] [PubMed] [Google Scholar]
- 42.Rabut G, Peter M. Function and regulation of protein neddylation. ‘Protein modifications: beyond the usual suspects’ review series. EMBO Rep. 2008;9:969–976. doi: 10.1038/embor.2008.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010;1:708–716. doi: 10.1177/1947601910382898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Soucy TA, Smith PG, Rolfe M. Targeting NEDD8-activated cullin-RING ligases for the treatment of cancer. Clin Cancer Res. 2009;15:3912–3916. doi: 10.1158/1078-0432.CCR-09-0343. [DOI] [PubMed] [Google Scholar]
- 45.Nawrocki ST, Griffin P, Kelly KR, Carew JS. MLN4924: a novel first-in-class inhibitor of NEDD8-activating enzyme for cancer therapy. Expert Opin Investig Drugs. 2012;21:1563–1573. doi: 10.1517/13543784.2012.707192. [DOI] [PubMed] [Google Scholar]
- 46.Shen TL, Park AY, Alcaraz A, Peng X, Jang I, Koni P, et al. Conditional knockout of focal adhesion kinase in endothelial cells reveals its role in angiogenesis and vascular development in late embryogenesis. J Cell Biol. 2005;169:941–952. doi: 10.1083/jcb.200411155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15:859–876. doi: 10.1101/gad.862101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhao X, Peng X, Sun S, Park AY, Guan JL. Role of kinase-independent and -dependent functions of FAK in endothelial cell survival and barrier function during embryonic development. J Cell Biol. 2010;189:955–965. doi: 10.1083/jcb.200912094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bockbrader KM, Tan M, Sun Y. A small molecule Smac-mimic compound induces apoptosis and sensitizes TRAIL- and etoposide-induced apoptosis in breast cancer cells. Oncogene. 2005;24:7381–7388. doi: 10.1038/sj.onc.1208888. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.