

Abstract
Aims/Introduction: Heparan sulfate (HS) mediates a variety of molecular recognition events that are essential for differentiation, morphogenesis and homeostasis through various HS forms that result from differential sulfate modification. Recently, we found that HS is localized exclusively around βß‐cells in islets of adult mice and is required for insulin secretion. The aim of this study was to examine the contribution of HS sulfate groups to insulin secretion.
Materials and Methods: Glucose‐induced insulin secretion (GIIS) was examined in mouse pancreatic islets, the mouse pancreatic β‐cell line MIN6 cells and its derivative MIN6T3 cells after removal of sulfate groups by sodium chlorate, a competitive inhibitor of glycosaminoglycan sulfation. Quantitative reverse transcription polymerase chain reaction was used for analyzing messenger ribonucleic acid (mRNA) expression of HS modification enzymes. Expression of HS 3‐O‐sulfotransferase isoform‐1 (Hs3st1) was silenced and GIIS was examined.
Results: Impaired insulin secretion by islets, MIN6 cells and MIN6T3 cells was observed after treatment with sodium chlorate. Sodium chlorate‐treatment upregulated the mRNA expression of sulfotransferases expressed in MIN6T3 cells. Expression of the Hs3st1 was strongly upregulated by sodium chlorate‐treatment, and its silencing by RNA interference reduced GIIS in MIN6T3 cells.
Conclusions: Our data suggest that the 3‐O‐sulfate group of HS that is modified by Hs3st1 plays a significant role(s) in the insulin secretory pathway, selectively through an interaction with factor(s) upstream of membrane depolarization in β‐cells. (J Diabetes Invest, doi: 10.1111/j.2040‐1124.2012.00205.x, 2012)
Keywords: Heparan sulfate, Insulin secretion, Sulfotransferase
Introduction
The predominant function of pancreatic islet β‐cells is insulin secretion, which is regulated by blood glucose levels. Glucose that is transported into β‐cells through the glucose transporters (Glut2 in rodents and Glut1 in humans)1 is metabolized by glycolysis to produce adenosine triphosphate (ATP). The resultant increase in the cytosolic ATP/adenosine diphosphate ratio closes ATP‐sensitive K+ channels in the plasma membrane2,3, leading to membrane depolarization. This depolarization activates voltage‐dependent Ca2+ channels and the consequent influx of Ca2+ triggers insulin secretion4–6.
We recently found that β‐cells in the islets of adult mice are surrounded exclusively by heparan sulfate (HS), and that HS is required for insulin secretion, β‐cell proliferation and islet morphogenesis7. Furthermore, a recent report found endogenous HS as a βß‐cell target for autoimmune damage in non‐obese diabetic mice, a model of type 1 diabetes in humans8. HS is a sulfated glycosaminoglycan that is distributed on the cell surface and in the extracellular matrix, and is involved in diverse cellular phenomena including differentiation, proliferation and homeostasis by interacting with various protein ligands and receptors9,10. The structure of HS is that of a linear polysaccharide, which consists of a repeating disaccharide unit backbone onto which is superimposed a complex pattern of modifications. The first modification to occur after synthesis is N‐deacetylation and subsequent N‐sulfation of glucosamine units, which is carried out by N‐deacetylase/N‐sulfotransferase (NDST)11,12. The enzyme C5‐epimerase converts some glucuronic acid (GlcUA) residues to iduronic acid (IdoUA), and finally, various types of sulfation are mediated by the catalysis of HS 2‐O‐, 6‐O‐ and 3‐O‐sulfotransferases (Hs2st, Hs6st and Hs3st)13,14. The resultant polymorphic sulfated structural motifs (a complex combination of N‐, 2‐O‐, 6‐O‐ and 3‐O‐sulfate groups) are responsible for binding to specific macromolecules and modulating their biological functions15–17. For example, 3‐O‐sulfation of HS is required for binding to antithrombin and herpes simplex virus type 1 (HSV‐1). However, additional, specific modification of the HS oligosaccharide is necessary for its binding. Thus, different oligosaccharide modifications are required for binding to antithrombin and to HSV‐118. These results show the importance of the fine structure of HS modification for its recognition specificity.
Our recent finding showed that HS is required by β‐cells for insulin secretion7. However, the contribution of HS sulfate groups and the fine structure of HS sulfate modification to β‐cell function is still unclear. To determine if the sulfate groups of HS are required for insulin secretion, we decreased the number of sulfate groups on HS by competitive inhibition of sulfation or by reduction of the expression of the sulfotransferase Hs3st1 through ribonucleic acid (RNA) interference, and then examined insulin secretion induced by glucose or KCl.
Materials and Methods
Cell Culture and Subcloning of MIN6 Cells
MIN6 cells19 were kindly donated by Professor J.‐I. Miyazaki (Osaka University), and were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 25 mmol/L glucose, 10% fetal calf serum, 50 μmol/L 2‐mercaptoethanol, 100 μg/mL streptomycin sulfate and 60.5 IU/mL penicillin G under humidified conditions, and 5% CO2–95% air at 37°C. Subcloning of MIN6 cells was carried out using the limiting dilution method20,21. The cells were then screened for and selected according to their insulin secretory response at 16.7 mmol/L glucose compared with that at 2.8 mmol/L glucose, as well as their expression of HS. The insulin concentration of the medium was determined with an insulin ELISA kit (Morinaga Institute of Biological Science, Yokohama, Japan) using mouse insulin as a standard. Western blotting detection of HS was carried out as described previously7.
Sodium Chlorate Treatment and Silencing of Hs3st1 Expression
Pancreatic islets isolated from 8‐week‐old Institute of Cancer Research mice were incubated either without or with 30 mmol/L sodium chlorate (SC) in RPMI1640 medium supplemented with 5.5 mmol/L glucose, 0.5% bovine serum albumin (BSA), 100 μg/mL streptomycin sulfate and 100 IU/mL penicillin G for 5 days at 37ºC1. MIN6 cells were incubated either without or with SC22 (≥99%, American Chemical Society reagent grade; Sigma‐Aldrich, St. Louis, MO, USA) and/or HS (pig mucosa, containing both low‐ and high‐sulfated species; Iduron, Manchester, UK) in DMEM for 5 days. For silencing of gene expression, the following 27‐mer oligoribonucleotides and their reverse sequence were synthesized by Integrated DNA Technologies (Coralville, IA, USA). Two independent Dicer‐substrate small interfering RNA (DsiRNA) were designed against different regions of the complete mouse Hs3st1 complementary deoxyribonucleic acid (cDNA; NCBI Accession NM_010474). The sequences of the DsiRNA are as follows: Hs3st1 (1): sense, 5′‐UGAAGGAACUUAACUAUUGCACUGA‐3′ and antisense, 5′‐UCAGUGCAAUAGUUAAGUUCCUUCAUA‐3′; Hs3st1 (2): sense, 5′‐AGUAUUAGAUUCACAGUUGCCAUAT‐3′ and antisense, 5′‐AUAUGGCAACUGUGAAUCUAAUACUGU‐3′; DsiRNA negative control (NC DsiRNA): sense, 5′‐CGUUAAUCGCGUAUAAUACGCGUAT‐3′ and antisense 5′‐AUACGCGUAUUAUACGCGAUUAACGAC‐3′. Cells that had reached 60% confluence were transfected with 100 nmol/L of control or Hs3st1 DsiRNA that had been complexed for 30 min with 3 μmol/L of Transductin (Integrated DNA Technologies).
Cell Growth Assay
Cells (2 × 104 cells/well, 96‐well plate) were seeded and precultured in DMEM for 2 days. Thereafter, cells were incubated with SC at several concentrations or transfected with DsiRNA. Five days later, cell growth and ATP content were measured colorimetrically using Cell Counting Kit‐8 (Dojindo, Kumamoto, Japan) and ATP assay system (Toyo Ink, Tokyo, Japan) following the manufacturer’s instructions. Assay of cell growth was based on the cleavage of the 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium, monosodium salt (WST‐8) by mitochondrial dehydrogenase in viable cells to a formazan dye. Assay of the ATP measurement was based on the ability of luciferase to convert luciferin to oxyluciferin in the presence of ATP23.
Semiquantitative and Real‐Time Polymerase Chain Reaction
Total RNA was extracted using the Fast Pure RNA kit (Takara BIO INC, Kyoto, Japan). Reverse transcription was carried out using the Primescript RT reagent Kit (Takara). Semiquantitative and real‐time polymerase chain reaction (PCR) were carried out in triplicate for every cDNA sample on a DNA Engine Opticon 2 Real‐Time PCR Detection System (MJ Research/Bio‐Rad, Hercules, CA, USA) using the SsoFast EvaGreen Supermix (Bio‐Rad). The sequences of the primers used are listed in Table 1.
Table 1. Sequences of the primers used for polymerase chain reaction.
| Gene | Sense primers | Antisense primers | Product size (bp) |
|---|---|---|---|
| Insulin 1 | 5′‐TAGTGACCAGCTATAATCAGAG‐3′ | 5′‐ACGCCAAGGTCTGAAGGTCC‐3′ | 289 |
| Insulin 2 | 5′‐CCCTGCTGGCCCTGCTCTT‐3′ | 5′‐AGGTCTGAAGGTCACCTGCT‐3′ | 213 |
| PC1/3 | 5′‐ATGGAGCAAAGAGGTTGGAC‐3′ | 5′‐GCTGCAGTCATTCTGGTATC‐3′ | 419 |
| PC2 | 5′‐TCGCCAAGTTGCAGCAGAAC‐3′ | 5′‐CTTCGGCCACGTTCAAGTCTA‐3′ | 314 |
| Ndst1 | 5′‐GCACAGAGGAGTACCCACAT‐3′ | 5′‐TGATCTTGTCCAGCTCACTG‐3′ | 156 |
| Ndst2 | 5′‐CTTTGTGGGCAAAGAAGGTA‐3′ | 5′‐CAGCATCCTCCTCTTCTGTC‐3′ | 155 |
| Ndst3 | 5′‐GCTCTTCACCCACACAATTT‐3′ | 5′‐CAGTCGGTCGTTTCCATAGT‐3′ | 154 |
| Ndst4 | 5′‐TGAATTCTGGTGGTTTCCTC‐3′ | 5′‐CCAGACTTTCTTCCAAGCTG‐3′ | 208 |
| Epimerase | 5′‐AGCAGAAGACAGGGACAGAA‐3′ | 5′‐CACAGACACACTCCCATTTG‐3′ | 214 |
| Hs2st1 c | 5′‐CGGAAACAAGGAGACAAAAA‐3′ | 5′‐GTGACTCCCACCAGGAAGTA‐3′ | 194 |
| Hs6st1 | 5′‐ACCGAACTCACCAACTGTGT‐3′ | 5′‐CAAGTTGCTCCTCTCTGGAC‐3′ | 172 |
| Hs6st2 | 5′‐CAGGTGGAGGATTTTTCAGA‐3′ | 5′‐CAAGTTGCTCCTCTCTGGAC‐3′ | 207 |
| Hs6st3 | 5′‐CCATCATGGAGAAGAAGGAT‐3′ | 5′‐GTAGGCAGCTCATCTGGTGT‐3′ | 186 |
| Hs3st1 | 5′‐ATGCACACATGCTGAACTGG‐3′ | 5′‐GCAGTAGAAGCCCTTGGTTTTG‐3′ | 179 |
| Hs3st2 | 5′‐AAGCCAGGCACCAAACG‐3′ | 5′‐TCCAGTGTGATCTGGGTCTCC‐3′ | 206 |
| Hs3st3a1 | 5′‐GACCCTGGCCTTACTTCTGG‐3′ | 5′‐AGGGTTCTGGGCATCAAATC‐3′ | 204 |
| Hs3st3b1 | 5′‐CAGTCCCATCTCCAGCTTCTTC‐3′ | 5′‐AGGGTTCTGGGCATCAAGTC‐3′ | 210 |
| Hs3st4 | 5′‐CATCGGGGTCAAGAAAGGAG‐3′ | 5′‐CCTCATTCGTCACAAAATAACTGG‐3′ | 203 |
| Hs3st5 | 5′‐CTGGGAAGCCTTGCTGTTG‐3′ | 5′‐GGAGGTGAACCTGCTCCTTG‐3′ | 205 |
| Hs3st6 | 5′‐CTTTCCTCAGGCGCTCATTG‐3′ | 5′‐ATGGTGATCTGCCCATCCAG‐3′ | 186 |
| β‐Actin | 5′‐ACAGCTTCTTTGCAGCTCCTTC‐3′ | 5′‐CCCATTCCCACCATCACAC‐3′ | 196 |
Hsst, heparan sulfate sulfotransferase; Ndst, N‐deacetylase/N‐sulfotransferase; PC, prohormone convertase.
Measurement of Insulin Secretion by Isolated Islets or Cells, and Their Insulin and Protein Content
Batches of 10 islets were pre‐incubated in Krebs Ringer bicarbonate buffer (KRB) containing 2.8 mmol/L glucose for 1 h at 37°C in 95% O2/5% CO2, and were then incubated in 0.1 mL of fresh KRB containing various concentrations of glucose for 1 h7. Cells (1 × 105 cells/well, 48‐well plate) were seeded and precultured in DMEM for 2 days, and cultured with/without SC or DsiRNA for the appropriate period as described earlier. On the day of the experiment, the cells were placed in 2‐(4‐[2‐hydroxyethyl]‐1‐piperazine) ethanesulfonic acid (HEPES)‐balanced KRB19 (Krebs–Ringer–HEPES [KRH]: 119 mmol/L NaCl, 4.7 mmol/L KCl, 2.5 mmol/L CaCl2, 1.2 mmol/L MgCl2, 1.2 mmol/L KH2PO4, 25 mmol/L NaHCO3 and 20 mmol/L HEPES, pH 7.4) supplemented with 5 mg/mL BSA and 2.8 mmol/L glucose for 2 h at 37°C, and were then incubated in fresh KRH containing various concentrations of glucose or KCl for 2 h. After incubation, the medium was removed from the islets or cells and the insulin concentration of the medium was determined using the insulin ELISA kit as describe earlier. The total protein level of the islets or cells was quantified using the Micro‐BCA Protein Assay Reagent (PIERCE Biotechnology Inc, Rockford, IL, USA). The insulin content of cultured cells was measured as described7.
Statistical Analysis
Data are presented as means ± standard error. Statistical significance was determined using Student’s t‐test. A P‐value of 0.05 was considered significant.
Results
Impaired Insulin Secretion in Sodium Chlorate‐Treated MIN6T3 Cells
Our previous report showed that β‐cells in islets are exclusively surrounded by HS, and that HS is required for islet insulin secretion and β‐cell proliferation7. To facilitate further analysis of this phenomenon, we examined the MIN6 cultured cell line, which is derived from a mouse insulinoma and its subclone, both of which retain the function of insulin secretion as described later. We first checked HS expression in MIN6 cells, as well as in pancreatic β‐cells. As shown by western blotting in Figure 1, HS was detected in MIN6 cells. However, MIN6 cells have been reported to show functional heterogeneity with increasing passage number, and clones that have functionally lost glucose‐induced insulin secretion (GIIS) have been isolated20,21. These reports suggested the possibility that some of the MIN6 cells might decrease or lose HS expression over time. Therefore, for analysis of the relationship of the sulfate fine structure of HS and β‐cell function, selection of a single optimal cell line was required. In order to obtain such a cell line, we subcloned MIN6 cells in order to obtain cell lines that retained the characteristics of β‐cells; that is, GIIS and strong HS expression. Of the 30 subclones obtained, two subclones showed a good response to glucose, as well as strong HS expression. One of these cell lines, designated as MIN6T3, was used for further analysis (Figure 1).
Figure 1.
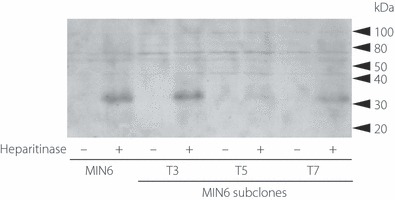
Expression of heparan sulfate (HS) in MIN6 and its subclone MIN6T3. MIN6 cells and its sublines, T3 (MIN6T3), T5 and T7, were treated without (−) or with (+) 0.1 U/mL heparitinase for 1 h. The cells were then directly lysed in sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS‐PAGE) sample buffer and were subjected to SDS‐PAGE followed by immunoblotting with monoclonal antibody (mAb) 3G10 that recognizes uronates on the HS stubs generated after heparitinase digestion. Molecular weight markers are shown on the right. Abundant expression of HS was observed in T3, whereas other cell lines (T5 and T7) showed weak expression of HS.
We first determined the contribution of the sulfate groups of HS to insulin secretion. For this purpose, we treated MIN6 cells in order to desulfate HS chains with 30 mmol/L SC, the concentration that effectively inhibits HS sulfation22, and then examined the GIIS and insulin content of the cells. As shown in Figures 2a,b, insulin secretion is time‐dependently decreased and insulin content is gradually increased by SC treatment. Viability of MIN6T3 cells around the range of concentration of SC so far used22,24,25 for inhibition of sulfation is shown in Figure 2c. The cell viability of 30 mmol/L SC treatment for 5 days was 90% compared with that of the control of 0 mmol/L SC treatment. The time‐dependent effect of SC treatment for cell proliferation was also examined in 30 mmol/L SC by WST‐8 cleavage (Figure 2d). ATP content in the cells treated with 30 mmol/L SC for 5 days showed a similar extent of decrease (Figure 2e). This growth inhibitory effect of SC might be caused in part by desulfation of 2‐O‐ and/or 6‐O‐sulfate groups in HS, which are functionally important for FGF signaling25–30. As shown in Figure 2f–h, treatment of mouse pancreatic islets or MIN6 cells with 30 mmol/L SC resulted in a decrease in GIIS both in islets and MIN6 similar to the result obtained in MIN6T3 cells. The addition of exogenous soluble HS to the culture recovered the GIIS of the SC‐treated MIN6T3 cells (Figure 2i). Furthermore, the addition of HS to the medium without SC also enhanced insulin secretion (Figure 2j).
Figure 2.
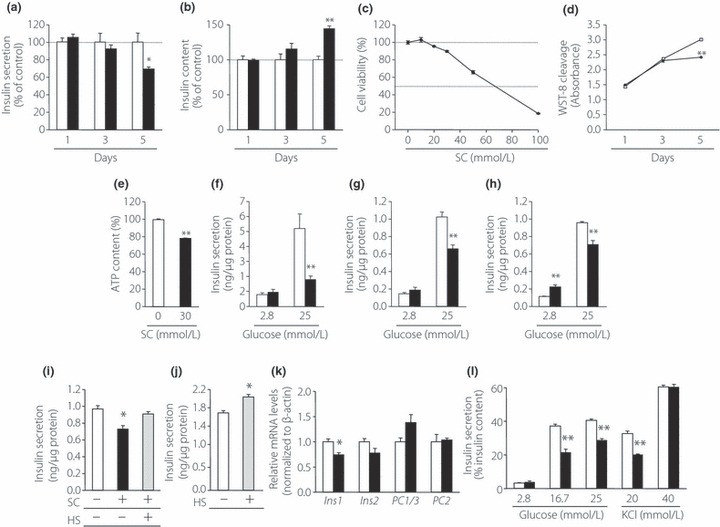
Inhibitory effects of sodium chlorate (SC) treatment on insulin secretion. (a) Insulin secretion of MIN6T3 cells after treatment of SC. (b) Effect of SC treatment on the insulin content of MIN6T3 cells (n = 4). (c) Cell viability of MIN6T3 cells treated with various concentration of SC for 5 days. Viability of cells without SC treatment was indicated as 100% (n = 5–8). (d) Effect of SC treatment on cell proliferation. MIN6T3 cells were incubated with or without 30 mmol/L SC, and 2‐(2‐methoxy‐4‐nitrophenyl)‐3‐(4‐nitrophenyl)‐5‐(2,4‐disulfophenyl)‐2H‐tetrazolium, monosodium salt (WST‐8) cleavage was measured (n = 5–10). (e) Effect of SC treatment on the adenosine triphosphate (ATP) content in MIN6T3 cells (n = 9). Insulin secretion from (f) isolated islets, (g) MIN6 cells (h) and MIN6T3 cells was measured after incubation for 5 days with or without 30 mmol/L SC (n = 3–5). (i) Recovery of insulin secretion from SC‐treated cells after the addition of heparan sulfate (HS). MIN6T3 cells were treated with SC for 5 days in the presence or absence of 100 μg/mL HS, as indicated. Insulin secretion was measured using 25 mmol/L glucose (n = 3). (j) Enhancement of insulin secretion by supplement of HS. MIN6T3 cells were cultured with or without 100 μg/mL HS for 5 days (n = 3). (k) The messenger ribonucleic acid (mRNA) levels of insulin‐related genes were determined using real‐time reverse transcription polymerase chain reaction after incubation for 5 days with SC treatment. The mRNA level of each gene was expressed relative to that in cells that were incubated without SC treatment, which was set at 1 (n = 3). (l) Effect of SC treatment on insulin secretion from MIN6T3 cells in response to glucose or KCl. Data are expressed as insulin secretion relative to basal secretion at 2.8 mmol/L glucose (which was arbitrarily set at 1; n = 3–4). Open bars or squares, non‐treated cells; filled bars or squares, SC‐treated cells; shaded bars, HS‐treated cells. *P < 0.05; **P < 0.01.
To investigate the cause of the decrease in GIIS after SC treatment, the insulin content and the expression of insulin related genes in SC‐treated MIN6T3 cells were examined. Whereas insulin content was increased in SC‐treated MIN6T3 cells compared with controls without SC‐treatment (Figure 2b), the expression levels of Insulin 1, Insulin 2, PC1/3 and PC2 genes, which encode proinsulin processing enzymes involved in insulin biosynthesis, were not significantly increased in these cells (Figure 2k). These findings suggested that impaired insulin secretion resulting from SC‐treatment contributes both to intracellular insulin accumulation and to subsequent increased insulin content. To take into account the fact that cellular insulin content was increased by SC‐treatment, effects on insulin secretion in response to glucose or KCl were expressed as % insulin content. As shown in Figure 2l, insulin secretion in response to 16.7 and 25 mmol/L glucose, or to 20 mmol/L KCl, was reduced in SC‐treated MIN6T3 cells to 58, 71 and 62%, respectively, of controls without SC‐treatment. In contrast, no significant decrease of insulin secretion was observed in SC‐treated cells in response to 40 mmol/L KCl.
Expression of HS Modification Enzymes in SC‐Treated MIN6T3 Cells
It was likely that SC treatment would result in an increase in the expression of HS modification enzymes, especially sulfation enzymes, in order to resynthesize or repair HS and thereby restore its function. To identify the genes responsible for HS modification in β‐cells, we examined the messenger (m)RNA expression of genes that encode the enzymes N‐deacetylase/N‐sulfotransferases, C5‐epimerase and HS sulfotransferases, which are involved in HS modification. Gene expression was analyzed in MIN6T3 cells, using semiquantitative reverse transcription PCR (RT–PCR; Figure 3a–c). Expression of Hs2st1 mRNA, which encodes the enzyme that is responsible for 2‐O‐sulfation in mice, was not detected (Figure 3b). In contrast, at least one isoform of each of the other modifying enzymes was expressed in MIN6T3 cells. After SC treatment of these cells, the mRNA levels of Ndst1, Hs6st1, and Hs3st1, 2, 5 and 6 were increased 1.5‐fold or more compared with their levels in non‐treated cells (Figure 3d). The mRNA that showed the highest increase was that of one of the HS sulfotransferase genes, the Hs3st1 gene, whose mRNA level was significantly increased more than 2.5‐fold compared with the control. The Hs3st1 enzyme has been reported to transfer sulfate to the C‐3 position of a GlcNS ± 6S (where GlcNS is glucosamine N‐sulfate and S is sulfate) to form 3‐O‐sulfated HS31. Few or no transcripts of the Ndst2, ‐3, ‐4, Hs2st1, Hs6st2, ‐3 or Hs3st3a1 and st‐4 genes were detected (Figure 3a–c).
Figure 3.
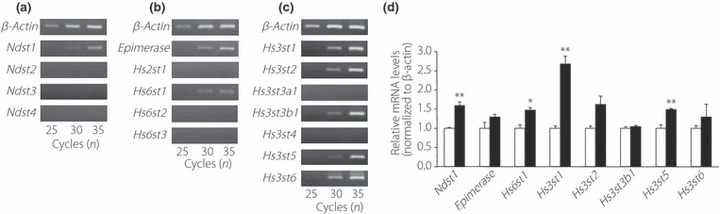
Expression of genes encoding enzymes involved in modification of heparan sulfate (HS) in MIN6T3 cells. Semiquantitative reverse transcription polymerase chain reaction (RT–PCR) analysis of the messenger ribonucleic acid (mRNA) expression in MIN6T3 cells of (a) the four isoforms of N‐deacetylase/N‐sulfotransferase (Ndst1, 2, 3 and 4); (b) Epimerase, HS 2‐O‐sulfotransferase isoform‐1 (Hs2st1) and the three isoforms of HS 6‐O‐sulfotransferase (Hs6st1, 2 and 3); (c) the seven isoforms of HS 3‐O‐sulfotransferase (Hs3st1, 2, 3a1, 3b1, 4, 5 and 6). After PCR reactions using 25, 30 or 35 amplification cycles, the samples were analyzed on a 2% agarose gel. The housekeeping gene, βß‐actin, was used as an internal control. (d) The mRNA levels of the indicated genes were determined using real‐time RT–PCR after incubation for 5 days with 30 mmol/L sodium chlorate (SC). The mRNA level of each gene was expressed relative to that in cells treated without SC, which was set at 1 (n = 3). Open bars, non‐treated cells; filled bars, SC‐treated cells. *P < 0.05; **P < 0.01.
Hs3st1 RNA Interference Impaired Insulin Secretion in MIN6T3 Cells
Hs3st1, the sulfotransferase that is markedly upregulated by SC treatment, might function in the sulfation of HS in β‐cells, which the present results suggest is involved in insulin secretion (Figure 2). To determine if Hs3st1 activity correlates with insulin secretion, and might therefore be mediated by 3‐O‐sulfation of HS made by Hs3st1, insulin secretion was analyzed in MIN6T3 cells in which expression of Hs3st1 was silenced. To silence Hs3st1, MIN6T3 cells were transfected with one of two different DsiRNA (1 and 2) targeted to the Hs3st1 gene or with a negative control sequence, NC DsiRNA. A significant decrease in Hs3st1 mRNA expression was observed after transfection of either Hs3st1 DsiRNA (1) or (2) (decrease to 0.43 and 0.38 of the level in control cells, respectively; Figure 4a). We examined mRNA expression of other Hs3st detected in MIN6T3 (Figure 4b for Hs3st1 DsiRNA [1] and Figure 4c for Hs3st1 DsiRNA [2]). Expression of no other Hs3st were commonly affected by DsiRNA (1) and (2), though Hs3st2 mRNA was slightly (85%) decreased by DsiRNA (1), and Hs3st6 was significantly upregulated (156%) by Hs3st1 DsiRNA (2). Next, we analyzed glucose‐induced and KCl‐induced insulin secretion and insulin content. GIIS of Hs3st1‐silenced MIN6T3 cells was reduced to 68 and 62%, respectively, of the level in control‐treated cells (Figure 4d). Insulin content was unaffected by silencing of Hs3st1 (Figure 4g). In contrast, insulin secretion induced by KCl in MIN6T3 cells was unaffected by DsiRNA‐silencing of Hs3st1 (Figure 4e,f), suggesting that Hs3st1‐mediated sulfation affects insulin secretion through an event that is upstream of membrane depolarization. We examined whether silencing of Hs3st1 affects proliferation of MIN6T3 cells, but transfection of DsiRNA shows no effect for WST‐8 cleavage or ATP content in the cells (Figure 4h,i).
Figure 4.
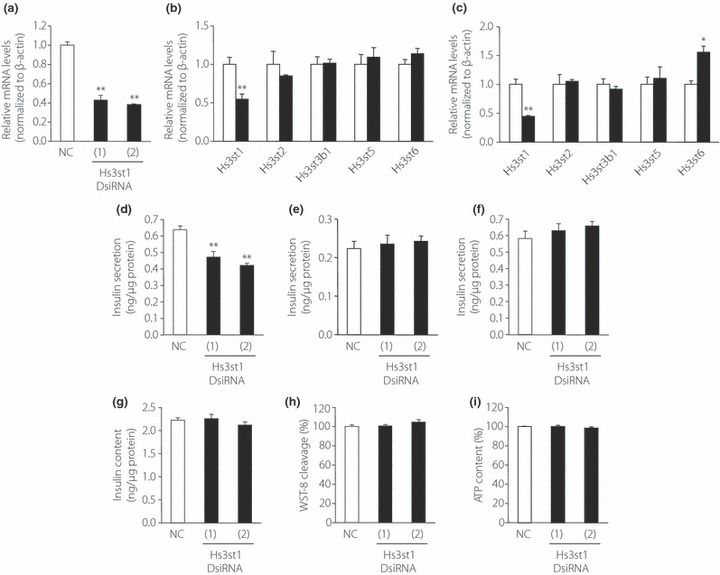
Heparan sulfate 3‐O‐sulfotransferase isoform‐1 (Hs3st1) knockdown by Dicer‐substrate small interfering RNA (DsiRNA) inhibits insulin secretion by MIN6T3 cells. MIN6T3 cells were transfected with negative control DsiRNA (NC) or with one of two different Hs3st1 DsiRNA (1 and 2). After 48 h incubation, total mRNA was isolated and the levels of (a) Hs3st1 mRNA and (b) mRNA of other Hs3sts (cells transfected with Hs3st1 DsiRNA[1], with (c) Hs3st1 DsiRNA[2]) were quantified by real‐time RT–PCR using β‐actin as an internal standard (n = 3). (d–i) Insulin secreted from MIN6T3 cells in response to (d) 25 mmol/L glucose, (e) 20 mmol/L KCl or (f) 40 mmol/L KCl was measured 5 days after transfection (n = 3–4). (g) Insulin content, (h) cell proliferation or (i) adenosine triphosphate (ATP) content was measured 5 days after transfection (n = 3–8). Open bars, NC transfected cells; filled bars, Hs3st1 DsiRNA (1) or (2) transfected cells. *P < 0.05; **P < 0.01.
Discussion
The results of this report show that the sulfate group(s) of HS is/are required for normal insulin secretion. A decrease in insulin secretion was observed after: (i) competitive inhibition of HS sulfation using SC treatment; and (ii) silencing of the expression of the 3‐O‐sulfotransferase, Hs3st1. HS interacts with a variety of macromolecules including growth factors, morphogens and extracellular matrix proteins, and these interactions are mediated by the sulfate groups of HS. Specific sulfate modifications of HS, which are generated by various sulfotransferases, contribute to its functional diversity and specificity. These modifications involve a combination of N‐sulfation, 2‐O‐sulfation, 6‐O‐sulfation and 3‐O‐sulfation. The present study also showed that the specific position of sulfate groups in HS plays functionally important roles in insulin secretion.
In the present study, we showed that silencing of the 3‐O‐sulfotransferase enzyme, Hs3st1, causes a decrease in insulin secretion, suggesting that 3‐O‐sulfation of HS is functionally important for insulin secretion by β‐cells. In addition, high expression of 3‐O‐sulfotransferases was noted in MIN6T3 cells. There are two distinct forms of 3‐O‐sulfated HS saccharides18. One form that is produced by several isoforms of Hs3st, but not by Hs3st1, can bind to HSV‐1 viral coat protein gD and is involved in virus entry into the cell32–39. The other 3‐O‐sulfated form of HS, which is generated by Hs3st1 or Hs3st5, can bind to antithrombin and inhibits blood coagulation (anticoagulant HS)38–43. It is known that Hs3st1 transfers sulfate to the C‐3 position of a GlcNS ± 6S that is linked to a GlcA (glucuronic acid) to form 3‐O‐sulfated HS that contains GlcA‐GlcNS3S ± 6S. HS modified in this manner functions as an anticoagulant31. It is therefore thought that 3‐O‐sulfated HS contains at least GlcA‐GlcNS3S ± 6S that is generated by Hs3st1, and that this modification of HS is required for proper insulin secretion. The high level of Hs3st1 mRNA induction observed after SC treatment suggests the presence of a possible feedback regulatory mechanism that maintains this form of 3‐O‐sulfation in HS for β‐cell function.
Modification of HS by 2‐O‐sulfation is less likely in β‐cells and is probably unnecessary for insulin secretion, because the present results show that mRNA expression of Hs2st1, which is the only known 2‐O‐sulfotransferase, was not detected in MIN6T3 cells. The contribution of other modifications of HS to insulin secretion, including N‐ and 6‐O‐sulfation, and the form of 3‐O‐sulfation that mediates HSV‐1 entry, remains unclear. Although their level of mRNA induction after SC‐treatment was not very high, the present results, which showed that random desulfation by SC treatment reduces insulin secretion, even that induced by KCl treatment, suggest that sulfation other than the 3‐O‐sulfation generated by Hs3st1 might affect insulin secretion downstream of depolarization in the insulin secretion pathway.
Another interesting question is how the sulfate groups of HS, in particular 3‐O‐sulfation of HS, function in the insulin secretion pathway. The present results showed that a reduction in 3‐O‐sulfation of HS affects events upstream of depolarization. This result suggests that 3‐O‐sulfate groups might interact with extracellular molecules to affect signaling pathways that influence insulin secretion, including pathways such as glucose uptake, subsequent glycolysis and ATP synthesis. Several studies have suggested that the sulfate groups of HS interact with intercellular signaling molecules. One of these signaling molecules, Notch, has been reported to interact with 3‐O‐sulfated HS, although the relationship between Notch signaling and insulin secretion is still unclear. In Drosophila, the levels of Notch proteins on the cell surface were markedly decreased and morphological defects were observed after RNA interference of Hs3st‐b, one of the 3‐O‐sulfotransferases44. However the interaction of Hs3st‐b and Notch is analyzed only genetically, and molecular interaction between the 3‐O‐sulfate group of HS and Notch and/or factor(s) in the Notch signaling pathway is still unclear. Alternatively, HS might directly interact with, and contribute to, the stability of membrane protein(s), such as the glucose transporter, (for example Glut2 in rodents or Glut1 in humans)1, in the insulin secretion pathway.
In conclusion, the present study showed that the sulfate groups of HS, in particular the 3‐O‐sulfate modification generated by Hs3st1, are necessary for maintaining normal GIIS. In addition, the present results suggest that examination of β‐cell dysfunctions in terms of HS function, especially the roles of sulfate groups in HS, will provide a novel perspective that might lead to a better understanding of diabetes mellitus. Furthermore, modification of the sulfate fine structure of HS in β‐cells could be a new target for the development of drugs and therapy to improve β‐cell functions in diabetes patients.
Acknowledgements
We are grateful to Professor Jun‐ichi Miyazaki (Division of Stem Cell Regulation Research, Osaka University Medical School, Suita, Japan) for providing the MIN6 cell line. This work was supported in part by Grants‐in‐Aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan. There is no financial support or relationships that may pose a conflict of interest.
References
- 1.De Vos A, Heimberg H, Quartier E, et al. Human and rat beta cells differ in glucose transporter but not in glucokinase gene expression. J Clin Invest 1995; 96: 2489–2495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cook DL, Hales CN. Intracellular ATP directly blocks K+ channels in pancreatic B‐cells. Nature 1984; 311: 271–273 [DOI] [PubMed] [Google Scholar]
- 3.Detimary P, Van den Berghe G, Henquin JC. Concentration dependence and time course of the effects of glucose on adenine and guanine nucleotides in mouse pancreatic islets. J Biol Chem 1996; 271: 20559–20565 [DOI] [PubMed] [Google Scholar]
- 4.Wollheim CB, Sharp GW. Regulation of insulin release by calcium. Physiol Rev 1981; 61: 914–973 [DOI] [PubMed] [Google Scholar]
- 5.Hoenig M, Sharp GW. Glucose induces insulin release and a rise in cytosolic calcium concentration in a transplantable rat insulinoma. Endocrinology 1986; 119: 2502–2507 [DOI] [PubMed] [Google Scholar]
- 6.Satin LS, Cook DL. Voltage‐gated Ca2+ current in pancreatic B‐cells. Pflugers Arch 1985; 404: 385–387 [DOI] [PubMed] [Google Scholar]
- 7.Takahashi I, Noguchi N, Nata K, et al. Important role of heparan sulfate in postnatal islet growth and insulin secretion. Biochem Biophys Res Commun 2009; 383: 113–118 [DOI] [PubMed] [Google Scholar]
- 8.Ziolkowski AF, Popp SK, Freeman C, et al. Heparan sulfate and heparanase play key roles in mouse βß‐cell survival and autoimmune diabetes. J Clin Invest 2012; 122: 132–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bernfield M, Götte M, Park PW, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem 1999; 68: 729–777 [DOI] [PubMed] [Google Scholar]
- 10.Kramer KL, Yost HJ. Heparan sulfate core proteins in cell‐cell signaling. Annu Rev Genet 2003; 37: 461–484 [DOI] [PubMed] [Google Scholar]
- 11.Grobe K, Ledin J, Ringvall M, et al. Heparan sulfate and development: differential roles of the N‐acetylglucosamine N‐deacetylase/N‐sulfotransferase isozymes. Biochim Biophys Acta 2002; 1573: 209–215 [DOI] [PubMed] [Google Scholar]
- 12.Kjellén L. Glucosaminyl N‐deacetylase/N‐sulphotransferases in heparan sulphate biosynthesis and biology. Biochem Soc Trans 2003; 31: 340–342 [DOI] [PubMed] [Google Scholar]
- 13.Habuchi O. Diversity and functions of glycosaminoglycan sulfotransferases. Biochim Biophys Acta 2000; 1474: 115–127 [DOI] [PubMed] [Google Scholar]
- 14.Rosenberg RD, Shworak NW, Liu J, et al. Heparan sulfate proteoglycans of the cardiovascular system. Specific structures emerge but how is synthesis regulated? J Clin Invest 1997; 100: S67–S75 [PubMed] [Google Scholar]
- 15.Lindahl U, Kusche‐Gullberg M, Kjellén L. Regulated diversity of heparan sulfate. J Biol Chem 1998; 273: 24979–24982 [DOI] [PubMed] [Google Scholar]
- 16.Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol 2001; 11: 75–82 [DOI] [PubMed] [Google Scholar]
- 17.Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest 2001; 108: 169–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Esko JD, Selleck SB. Order out of chaos: assembly of ligand binding sites in heparan sulfate. Annu Rev Biochem 2002; 71: 435–471 [DOI] [PubMed] [Google Scholar]
- 19.Miyazaki JI, Araki K, Yamato E, et al. Establishment of a pancreatic beta cell line that retains glucose‐inducible insulin secretion: special reference to expression of glucose transporter isoforms. Endocrinology 1990; 127: 126–132 [DOI] [PubMed] [Google Scholar]
- 20.Minami K, Yano H, Miki T, et al. Insulin secretion and differential gene expression in glucose‐responsive and ‐unresponsive MIN6 sublines. Am J Physiol Endocrinol Metab 2000; 279: E773–E781 [DOI] [PubMed] [Google Scholar]
- 21.Lilla V, Webb G, Rickenbach K, et al. Differential gene expression in well‐regulated and dysregulated pancreatic beta‐cell (MIN6) sublines. Endocrinology 2003; 144: 1368–1379 [DOI] [PubMed] [Google Scholar]
- 22.Humphries DE, Silbert JE. Chlorate: a reversible inhibitor of proteoglycan sulfation. Biochem Biophys Res Commun 1988; 154: 365–371 [DOI] [PubMed] [Google Scholar]
- 23.Lundin A, Thore A. Analytical information obtainable by evaluation of the time course of fir e fly bioluminescence in the assay of ATP. Anal Biochem 1975; 66: 47–63 [DOI] [PubMed] [Google Scholar]
- 24.Safaiyan F, Kolset SO, Prydz K, et al. Selective effects of sodium chlorate treatment on the sulphation of heparan sulphate. J Biol Chem 1999; 274: 36267–36273 [DOI] [PubMed] [Google Scholar]
- 25.Rapraeger AC, Krufka A, Olwin BB. Requirement of heparan sulfate for bFGF‐mediated fibroblast growth and myoblast differentiation. Science 1991; 252: 1705–1708 [DOI] [PubMed] [Google Scholar]
- 26.Gallagher JT. Heparan sulfate: growth control with a restricted sequence menu. J Clin Invest 2001; 108: 357–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guimond S, Maccarana M, Olwin BB, et al. Activating and inhibitory heparin sequences for FGF‐2 (basic FGF). Distinct requirements for FGF‐1, FGF‐2, and FGF‐4. J Biol Chem 1993; 268: 23906–23914 [PubMed] [Google Scholar]
- 28.Ishihara M, Takano R, Kanda T, et al. Importance of 6‐O‐sulfate groups of glucosamine residues in heparin for activation of FGF‐1 and FGF‐2. J Biochem 1995; 118: 1255–1260 [DOI] [PubMed] [Google Scholar]
- 29.Ishihara M, Kariya Y, Kikuchi H, et al. Importance of 2‐O‐sulfate groups of uronate residues in heparin for activation of FGF‐1 and FGF‐2. J Biochem 1997; 121: 345–349 [DOI] [PubMed] [Google Scholar]
- 30.Ostrovsky O, Berman B, Gallagher J, et al. Differential effects of heparin saccharides on the formation of specific fibroblast growth factor (FGF) and FGF receptor complexes. J Biol Chem 2002; 277: 2444–2453 [DOI] [PubMed] [Google Scholar]
- 31.Lawrence R, Kuberan B, Lech M, et al. Mapping critical biological motifs and biosynthetic pathways of heparan sulfate. Glycobiology 2004; 14: 467–479 [DOI] [PubMed] [Google Scholar]
- 32.Shukla D, Liu J, Blaiklock P, et al. A novel role for 3‐O‐sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 1999; 99: 13–22 [DOI] [PubMed] [Google Scholar]
- 33.Shukla D, Spear PG. Herpes viruses and heparan sulfate: an intimate relationship in aid of viral entry. J Clin Invest 2001; 108: 503–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tiwari V, Clement C, Duncan MB, et al. A role for 3‐O‐sulfated heparan sulfate in cell fusion induced by herpes simplex virus type 1. J Gen Virol 2004; 85: 805–809 [DOI] [PubMed] [Google Scholar]
- 35.Xu D, Tiwari V, Xia G, et al. Characterization of heparan sulphate 3‐O‐sulphotransferase isoform 6 and its role in assisting the entry of herpes simplex virus type 1. Biochem J 2005; 385: 451–459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tiwari V, O’Donnell CD, Oh MJ, et al. A role for 3‐O‐sulfotransferase isoform‐4 in assisting HSV‐1 entry and spread. Biochem Biophys Res Commun 2005; 338: 930–937 [DOI] [PubMed] [Google Scholar]
- 37.O’Donnell CD, Tiwari V, Oh MJ, et al. A role for heparan sulfate 3‐O‐sulfotransferase isoform 2 in herpes simplex virus type 1 entry and spread. Virology 2006; 346: 452–459 [DOI] [PubMed] [Google Scholar]
- 38.Xia G, Chen J, Tiwari V, et al. Heparan sulfate 3‐O‐sulfotransferase isoform 5 generates both an antithrombin‐binding site and an entry receptor for herpes simplex virus, type 1. J Biol Chem 2002; 277: 37912–37919 [DOI] [PubMed] [Google Scholar]
- 39.Chen J, Duncan MB, Carrick K, et al. Biosynthesis of 3‐O‐sulfated heparan sulfate: unique substrate specificity of heparan sulfate 3‐O‐sulfotransferase isoform 5. Glycobiology 2003; 13: 785–794 [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Shworak NW, Fritze LM, et al. Purification of heparan sulfate D‐glucosaminyl 3‐O‐sulfotransferase. J Biol Chem 1996; 271: 27072–27082 [DOI] [PubMed] [Google Scholar]
- 41.Zhang L, Lawrence R, Schwartz JJ, et al. The effect of precursor structures on the action of glucosaminyl 3‐O‐sulfotransferase‐1 and the biosynthesis of anticoagulant heparan sulfate. J Biol Chem 2001; 276: 28806–28813 [DOI] [PubMed] [Google Scholar]
- 42.Petitou M, Casu B, Lindahl U. 1976‐1983, a critical period in the history of heparin: the discovery of the antithrombin binding site. Biochimie 2003; 85: 83–89 [DOI] [PubMed] [Google Scholar]
- 43.Duncan MB, Chen J, Krise JP, et al. The biosynthesis of anticoagulant heparan sulfate by the heparan sulfate 3‐O‐sulfotransferase isoform 5. Biochim Biophys Acta 2004; 1671: 34–43 [DOI] [PubMed] [Google Scholar]
- 44.Kamimura K, Rhodes JM, Ueda R, et al. Regulation of Notch signaling by Drosophila heparan sulfate 3‐O sulfotransferase. J Cell Biol 2004; 166: 1069–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]