
Table 2. Peripheral lipid-mediated signaling as a source of new analgesic medicines.
The current therapy of chronic pain relies heavily on opiates (e.g., morphine and oxycodone) and anticonvulsants (e.g., gabapentin and pregabalin), two classes of drugs that primarily act on neural circuits of the brain and spinal cord. These agents help many, but their use is associated with central side effects and abuse potential. The existence of peripheral lipid-mediated mechanisms that regulate the flow of nociceptive signals to the central nervous system (CNS) offers the opportunity to develop medications that control pain without producing undesired central effects. Activation of CB1 cannabinoid receptors in the brain is psychotropic, but peripherally restricted CB1 agonists or globally active CB2 agonists (e.g., AM1241) reduce nociceptive responses in rodent models without eliciting overt signs of cannabinoid intoxication. Marked anti-nociceptive actions, mediated by CB1 and/or CB2 receptors, are also observed with inhibitors of the endocannabinoid-hydrolyzing enzymes, fatty acid amide hydrolase (FAAH, e.g. URB597 or URB937) and monoacylglycerol lipase (MGL, e.g. JZL184). PPAR-α-mediated antinociception can be achieved either using direct PPAR-α agonists, such as the endogenous ligand palmitoylethanolamide (PEA), or inhibitors of the PEA-hydrolyzing enzyme N-acylethanolamine acid amidase (NAAA, e.g. ARN077). Similar approaches may be used to exploit the analgesic properties of endogenous bioactive lipids generated by oxygenation of polyunsaturated fatty acids (PUFAs): inhibition of soluble epoxide hydrolase (sEH), which converts epoxygenated PUFAs into inactive dihydroxy-acids, and administration of resolvins both produce marked antinociception in rodent models.
| Molecular Target | Representative Probe | Chemical Structure | Select Ref. |
|---|---|---|---|
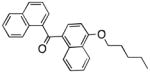
| Peripheral CB1 receptor | Naphthalen-1-yl-(4-pentyloxynaphthalen-1-yl)methanone |
|
32 |
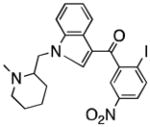
| CB2 receptor | AM1241 |
|
41 |
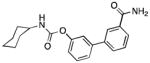
| FAAH | URB597 |
|
98 |
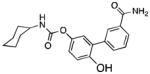
| Peripheral FAAH | URB937 |
|
57 |
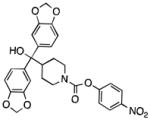
| MGL | JZL184 |
|
99 |
| PPAR-α | Palmitoylethanolamide |
|
76 |
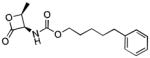
| NAAA | ARN077 |
|
100 |
| Soluble EH | t-AUCB |
|
89 |
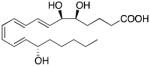
| CB1 receptor (Allosteric Modulation) | Lipoxin A4 |
|
92 |
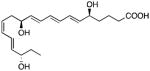
| ChemR32 | Resolvin E1 |
|
88 |