

Abstract
Schwannomatosis is the third major form of neurofibromatosis and is characterized by the development of multiple schwannomas in the absence of bilateral vestibular schwannomas. The 2011 Schwannomatosis Update was organized by the Children’s Tumor Foundation (www.ctf.org) and held in Los Angeles, CA, from June 5–8, 2011. This article summarizes the highlights presented at the Conference and represents the “state-of-the-field” in 2011. Genetic studies indicate that constitutional mutations in the SMARCB1 tumor suppressor gene occur in 40–50% of familial cases and in 8–10% of sporadic cases of schwannomatosis. Tumorigenesis is thought to occur through a four-hit, three-step model, beginning with a germline mutation in SMARCB1 (hit 1), followed by loss of a portion of chromosome 22 that contains the second SMARCB1 allele and one NF2 allele (hits 2 and 3), followed by mutation of the remaining wild-type NF2 allele (hit 4). Insights from research on HIV and pediatric rhabdoid tumors have shed light on potential molecular pathways that are dysregulated in schwannomatosis-related schwannomas. Mouse models of schwannomatosis have been developed and promise to further expand our understanding of tumorigenesis and the tumor microenvironment. Clinical reports have described the occurrence of intracranial meningiomas in schwannomatosis patients and in families with germline SMARCB1 mutations. The authors propose updated diagnostic criteria to incorporate new clinical and genetic findings since 2005. In the next 5 years, the authors expect that advances in basic research in the pathogenesis of schwannomatosis will lead toward clinical investigations of potential drug therapies.
Keywords: neurofibromatoses, schwannoma, rhabdoid tumor
INTRODUCTION
Schwannomatosis is a form of neurofibromatosis that is clinically and genetically distinct from neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). Schwannomatosis is characterized by the predisposition to develop multiple schwannomas, but the clinical spectrum continues to expand as more cases are reported. Because the phenotype of schwannomatosis overlaps with NF2, the first published reports did not appear in the literature until the 1990s. By 2003, genetic studies showed that the NF2 locus was excluded as the cause for familial schwannomatosis. The candidate region for the causative gene was narrowed during the 2000s, and in 2007, Hulsebos et al. reported a constitutional SMARCB1 mutation in a family affected by schwannomatosis. Since 2007, research on the genetic and molecular basis of schwannomatosis has expanded. The Children’s Tumor Foundation sponsored a conference held in Los Angeles, CA, on June 5–8, 2011 in which about 30 schwannomatosis researchers and clinicians discussed recent advances in the genetics, biology, and clinical description of schwannomatosis. This article provides a synopsis of the highlights presented at the Conference and as such, is a “state-of-the-field” for schwannomatosis research in 2011.
MEETING UPDATE
Genetics
Genetic identification of SMARCB1
Schwannomatosis is characterized by the development of multiple schwannomas. Only about 15 years ago, schwannomatosis was recognized as a clinical entity that is distinct from neurofibromatosis type 2 (NF2) [MacCollin et al., 1996; Evans et al., 1997]. Genetic evidence that schwannomatosis is different from NF2 was initially provided by the molecular analysis of schwannomas of schwannomatosis patients. These studies revealed that the multiple tumors of individual patients harbored independent mutations in the NF2 gene, which were not present in their respective constitutional DNAs [Jacoby et al., 1997; Kaufman et al., 2003]. Subsequent linkage analyses in schwannomatosis families excluded the NF2 gene as the schwannomatosis-predisposing gene and suggested a location of this gene near marker D22S1174, which is about 6 cM centromeric to the NF2 gene on chromosome 22. Hulsebos et al. [2007] considered the SMARCB1 gene to be an attractive candidate gene for schwannomatosis, because it was a known tumor suppressor gene and located at a very short distance from marker D22S1174. Indeed, they identified an inactivating germline mutation in exon 1 of the SMARCB1 gene in a father and daughter who both had schwannomatosis. In addition, in accordance with the tumor suppressor gene model, they found inactivation of the wild-type copy of the SMARCB1 gene, by a second inactivating mutation or by deletion, in schwannomas of the patients. These findings identified the SMARCB1 gene as a predisposing gene in schwannomatosis.
Studies by others have confirmed the involvement of the SMARCB1 gene in 40–50% of familial and less than 10% of sporadic cases of schwannomatosis [Boyd et al., 2008; Hadfield et al., 2008; Sestini et al., 2008; Rousseau et al., 2011; Smith et al., 2012]. In these studies, additional somatic inactivation of the NF2 gene was reported for the schwannomas. In SMARCB1 mutation-positive schwannomas, the deletions always involved loss of one copy of chromosome 22 [Hadfield et al., 2010b]. These observations suggest a four-hit, three-step model of tumorigenesis, in which the mutated germline SMARCB1 gene copy is retained in the tumor (hit 1), whereas chromosome 22, or at least a segment containing the wild type SMARCB1 gene copy and a wild-type copy of the NF2 gene, is lost (hits 2 and 3), followed by mutation of the remaining wild-type NF2 gene copy (hit 4) (Fig. 1).
FIG. 1.
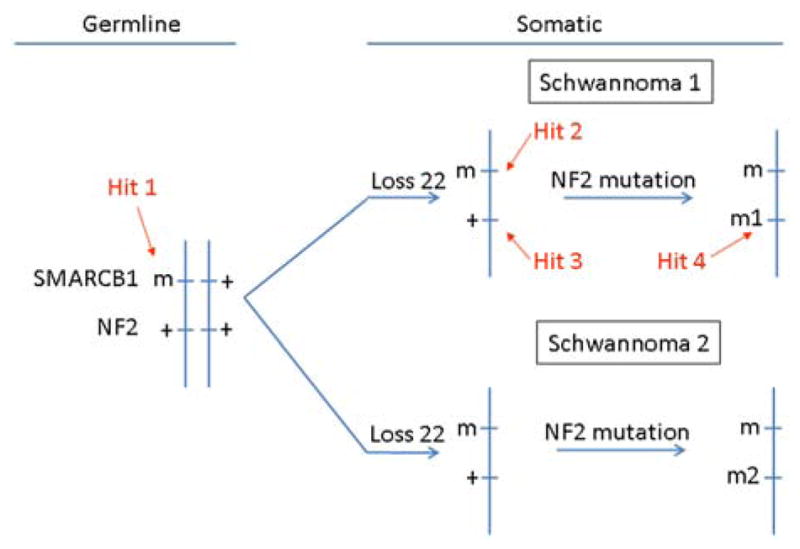
Four-hit, three-step mechanism for SMARCB1 and NF2 inactivation in multiple schwannomas of a SMARCB1-mutation-positive schwannomatosis patient. Tumorigenesis begins with a germline mutation in SMARCB1 (hit 1), and is followed by loss of a portion of chromosome 22 that contains the second SMARCB1 allele and one NF2 allele (hits 2 and 3), and by mutation of the remaining wild-type NF2 allele (hit 4).
Conflicting evidence exists with regard to the involvement of the SMARCB1 gene in the development of multiple meningiomas. Somatic mutations in the SMARCB1 gene have been shown to occur in sporadic meningiomas, although at low frequency (<3% of cases) [Bruder et al., 1999; Schmitz et al., 2001; Rieske et al., 2003]. However, germline mutations in the SMARCB1 gene could not be detected in 47 patients with multiple meningiomas, including eight familial cases [Hadfield et al., 2010a]. On the other hand, the inheritance of a germline mutation in the SMARCB1 gene was found in two families with schwannomatosis and multiple meningiomas [Bacci et al., 2010; Christiaans et al., 2011]. For the meningiomas in one of these families, it could be demonstrated that the four-hit, three step model also applies to these tumors, and that the cranial meningiomas were preferentially located at the falx cerebri [van den Munckhof et al., 2012].
Despite the frequent combination of germline SMARCB1 and somatic NF2 mutations, a large proportion of familial schwannomatosis patients and the majority of sporadic schwannomatosis patients have no known causative mutation [Boyd et al., 2008; Hadfield et al., 2008; Sestini et al., 2008; Rousseau et al., 2011; Smith et al., 2012]. In addition, there are currently no known causative genes for multiple meningiomas unrelated to NF2 or schwannomatosis. This leaves open the possibility of discovering further predisposing, or modifier, genes. Recent developments in technologies such as whole genome and whole exome sequencing analysis have now made them accessible for use in identification of these novel genes, which will help to elucidate the overall affected cellular pathways involved. This will then indicate targets for the development of novel therapies.
GENOTYPE–PHENOTYPE CORRELATIONS
In addition to schwannomatosis, germline mutations in the SMARCB1 gene cause the inherited prediposition to rhabdoid tumors characterized by the presence of malignant rhabdoid tumors of the kidney, atypical teratoid/rhabdoid tumors (AT/RT) of CNS, and extra-renal rhabdoid tumors that develop in childhood [Versteege et al., 1998; Hulsebos et al., 2007]. The factors determining which patients will develop schwannomatosis and which will develop rhabdoid tumors are unclear. Non-penetrance of rhabdoid tumors in patients with germline mutations of SMARCB1 leaves open the possibility that surviving mutation carriers will develop schwannomas later in life. However, it is extremely rare to find both phenotypes in the same family. Currently, there are only three reported families in which either rhabdoid tumors or schwannomas are seen in different family members with the same inherited mutation. The schwannomas seen in these patients seem to differ histologically from those typically found in schwannomatosis [Swensen et al., 2009; Eaton et al., 2011a; Carter et al., 2012]. The causative mutations in these families are predicted to cause truncation of the protein.
There is a direct correlation between rhabdoid tumors and SMARCB1 inactivation, as >95% of tumors show biallelic loss of this gene due to deletions, nonsense and frameshift mutations, or lack of mRNA and protein. Germline mutations in SMARCB1 are found in one third of patients with rhabdoid tumors and tend to be truncating (frameshift/nonsense) mutations, or deletions of one or more exons, leading to a complete knockout of the SMARCB1 gene [Bourdeaut et al., 2011; Eaton et al., 2011b]. This is coupled with a somatic mutation of the second allele, leading to biallelic inactivation of SMARCB1.
In contrast, inherited mutations found in familial schwannomatosis are more likely to be non-truncating (missense or splice-site) mutations [Boyd et al., 2008; Hadfield et al., 2008], which might account for the milder phenotype as these are potentially hypomorphic. However, sporadic schwannomatosis patients may carry truncating (frameshift or nonsense) or non-truncating (missense/splice-site) mutations [Rousseau et al., 2011; Smith et al., 2012], which are predicted to knock out the protein product. Schwannomatosis-associated SMARCB1 mutations do seem to be more frequent at either end of the gene whereas mutations associated with the inherited prediposition to rhabdoid tumors are more centrally located. It is possible that the specific combination of resulting somatic mutations, including the frequent co-mutation of the NF2 gene in tumors from schwannomatosis patients [Boyd et al., 2008; Hadfield et al., 2008], may regulate the severity of the resulting phenotype. However, given the relatively small number of families published with each phenotype more work is required to understand why the mutation patterns in schwannomatosis families are distinct and usually separate from patients with the inherited prediposition to rhabdoid tumors.
INHERITANCE
The majority of cases of schwannomatosis are sporadic [Evans et al., 1997; Antinheimo et al., 2000; Gonzalvo et al., 2011]. Case series suggest that only around 15–25% of cases are inherited from an affected parent. Confirmation of a germline SMARCB1 mutation will prove the inherited nature of disease and confirm a 50% risk of transmission to the next generation. However, only about 10% of sporadic cases and 40–60% of inherited cases have an identifiable mutation [Boyd et al., 2008; Hadfield et al., 2008; Rousseau et al., 2011]. The transmission risk can be assumed to be 50% in an individual with schwannomas who has a proven family history regardless of SMARCB1 status, but the risks for a sporadic case are less clear. Even with exclusion of SMARCB1, it must be assumed that there is still a small risk of transmission to offspring. Quantifying transmission risk is also complicated by phenotypic overlap between patients with sporadic schwannomatosis and mosaic NF2, since the latter patients may fulfill current schwannomatosis criteria [Baser et al., 2006; Murray et al., 2006]. Mosaicism also exists for SMARCB1 [Hulsebos et al., 2010], although the frequency of SMARCB1 mosaicism is still not clear. Nonetheless, mosaicism may account for a further proportion of sporadic schwannomatosis cases. Non-penetrance has been described in schwannomatosis although this appears at present to be less likely in SMARCB1 families. Ideally, molecular analysis of the NF2 and SMARCB1 genes from two separate tumors from an individual are required to confirm an underlying diagnosis.
CELL BIOLOGY
Function of SMARCB1
SMARCB1 and the SWI/SNF complex
Eukaryotic epigenetic regulation is mediated by chromatin remodeling factors that are high molecular weight multi-subunit protein complexes [Martens and Winston, 2003; Cairns, 2007; Osley et al., 2007; Wang et al., 2007; van Vugt et al., 2007; Racki and Narlikar, 2008]. These complexes can be divided into two general groups: Those that covalently modify histones and DNA by acetylation/deacetylation or methylation/demethylation, and those that use the energy of ATP to reorganize/reposition the nucleosomes. The latter complexes do not modify the chromatin in a covalent manner although they may indirectly influence such modifications [Banine et al., 2005; Datta et al., 2005].
SMARCB1 (also called hSNF5, INI1, and BAF47) is a component of the mammalian SWI/SNF complex, a prototypical ATP-dependent chromatin remodeling complex consisting of at least nine subunits that are conserved among eukaryotes (Fig. 2) [Wang et al., 1996a; Wang et al., 1996b; Narlikar et al., 2002; Martens and Winston, 2003]. Among these are four core subunits required for chromatin remodeling, including an ATPase (SWI2/SNF2/BRG1 or BRM1), SMARCB1, BAF155, and BAF170 [Martens and Winston, 2003]. Among these, BRG1 and BAF155 are known to play critical roles in mammalian nervous system development [Kim et al., 2001; Seo et al., 2005; Matsumoto et al., 2006; Lessard et al., 2007].
FIG. 2.
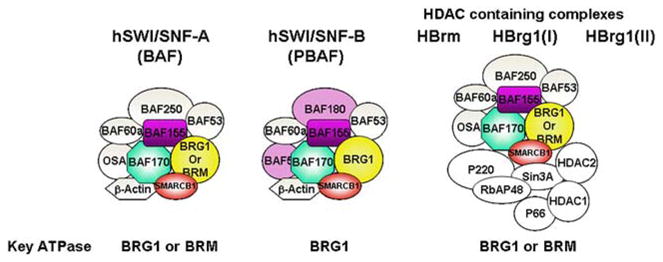
Schematic representation of multi-subunit SWI/SNF complexes. SMARCB1 (in shaded red) is present in all complexes.
Several functionally distinct classes of SWI/SNF complexes have been recognized in eukaryotes. Two major classes include hSWI/SNF-A or BAF (BRG1-associated Factor) complex and hSWI/SNF-B or PBAF complex. The two classes of complexes are distinguished by the presence of specific subunits [Martens and Winston, 2003; van Vugt et al., 2007]. Additional complexes containing mixtures of components of SWI/SNF and HDAC1 complexes have also been identified (See Fig. 2) [Martens and Winston, 2003]. Since SMARCB1 is present in all of these complexes, understanding the distinct roles played by these various complexes will be essential for deciphering the role of SMARCB1 in schwannomatosis.
The functions of non-catalytic subunits of SWI/SNF complexes, including SMARCB1, are not completely understood. The components of the SWI/SNF complex do not have sequence-specific DNA binding activity and hence they are targeted to specific promoter regions via protein–protein interactions with sequence-specific transcription factors, either activators or repressors [Cairns, 1998; Cheng et al., 1999; Kingston and Narlikar, 1999; Martens and Winston, 2003; Cairns, 2007; Racki and Narlikar, 2008].
Structure/function analysis of SMARCB1
SMARCB1 was originally called INI1 and was originally identified as an interacting protein for HIV-1 integrase (IN) [Kalpana et al., 1994]. Structure/function analysis has revealed that SMARCB1 has three conserved domains. Two of these domains (Rpt I and Rpt II) are direct and imperfect repeats of each other, and the Rpt I domain is necessary and sufficient to bind to HIV-1 IN in vitro and in vivo [Morozov et al., 1998; Yung et al., 2001]. SMARCB1 is also engaged in protein–protein interactions with other viral and cellular proteins (Fig. 3) [Wu et al., 1996; Rozenblatt-Rosen et al., 1998; Cheng et al., 1999; Lee et al., 1999]. Viral proteins include the E1 protein of human papilloma virus 18 and the EBNA-2 protein of Epstein–Barr virus [Wu et al., 1996; Lee et al., 1999; Wu et al., 2000]. The cellular proteins include MYC, MLL (HXR), GADD34, and AKT [Rozenblatt-Rosen et al., 1998; Cheng et al., 1999; Wu et al., 2002; Foster et al., 2006]. Furthermore, SMARCB1 harbors additional functional domains including a nuclear export signal that is constitutively activated by the deletion of the C-terminal regions, a homology region III, a non-specific DNA binding domain, and a region involved in nuclear import (Fig. 3) [Morozov et al., 1998; Craig et al., 2002]. Mutations of SMARCB1 in schwannomatosis span all these domains and hence understanding of specific roles played by these domains will provide insight into its function.
FIG. 3.
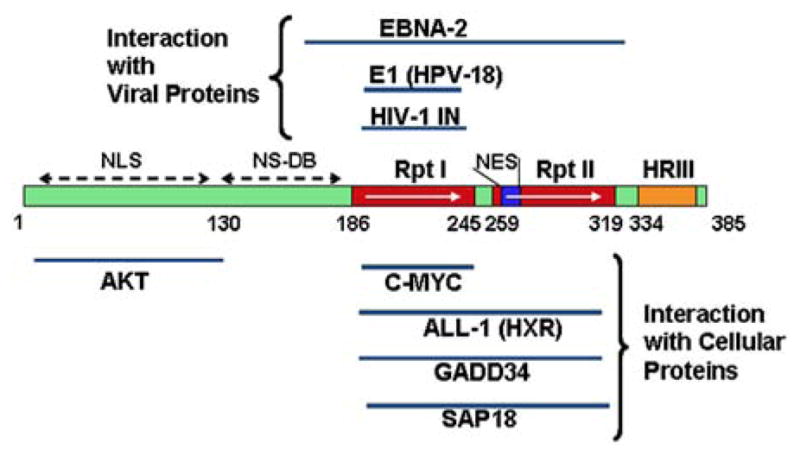
Schematic representation of different domains of SMARCB1/hSNF5 and their function. Rpt, repeat; HRIII, homology region III; NES, nuclear export signal; NLS, nuclear localization signal, and NS-DB, nonspecific DNA binding.
Role of SMARCB1 in HIV-1 replication
Understanding its interactions with HIV-1 IN may reveal clues about the functions of SMARCB1. It has been reported that SMARCB1 has a multifaceted role in HIV-1 replication. Binding of IN to SMARCB1 may facilitate targeting of incoming HIV-1 viral DNA into open chromatin regions in infected cells [Kalpana et al., 1994; Lesbats et al., 2011]. SMARCB1 is also required for post-integrational events of HIV-1 viral assembly and particle production in the cytoplasm [Yung et al., 2001; Yung et al., 2004; Sorin and Kalpana, 2006]. These studies indicate an undefined function for SMARCB1 outside the nucleus in the cell, and understanding these roles may provide novel insight into its role in schwannomatosis and other cancers. Furthermore, SMARCB1 also has been demonstrated to bind to the HIV-1 trans-activator protein, Tat, and to activate transcription of the HIV-1 promoter within its long terminal repeats [Ariumi et al., 2006; Mahmoudi et al., 2006].
Crosstalk between merlin and the HIV-1 host proteins
Studies indicate a four-hit mechanism for genesis of schwannomatosis where mutations in both NF2 and SMARCB1 genes lead to pathogenesis. Currently, the functional interaction and relationship between SMARCB1 and NF2 are unknown. However, it is intriguing to note that there is interplay between NF2 and the cellular factors that influence HIV-1 replication.
CLR4DCAF1 or VprBP is an E3 ubiquitin ligase that binds to an HIV-1 encoded accessory protein called Vpr. This association of Vpr with CLR4DCAF1 appears to be required for inducing G2/M arrest [DeHart et al., 2007; Wen et al., 2012]. Recent studies indicate that merlin also associates with CLR4DCAF1 in the nucleus and suppresses its activity and tumorigenesis [Li et al., 2010].
TRBP (transactivator-responsive RNA binding protein) is another HIV-1 host protein that binds to HIV-1 encoded Tat [Bannwarth and Gatignol, 2005; Corbeau, 2008; Clerzius et al., 2011]. TRBP is a Dicer co-factor required for cellular RNA interference (RNAi). HIV-1 Tat suppresses the RNAi by sequestration of TRBP away from the Dicer complex. Merlin also seems to inhibit TRBP, which has growth-promoting potential, by ubiquitination and degradation of TRBP protein [Lee et al., 2004]. Whether or not these interactions of merlin with HIV-1 host proteins have anything to do with SMARCB1 is unclear at this point.
Tumor suppressor function ofSMARCB1
Various studies have indicated that SMARCB1 exerts its tumor suppressor function by: (i) inducing G1 arrest; (ii) inducing mitotic arrest; (iii) inhibiting aneuploidy and causing diploidization; and (iv) inducing senescence [Kalpana and Smith, 2010; Wilson and Roberts, 2011]. Induction of G0/G1 arrest by SMARCB1 is correlated with down-regulation of cyclin D1 [Zhang et al., 2002] and up-regulation of p16INK4a [Betz et al., 2002]. The cyclin D1 gene appears to be essential for genesis of tumors due to loss of Smarcb1 in mouse models as Smarcb1 +/− heterozygous mice in combination with cyclin D1−/− null genotype resulted in no tumor formation [Tsikitis et al., 2005]. Furthermore, cyclin D1 appears to be essential for survival of established tumors. Knocking-down cyclin D1 by siRNA in rhabdoid tumor cells induced cell cycle arrest and apoptosis in vitro [Alarcon-Vargas et al., 2006]. Interestingly enough it has been postulated that merlin exerts its tumor suppressor function via repression of PAK-induced cyclin D1 expression [Xiao et al., 2005]. It is possible then that SMARCB1 and merlin loss could co-operate in cyclin D1 induction. Small molecular weight pan-cyclin-dependent kinase inhibitors such as Flavopyridol were effective at nanomolar concentrations to inhibit rhabdoid tumor cells in vitro and primary tumor models in vivo [Smith et al., 2008; Smith et al., 2011]. These studies suggest that targeting cyclin D1 is an effective strategy to inhibit at least a subset of tumors arising due to loss of SMARCB1. Whether this strategy would also be effective for inhibiting schwannoma growth in schwannomatosis animal models and patients should be investigated.
In addition to regulating the cell cycle, SMARCB1 and the SWI/SNF components regulate lineage specific gene expression and embryonic stem cell programming. SMARCB1 and SWI/SNF complexes have been shown to be involved in the control of neurogenesis, myogenesis, adipogenesis, osteogenesis, and hematopoiesis [Young et al., 2005; Caramel et al., 2008; Yoo and Crabtree, 2009; Krosl et al., 2010; Albini and Puri, 2010; Nowak et al., 2012]. SMARCB1 and SWI/SNF complexes also regulate embryonic stem cell programming by antagonizing the activity of polycomb group proteins [Wilson et al., 2010].
Downstream pathways affected by SMARCB1
SWI/SNF complexes are involved in both activation and repression of a selective subset of genes [Martens and Winston, 2003]. The components of SWI/SNF complex do not posses sequence-specific DNA binding activity and hence are recruited to the promoter region by interaction of one of the subunits with gene-specific transcription factors [Cairns, 1998; Kingston and Narlikar, 1999; Cairns, 2007; Racki and Narlikar, 2008]. SMARCB1 appears to be a tethering protein that recruits SWI/SNF complexes to promoter sites by its interaction with an activator (for example, MYC) or repressor proteins bound to the promoter [Cheng et al., 1999; Martens and Winston, 2003].
Several studies that compared the gene expression profile in rhabdoid cells when SMARCB1 is re-expressed have lead to the identification of a number of downstream pathways that are directly or indirectly affected by SMARCB1. As described above, SMARCB1 affects cyclin D1 and p16 and hence regulates p16-cyclin D1/CDK4-pRb-E2F. SMARCB1 also appears to affect (i) mitotic spindle checkpoint; (ii) the interferon signaling pathway; and (iii) the Gli-Hedgehog pathway [Medjkane et al., 2004; Vries et al., 2005; Morozov et al., 2007; Jagani et al., 2010; Lee et al., 2011]. For example, SMARCB1 and the SWI/SNF complexes are involved in induction of interferon signaling [Agalioti et al., 2002; Liu et al., 2002; Pattenden et al., 2002; Cui et al., 2004; Morozov et al., 2007]. Reintroduction of SMARCB1 into SMARCB1−/− MON cells results in the activation of a statistically significant high abundance of IFN signal induced genes (ISGs) [Morozov et al., 2007].
In addition to inducing the ISGs, SMARCB1 repressed a set of mitotic genes including PLK1 and Aurora A [Morozov et al., 2007; Lee et al., 2011]. Aurora A and PLK1 are serine/threonine kinases that play multiple roles in mitosis. Aurora A is frequently over-expressed in many cancers and is indicative of poor prognosis [Nadler et al., 2008; Wang et al., 2009; Zhang et al., 2009]. Aurora A is an attractive target for developing anti-cancer therapy and, many Aurora A kinase inhibitors are currently under investigation in phase I clinical trials [Pollard and Mortimore, 2009; Dar et al., 2010; Kitzen et al., 2010; Wurzenberger and Gerlich, 2011; Yan et al., 2011]. Aurora A is a repressed downstream target of SMARCB1 and SMARCB1 associates with Aurora A promoter in RT and normal cells but not in non-RT cells [Lee et al., 2011]. Loss of SMARCB1 leads to aberrant over-expression of Aurora A in mouse and human tumors and knocking down Aurora A leads to inhibition of rhabdoid cell growth [Lee et al., 2011]. Targeting expression or activity of this gene is a novel therapeutic strategy for these tumors. At this point it is unclear if Aurora A is de-repressed in schwannomatosis due to SMARCB1 mutations.
MOUSE MODELS
A number of mice have been generated in which particular proteins within the SWI/SNF complex have been disrupted. The functional complexity of the SWI/SNF unit is reflected by the distinct phenotypes observed in mice lacking different subunits. For example, mice lacking Brm, an ATPase subunit, develop normally but demonstrate hyperproliferative phenotypes [Reyes et al., 1998]. In contrast, mice lacking Brg1 (the other SWI/SNF ATPase), Smarcb1 or BAF155 die at the peri-implantation stage [Klochendler-Yeivin et al., 2000; Roberts et al., 2000; Guidi et al., 2001; Tsikitis et al., 2005; Bultman et al., 2008]. In all three of these knockout mice, inner cell mass development is altered, and Brg1 and Smarcb1 inactivation also affects the trophectoderm lineage.
Mice haploinsufficient for SWI/SNF subunits demonstrate a number of distinct phenotypes. For example, Brg1 heterozygotes are susceptible to mammary carcinomas without evidence of loss of heterozygosity [Bultman et al., 2008]. In contrast, Smarcb1 heterozygotes undergo normal development and 5–35% later present with rhabdoid tumors and other tumor types, many of which are highly aggressive and metastatic [Klochendler-Yeivin et al., 2000; Roberts et al., 2000; Guidi et al., 2001; Tsikitis et al., 2005]. Where examined, tumorigenesis in these mice correlates with loss or inactivation of the remaining wild-type Smarcb1 allele. Roberts et al. [2002] generated both an inactivating-conditional Smarcb1 allele and a reversibly inactivating-conditional Smarcb1 allele in mice. Inhibition of Smarcb1 expression in a variety of adult tissues resulted in bone marrow failure and rapid death, whereas sporadic inactivation of Smarcb1 in hematopoietic tissues and in other organs resulted in rapid onset of lymphomas and rhabdoid tumors. Neither Brg1 nor Smarcb1 heterozygotes have been reported to develop schwannomas.
The phenotypes of animals with other conditional mutations of SWI/SNF subunits demonstrate distinct roles for these proteins that were not predicted from null or haploinsufficient mice. Matsumoto et al. [2006], for example, demonstrated that loss of Brg1 in neural progenitor cell populations results in precocious neuronal differentiation at the expense of glial cell differentiation in the brain and spinal cord. These effects are linked to a switch in neuron-associated SWI/SNF subunits [Lessard et al., 2007]. Gresh et al. [2005] specifically inactivated Smarcb1 in the developing liver, resulting in failed hepatic cell differentiation. Collectively, these mouse studies demonstrate that SWI/SNF factors, including Smarcb1, suppress tumor formation and are absolutely required for the survival and differentiation of non-malignant cells. These studies also highlight the diverse functions of SWI/SNF subunits and the need to examine these proteins using conditional knock-out approaches.
Phenotypic analysis of Smarcb1-deficient Schwann cells is hindered by the fact that homozygous Smarcb1 mutant embryos lack normal extra-embryonic development and fail at the peri-implantation stage. To circumvent this early embryonic-lethal phenotype, and to examine whether loss of Smarcb1 in Schwann cells confers a growth advantage that is sufficient to induce schwannoma formation or other schwannomatosis-related phenotypes (e.g., pain) in vivo, a number of groups are developing Smarcb1 conditional knockout mice to target Schwann cells.
One of the promoters used to generate Schwann cell-targeted disruption of Smarcb1 is the protein zero (P0) gene, one of the major proteins of peripheral myelin. A 1.1 kb stretch of the 5′ flanking sequence of the rat P0 gene is sufficient to direct expression of heterologous genes to Schwann cells in vivo [Messing et al., 1992], although expression in neural crest cells has also been reported [Yamauchi et al., 1999]. This expression follows the developmental schedule of the endogenous P0 gene showing a dramatic increase during the first week after birth. Moreover, transgenic mice expressing either SV40 large T-antigen or a naturally occurring mutant Nf2 allele under the rat P0 promoter develop schwannomas [Messing et al., 1994; Giovannini et al., 1999]. Therefore, this P0 promoter efficiently targets schwannoma precursor cells. Based on these data, P0-Cre;Smarcb1flox/flox mice are being generated to direct Cre expression to the schwannoma precursor cells.
Another strategy to assess the function of Smarcb1 in Schwann cells is to utilize mouse lines in which gene loss can be induced in Schwann cells. Mice are therefore being generated in which Smarcb1 disruption is induced using the proteolipid protein gene following injection of post-natal or adult animals with tamoxifen. This strategy has been used successfully to disrupt genes in Schwann cells in embryonic, post-natal and adult mice [Doerflinger et al., 2003]. Utilizing both of these strategies will likely result in complementary mouse models that define how loss of Smarcb1 in Schwann cells influences both developmental and adult phenotypes.
Additional somatic inactivation of NF2 or other genes may play a role in the development of schwannomas in schwannomatosis patients. Although Hulsebos et al. [2007] were unable to detect NF2 gene aberrations in their patients, others found NF2 gene mutations and loss in a considerable fraction of the investigated tumors from schwannomatosis patients [Jacoby et al., 1997; Kaufman et al., 2003; MacCollin et al., 2003; Boyd et al., 2008; Hadfield et al., 2008; Sestini et al., 2008]. Analysis of mice carrying both conditional mutant Nf2 and Smarcb1 alleles will be critical to assess how inactivation of Snf5/Nf2 influences the development of schwannomatosis-associated phenotypes.
CLINICAL FEATURES
Diagnostic Criteria
The diagnostic criteria set forth in 2005 [MacCollin et al., 2005] predated the ability to perform molecular testing for schwannomatosis, and also did not take account of the possibility of multiple meningiomas as a presenting feature. Given current knowledge of the disorder, we propose the following new criteria for diagnosis:
Molecular Diagnosis:
Two or more pathologically proved schwannomas or meningiomas AND genetic studies of at least two tumors with loss of heterozygosity (LOH) for chromosome 22 and two different NF2 mutations; if there is a common SMARCB1 mutation, this defines SMARCB1-associated schwannomatosis
One pathologically proved schwannoma or meningioma AND germline SMARCB1 pathogenic mutation
Clinical Diagnosis:
Two or more non-intradermal schwannomas, one with pathological confirmation, including no bilateral vestibular schwannoma by high-quality MRI (detailed study of internal auditory canal with slices no more than 3 mm thick). Recognize that some mosaic NF2 patients will be included in this diagnosis at a young age and that some schwannomatosis patients have been reported to have unilateral vestibular schwannomas or multiple meningiomas.
One pathologically confirmed schwannoma or intracranial meningioma AND affected first-degree relative
Consider as possible diagnosis if there are two or more non-intradermal tumors but none has been pathologically proven to be a schwannoma; the occurrence of chronic pain in association with the tumor(s) increase the likelihood of schwannomatosis
Patients with the following characteristics do not fulfill diagnosis for schwannomatosis:
Germline pathogenic NF2 mutation
Fulfill diagnostic criteria for NF2
First-degree relative with NF2
Schwannomas in previous field of radiation therapy only
PATHOLOGIC DIAGNOSIS OF SCHWANNOMATOSIS-ASSOCIATED SCHWANNOMAS
Several studies have documented the presence of hybrid tumors (benign nerve sheath tumors with characteristic features of both neurofibromas and schwannomas) in the setting of NF1, NF2, and schwannomatosis [MacCollin et al., 2005; Harder et al., 2012]. The presence of abundant myxoid stroma in a schwannoma should alert the pathologist to the possibility of an association with any of the neurofibromatoses (as opposed to sporadic schwannoma in a non-syndromic person). In addition to histological features (hybrid tumors, myxoid schwannomas), a mosaic pattern of staining for SMARCB1/INI1 is suggestive of a tumor related to NF2 or schwannomatosis. In an immunohistochemical study of a large cohort of schwannomas, a mosaic pattern of SMARCB1 expression was noted in tumors resected from patients with NF2 and familial schwannomatosis (>90% of tumors). In contrast, a mosaic pattern of expression is unusual (5%) in sporadic, non-syndromic tumors [Patil et al., 2008]. Thus, the finding of mosaic SMARCB1 expression on immunohistochemistry and/or hybrid histology suggests that a schwannoma may be associated with a form of neurofibromatosis. In these cases, the pathologic findings may direct the clinican to refer a patient with a solitary schwannoma for evaluation of neurofibromatosis.
TUMOR BURDEN IN SCHWANNOMATOSIS
Time and financial constraints limit the usefulness of regional MRI to comprehensively phenotype tumor burden in patients with schwannomatosis. For this reason, important features of schwannomatosis including whole-body tumor burden and distribution of schwannomas across body parts are still not well understood. To better understand the clinical phenotype of schwannomatosis, teams at Massachusetts General Hospital and University Hospital, Hamburg, have used whole-body MRI (WBRMI) to assess patients with schwannomatosis [Plotkin et al., 2012]. This technique involves the use of a standard MRI scanner with commercially available software to create a single DICOM image that extends from head to ankle in most patients. No contrast is required and a patient can be scanned in about 45 min without the need for ionizing radiation.
To date, 51 patients with schwannomatosis have been scanned. The mean age at diagnosis was 42 years; the mean age at the time of MRI scan was 48.5 years (range: 25–97 years). Fifty-one percent were male, and 16% had familial disease. WBMRI identified internal nerve sheath tumors in 71% of patients, including seven of eight patients that did not have a history of internal tumors. Internal tumors were classified radiologically as either discrete (locally circumscribed) or plexiform (invasive tumors or those that involved multiple nerve roots). In patients with tumors on WBMRI, 29/36 (81%) had only discrete appearing tumors, 3/36 (8%) had only plexiform appearing tumors, and 4/36 (11%) had both types. For patients with tumors, the median tumor count was 4 (range: 1–27 tumors) and the median whole-body tumor volume was 39.4 ml (range: 7.0–1371.5 ml). Median volume of plexiform appearing tumors was smaller in schwannomatosis patients (32 ml) than in a group of similarly imaged NF1 and NF2 patients, who had median tumor volumes of 206 and 104 ml, respectively. The distribution of tumors across body parts was measured using the distribution coefficient which ranges from 0 (representing tumor volume that is equally distributed throughout the body) to 1 (representing tumor volume located in only one body area) [Plotkin et al., 2012]. In the cohort of schwannomatosis patients, the median distribution coefficient was 0.9 (range: 0.26–1), indicating that total tumor volume is not distributed evenly across the body. This finding may reflect the relatively high frequency of anatomically limited schwannomatosis or the relatively low number of tumors in most patients.
In order to explore the relationship between tumor burden and pain, schwannomatosis patients were asked to rate their pain level at the time of imaging using a visual analog pain scale [scores ranges from 0 (no pain) to 10 (most pain)]. The median pain score for all schwannomatosis patients was 1.7. Surprisingly, the median pain score for patients with familial schwannomatosis was 0.5 whereas the median score for patients with sporadic schwannomatosis was 2.5.
CURRENT TREATMENT OPTIONS
Current Treatment Options
Pain medications
Chronic pain is common in patients with schwannomatosis [Merker et al., 2012]. The etiology of pain in schwannomatosis is unclear: there is no clear relationship between tumor number, size, location, and the intensity of pain, and the pain has both neuropathic and nociceptive features. Perhaps for this reason, there is no widely accepted approach for treating pain in these patients. Conference attendees supported the use of neuropathic pain medication without favoring a single approach for schwannomatosis. Reasonable options include calcium channel alpha 2-delta ligands (e.g., gabapentin, pregabalin). In addition, several medications designated as antidepressants (e.g., amitriptyline, nortriptyline, duloxetine) can have potent anti-neuropathic pain effects. Medications used for mood stabilization (e.g., lamotrigine, valproate) can also be effective in chronic pain scenarios. It is not clear whether these drugs directly modulate the pain mechanism or treat the depression and anxiety associated with chronic pain.
Surgical considerations
Surgery is the treatment of choice for symptomatic schwannomas and, in many patients, can relieve local pain or symptoms arising from compression of neighboring tissues. The major risk of surgery is secondary nerve injury and hence, surgeons experienced with nerve-sparing surgery should be involved when considering a schwannoma resection. Anecdotal experience with schwannomatosis patients suggests that surgery can occasionally result in worsening of the global pain symptom. In this setting, repeated surgeries appear to offer less benefit in pain control and may contribute to worsening of the pain syndrome. This experience has raised the hypothesis that circulating factors (e.g., cytokines) that are released during surgery can potentiate painful stimuli. There is clearly a need to better understand surgical outcomes for patients with schwannomatosis.
Radiation
Experience with radiation therapy for management of schwannomatosis-related schwannomas is limited. Over the past two decades, there has been increasing experience with stereotactic radiation for sporadic vestibular schwannomas and spinal schwannomas [Niranjan et al., 2008; Gerszten et al., 2012]. Early results suggest that this modality is safe and effective for individuals without an underlying tumor suppressor syndrome. However, there is a theoretical risk that radiation exposure could increase the risk for malignant transformation in patients with schwannomatosis, as has been reported for NF1 and NF2 [Evans et al., 2006]. To date, there is no available data on the risk of secondary malignant transformation of tumors in schwannomatosis patients. At this time, most experts reserve the use of radiation for patients who require treatment for growing schwannomas that cannot be treated with surgery. The role of radiation for symptomatic (i.e., painful) schwannomas remains unclear.
Chemotherapy options
Currently, there are no approved drugs for treatment of schwannomas. Since SMARCB1 directly represses cyclin D1 and since tumors that arise due to loss of SMARCB1 are dependent on cyclin D1, preclinical studies were conducted to test the efficacy of drugs that target the cyclin D1/Cdk pathway [Zhang et al., 2002; Tsikitis et al., 2005]. Flavopiridol is a pan-cdk inhibitor and it represses cyclin D1 at the transcriptional level [Smith et al., 2008; Smith et al., 2011]. In preclinical models of primary rhabdoid tumors that arise due to loss of hSNF5, flavoporidol has shown efficacy in eliminating tumors. In a mouse model with transgenic SMARCB1 mutation with biallelic somatic inactivation of NF2, there is activation of the cyclin D1 and mTOR pathways. Possible additional molecular drug targets are those deregulated due to loss of SMARCB1, including mitotic genes, PLK1 and Aurora A that are upregualted/de-repressed in tumors, and interferon pathway [Lee et al., 2011]. Hence, the pathways consisting of cyclin D1, PLK1, Aurora A, p16, and interferons are all possible targets for tumors harboring SMARCB1 mutations. Finally, in a series of two cases (personal communication, J Blakeley, S Huson and DG Evans), the VEGF inhibitor bevacizumab has shown some benefit in pain control and improvement in function in two patients with life-threatening complications of schwannomatosis. In summary, at this time, there is no drug therapy that can be recommended for the treatment of the tumors in schwannomatosis. However, ongoing preclinical work suggests that early clinical studies (i.e., translational and dose finding studies) should be available soon for schwannomatosis patients.
CONCLUSIONS
There has been considerable progress in our understanding of the molecular and genetic basis of schwannomatosis since 2007. This has led to the development of refined diagnostic critera for schwannomatosis. The participants of the workshop identified priorities for continued research over the next 5 years. These priorities include identifying additional genes that predispose to familial and sporadic schwannomatosis, understanding the molecular basis for instability of chromosome 22 in schwannoma formation, characterizing genotype/phenotype correlations for schwannomatosis/inherited prediposition to rhabdoid tumors, uncovering the mechanism of pain in schwannomatosis, and preclinical testing of drugs effecting in schwannomatosis mouse models. Ultimately, these results will open the door for clinical trials to treat patients who are not good candidates for surgery. In the meantime, the International Schwannomatosis Registry (www.schwannomatosis.org) is prospectively enrolling patients into a database to assist researchers who wish to identify patients for these clinical investigations.
Acknowledgments
We thank all speakers and attendees who contributed to the 2011 Schwannomatosis Update and to this summary, and to Ms. Min Wong for her outstanding organization of the meeting. We also thank the donors whose enduring support makes this annual meeting possible.
References
- Agalioti T, Chen G, Thanos D. Deciphering the transcriptional histone acetylation code for a human gene. Cell. 2002;111:381–392. doi: 10.1016/s0092-8674(02)01077-2. [DOI] [PubMed] [Google Scholar]
- Alarcon-Vargas D, Zhang Z, Agarwal B, Challagulla K, Mani S, Kalpana GV. Targeting cyclin D1, a downstream effector of INI1/hSNF5, in rhabdoid tumors. Oncogene. 2006;25:722–734. doi: 10.1038/sj.onc.1209112. [DOI] [PubMed] [Google Scholar]
- Albini S, Puri PL. SWI/SNF complexes, chromatin remodeling and skeletal myogenesis: It’s time to exchange! Exp Cell Res. 2010;316:3073–3080. doi: 10.1016/j.yexcr.2010.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antinheimo J, Sankila R, Carpen O, Pukkala E, Sainio M, Jaaskelainen J. Population-based analysis of sporadic and type 2 neurofibromatosis-associated meningiomas and schwannomas. Neurology. 2000;54:71–76. doi: 10.1212/wnl.54.1.71. [DOI] [PubMed] [Google Scholar]
- Ariumi Y, Serhan F, Turelli P, Telenti A, Trono D. The integrase interactor 1 (INI1) proteins facilitate Tat-mediated human immunodeficiency virus type 1 transcription. Retrovirology. 2006;3:47. doi: 10.1186/1742-4690-3-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci C, Sestini R, Provenzano A, Paganini I, Mancini I, Porfirio B, Vivarelli R, Genuardi M, Papi L. Schwannomatosis associated with multiple meningiomas due to a familial SMARCB1 mutation. Neurogenetics. 2010;11:73–80. doi: 10.1007/s10048-009-0204-2. [DOI] [PubMed] [Google Scholar]
- Banine F, Bartlett C, Gunawardena R, Muchardt C, Yaniv M, Knudsen ES, Weissman BE, Sherman LS. SWI/SNF chromatin-remodeling factors induce changes in DNA methylation to promote transcriptional activation. Cancer Res. 2005;65:3542–3547. doi: 10.1158/0008-5472.CAN-04-3554. [DOI] [PubMed] [Google Scholar]
- Bannwarth S, Gatignol A. HIV-1 TAR RNA: The target of molecular interactions between the virus and its host. Curr HIV Res. 2005;3:61–71. doi: 10.2174/1570162052772924. [DOI] [PubMed] [Google Scholar]
- Baser ME, Friedman JM, Evans DG. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology. 2006;66:730–732. doi: 10.1212/01.wnl.0000201190.89751.41. [DOI] [PubMed] [Google Scholar]
- Betz BL, Strobeck MW, Reisman DN, Knudsen ES, Weissman BE. Re-expression of hSNF5/INI1/BAF47 in pediatric tumor cells leads to G1 arrest associated with induction of p16ink4a and activation of RB. Oncogene. 2002;21:5193–5203. doi: 10.1038/sj.onc.1205706. [DOI] [PubMed] [Google Scholar]
- Bourdeaut F, Lequin D, Brugieres L, Reynaud S, Dufour C, Doz F, Andre N, Stephan JL, Perel Y, Oberlin O, Orbach D, Bergeron C, Rialland X, Freneaux P, Ranchere D, Figarella-Branger D, Audry G, Puget S, Evans DG, Pinas JC, Capra V, Mosseri V, Coupier I, Gauthier-Villars M, Pierron G, Delattre O. Frequent hSNF5/INI1 Germline Mutations in Patients with Rhabdoid Tumor. Clin Cancer Res. 2011;17:31–38. doi: 10.1158/1078-0432.CCR-10-1795. [DOI] [PubMed] [Google Scholar]
- Boyd C, Smith MJ, Kluwe L, Balogh A, MacCollin M, Plotkin SR. Alterations in the SMARCB1 (INI1) tumor suppressor gene in familial schwannomatosis. Clin Genet. 2008;74:358–366. doi: 10.1111/j.1399-0004.2008.01060.x. [DOI] [PubMed] [Google Scholar]
- Bruder CE, Dumanski JP, Kedra D. The mouse ortholog of the human SMARCB1 gene encodes two splice forms. Biochem Biophys Res Commun. 1999;257:886–890. doi: 10.1006/bbrc.1999.0563. [DOI] [PubMed] [Google Scholar]
- Bultman SJ, Herschkowitz JI, Godfrey V, Gebuhr TC, Yaniv M, Perou CM, Magnuson T. Characterization of mammary tumors from Brg1 heterozygous mice. Oncogene. 2008;27:460–468. doi: 10.1038/sj.onc.1210664. [DOI] [PubMed] [Google Scholar]
- Cairns BR. Chromatin remodeling machines: Similar motors, ulterior motives. Trends Biochem Sci. 1998;23:20–25. doi: 10.1016/s0968-0004(97)01160-2. [DOI] [PubMed] [Google Scholar]
- Cairns BR. Chromatin remodeling: Insights and intrigue from single-molecule studies. Nat Struct Mol Biol. 2007;14:989–996. doi: 10.1038/nsmb1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caramel J, Medjkane S, Quignon F, Delattre O. The requirement for SNF5/INI1 in adipocyte differentiation highlights new features of malignant rhabdoid tumors. Oncogene. 2008;27:2035–2044. doi: 10.1038/sj.onc.1210847. [DOI] [PubMed] [Google Scholar]
- Carter JM, O’Hara C, Dundas G, Gilchrist D, Collins MS, Eaton K, Judkins AR, Biegel JA, Folpe AL. Epithelioid malignant peripheral nerve sheath tumor arising in a schwannoma, in a patient with “neuroblastoma-like” schwannomatosis and a novel germline SMARCB1 mutation. Am J Surg Pathol. 2012;36:154–160. doi: 10.1097/PAS.0b013e3182380802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SW, Davies KP, Yung E, Beltran RJ, Yu J, Kalpana GV. c-MYC interacts with INI1/hSNF5 and requires the SWI/SNF complex for transactivation function. Nat Genet. 1999;22:102–105. doi: 10.1038/8811. [DOI] [PubMed] [Google Scholar]
- Christiaans I, Kenter SB, Brink HC, van Os TA, Bass F, van den Munckhof P, Kidd AM, Hulsebos TJ. Germline SMARCB1 mutation and somatic NF2 mutations in familial multiple meningiomas. J Med Genet. 2011;2011:93–97. doi: 10.1136/jmg.2010.082420. [DOI] [PubMed] [Google Scholar]
- Clerzius G, Gelinas JF, Gatignol A. Multiple levels of PKR inhibition during HIV-1 replication. Rev Med Virol. 2011;21:42–53. doi: 10.1002/rmv.674. [DOI] [PubMed] [Google Scholar]
- Corbeau P. Interfering RNA and HIV: Reciprocal interferences. PLoS Pathog. 2008;4:e1000162. doi: 10.1371/journal.ppat.1000162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig E, Zhang ZK, Davies KP, Kalpana GV. A masked NES in INI1/hSNF5 mediates hCRM1-dependent nuclear export: Implications for tumorigenesis. EMBO J. 2002;21:31–42. doi: 10.1093/emboj/21.1.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui K, Tailor P, Liu H, Chen X, Ozato K, Zhao K. The chromatin-remodeling BAF complex mediates cellular antiviral activities by promoter priming. Mol Cell Biol. 2004;24:4476–4486. doi: 10.1128/MCB.24.10.4476-4486.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar AA, Goff LW, Majid S, Berlin J, El-Rifai W. Aurora kinase inhibitors–rising stars in cancer therapeutics? Mol Cancer Ther. 2010;9:268–278. doi: 10.1158/1535-7163.MCT-09-0765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta J, Majumder S, Bai S, Ghoshal K, Kutay H, Smith DS, Crabb JW, Jacob ST. Physical and functional interaction of DNA methyltransferase 3A with Mbd3 and Brg1 in mouse lymphosarcoma cells. Cancer Res. 2005;65:10891–10900. doi: 10.1158/0008-5472.CAN-05-1455. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- DeHart JL, Zimmerman ES, Ardon O, Monteiro-Filho CM, Arganaraz ER, Planelles V. HIV-1 Vpr activates the G2 checkpoint through manipulation of the ubiquitin proteasome system. Virol J. 2007;4:57–57. doi: 10.1186/1743-422X-4-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doerflinger NH, Macklin WB, Popko B. Inducible site-specific recombination in myelinating cells. Genesis. 2003;35:63–72. doi: 10.1002/gene.10154. [DOI] [PubMed] [Google Scholar]
- Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 mutations in familial and sporadic rhabdoid tumors. Pediatr Blood Cancer. 2011a;56:7–15. doi: 10.1002/pbc.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton KW, Tooke LS, Wainwright LM, Judkins AR, Biegel JA. Spectrum of SMARCB1/INI1 Mutations in Familial and Sporadic Rhabdoid Tumors. Pediatr Blood Cancer. 2011b;56:7–15. doi: 10.1002/pbc.22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG, Birch JM, Ramsden RT, Sharif S, Baser ME. Malignant transformation and new primary tumours after therapeutic radiation for benign disease: Substantial risks in certain tumour prone syndromes. J Med Genet. 2006;43:289–294. doi: 10.1136/jmg.2005.036319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans DG, Mason S, Huson SM, Ponder M, Harding AE, Strachan T. Spinal and cutaneous schwannomatosis is a variant form of type 2 neurofibromatosis: A clinical and molecular study. J Neurol Neurosurg Psychiatry. 1997;62:361–366. doi: 10.1136/jnnp.62.4.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster KS, McCrary WJ, Ross JS, Wright CF. Members of the hSWI/SNF chromatin remodeling complex associate with and are phosphorylated by protein kinase B/Akt. Oncogene. 2006;25:4605–4612. doi: 10.1038/sj.onc.1209496. [DOI] [PubMed] [Google Scholar]
- Gerszten PC, Quader M, Novotny J, Flickinger JC. Radiosurgery for benign tumors of the spine: Clinical experience and current trends. Technol Cancer Res Treat. 2012;11:133–139. doi: 10.7785/tcrt.2012.500242. [DOI] [PubMed] [Google Scholar]
- Giovannini M, Robanus-Maandag E, Niwa-Kawakita M, van der Valk M, Woodruff JM, Goutebroze L, Merel P, Berns A, Thomas G. Schwann cell hyperplasia and tumors in transgenic mice expressing a naturally occurring mutant NF2 protein. Genes Dev. 1999;13:978–986. doi: 10.1101/gad.13.8.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalvo A, Fowler A, Cook RJ, Little NS, Wheeler H, McDonald KL, Biggs MT. Schwannomatosis, sporadic schwannomatosis, and familial schwannomatosis: A surgical series with long-term follow-up. J Neurosurg. 2011;114:756–762. doi: 10.3171/2010.8.JNS091900. [DOI] [PubMed] [Google Scholar]
- Gresh L, Bourachot B, Reimann A, Guigas B, Fiette L, Garbay S, Muchardt C, Hue L, Pontoglio M, Yaniv M, Klochendler-Yeivin A. The SWI/SNF chromatin-remodeling complex subunit SNF5 is essential for hepatocyte differentiation. EMBO J. 2005;24:3313–3324. doi: 10.1038/sj.emboj.7600802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidi CJ, Sands AT, Zambrowicz BP, Turner TK, Demers DA, Webster W, Smith TW, Imbalzano AN, Jones SN. Disruption of Ini1 leads to peri-implantation lethality and tumorigenesis in mice. Mol Cell Biol. 2001;21:3598–3603. doi: 10.1128/MCB.21.10.3598-3603.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadfield KD, Newman WG, Bowers NL, Wallace A, Bolger C, Colley A, McCann E, Trump D, Prescott T, Evans DG. Molecular characterisation of SMARCB1 and NF2 in familial and sporadic schwannomatosis. J Med Genet. 2008;45:332–339. doi: 10.1136/jmg.2007.056499. [DOI] [PubMed] [Google Scholar]
- Hadfield KD, Smith MJ, Trump D, Newman WG, Evans DG. SMARCB1 mutations are not a common cause of multiple meningiomas. J Med Genet. 2010a;47:568. doi: 10.1136/jmg.2009.075721. [DOI] [PubMed] [Google Scholar]
- Hadfield KD, Smith MJ, Urquhart JE, Wallace AJ, Bowers NL, King AT, Rutherford SA, Trump D, Newman WG, Evans DG. Rates of loss of heterozygosity and mitotic recombination in NF2 schwannomas, sporadic vestibular schwannomas and schwannomatosis schwannomas. Oncogene. 2010b;29:6216–6221. doi: 10.1038/onc.2010.363. [DOI] [PubMed] [Google Scholar]
- Harder A, Wesemann M, Hagel C, Schittenhelm J, Fischer S, Tatagiba M, Nagel C, Jeibmann A, Bohring A, Mautner VF, Paulus W. Hybrid neurofibroma/schwannoma is overrepresented among schwannomatosis and neurofibromatosis patients. Am J Surg Pathol. 2012;36:702–709. doi: 10.1097/PAS.0b013e31824d3155. [DOI] [PubMed] [Google Scholar]
- Hulsebos TJ, Kenter SB, Jakobs ME, Baas F, Chong B, Delatycki MB. SMARCB1/INI1 maternal germ line mosaicism in schwannomatosis. Clin Genet. 2010;77:86–91. doi: 10.1111/j.1399-0004.2009.01249.x. [DOI] [PubMed] [Google Scholar]
- Hulsebos TJ, Plomp AS, Wolterman RA, Robanus-Maandag EC, Baas F, Wesseling P. Germline mutation of INI1/SMARCB1 in familial schwannomatosis. Am J Hum Genet. 2007;80:805–810. doi: 10.1086/513207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacoby LB, Jones D, Davis K, Kronn D, Short MP, Gusella J, MacCollin M. Molecular analysis of the NF2 tumor-suppressor gene in schwannomatosis. Am J Hum Genet. 1997;61:1293–1302. doi: 10.1086/301633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagani Z, Mora-Blanco EL, Sansam CG, McKenna ES, Wilson B, Chen D, Klekota J, Tamayo P, Nguyen PT, Tolstorukov M, Park PJ, Cho YJ, Hsiao K, Buonamici S, Pomeroy SL, Mesirov JP, Ruffner H, Bouwmeester T, Luchansky SJ, Murtie J, Kelleher JF, Warmuth M, Sellers WR, Roberts CW, Dorsch M. Loss of the tumor suppressor Snf5 leads to aberrant activation of the Hedgehog-Gli pathway. Nat Med. 2010;16:1429–1433. doi: 10.1038/nm.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalpana GV, Marmon S, Wang W, Crabtree GR, Goff SP. Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5. Science. 1994;266:2002–2006. doi: 10.1126/science.7801128. [DOI] [PubMed] [Google Scholar]
- Kalpana GV, Smith ME. Development of targeted therapies for rhabdoid tumors based on the functions of INI1/hSNF5 tumor suppressor. In: Houghton Peter J, Arceci Robert J., editors. Molecularly targeted therapy for childhood cancer. New York: Springer; 2010. pp. 305–330. [Google Scholar]
- Kaufman DL, Heinrich BS, Willett C, Perry A, Finseth F, Sobel RA, MacCollin M. Somatic instability of the NF2 gene in schwannomatosis. Arch Neurol. 2003;60:1317–1320. doi: 10.1001/archneur.60.9.1317. [DOI] [PubMed] [Google Scholar]
- Kim JK, Huh SO, Choi H, Lee KS, Shin D, Lee C, Nam JS, Kim H, Chung H, Lee HW, Park SD, Seong RH. Srg3, a mouse homolog of yeast SWI3, is essential for early embryogenesis and involved in brain development. Mol Cell Biol. 2001;21:7787–7795. doi: 10.1128/MCB.21.22.7787-7795.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingston RE, Narlikar GJ. ATP-dependent remodeling and acetylation as regulators of chromatin fluidity. Genes Dev. 1999;13:2339–2352. doi: 10.1101/gad.13.18.2339. [DOI] [PubMed] [Google Scholar]
- Kitzen JJ, de Jonge MJ, Verweij J. Aurora kinase inhibitors. Crit Rev Oncol Hematol. 2010;73:99–110. doi: 10.1016/j.critrevonc.2009.03.009. [DOI] [PubMed] [Google Scholar]
- Klochendler-Yeivin A, Fiette L, Barra J, Muchardt C, Babinet C, Yaniv M. The murine SNF5/INI1 chromatin remodeling factor is essential for embryonic development and tumor suppression. EMBO Rep. 2000;1:500–506. doi: 10.1093/embo-reports/kvd129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krosl J, Mamo A, Chagraoui J, Wilhelm BT, Girard S, Louis I, Lessard J, Perreault C, Sauvageau G. A mutant allele of the Swi/Snf member BAF250a determines the pool size of fetal liver hemopoietic stem cell populations. Blood. 2010;116:1678–1684. doi: 10.1182/blood-2010-03-273862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D, Sohn H, Kalpana GV, Choe J. Interaction of E1 and hSNF5 proteins stimulates replication of human papillomavirus DNA. Nature. 1999;399:487–491. doi: 10.1038/20966. [DOI] [PubMed] [Google Scholar]
- Lee JY, Kim H, Ryu CH, Kim JY, Choi BH, Lim Y, Huh PW, Kim YH, Lee KH, Jun TY, Rha HK, Kang JK, Choi CR. Merlin, a tumor suppressor, interacts with transactivation-responsive RNA-binding protein and inhibits its oncogenic activity. J Biol Chem. 2004;279:30265–30273. doi: 10.1074/jbc.M312083200. [DOI] [PubMed] [Google Scholar]
- Lee S, Cimica V, Ramachandra N, Zagzag D, Kalpana GV. Aurora A is a repressed effector target of the chromatin remodeling protein INI1/hSNF5 required for rhabdoid tumor cell survival. Cancer Res. 2011;71:3225–3235. doi: 10.1158/0008-5472.CAN-10-2167. [DOI] [PubMed] [Google Scholar]
- Lesbats P, Botbol Y, Chevereau G, Vaillant C, Calmels C, Arneodo A, Andreola ML, Lavigne M, Parissi V. Functional coupling between HIV-1 integrase and the SWI/SNF chromatin remodeling complex for efficient in vitro integration into stable nucleosomes. PLoS Pathog. 2011;7:e1001280. doi: 10.1371/journal.ppat.1001280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessard J, Wu JI, Ranish JA, Wan M, Winslow MM, Staahl BT, Wu H, Aebersold R, Graef IA, Crabtree GR. An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 2007;55:201–215. doi: 10.1016/j.neuron.2007.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You L, Cooper J, Schiavon G, Pepe-Caprio A, Zhou L, Ishii R, Giovannini M, Hanemann CO, Long SB, Erdjument-Bromage H, Zhou P, Tempst P, Giancotti FG. Merlin/NF2 suppresses tumorigenesis by inhibiting the E3 ubiquitin ligase CRL4(DCAF1) in the nucleus. Cell. 2010;140:477–490. doi: 10.1016/j.cell.2010.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Kang H, Liu R, Chen X, Zhao K. Maximal induction of a subset of interferon target genes requires the chromatin-remodeling activity of the BAF complex. Mol Cell Biol. 2002;22:6471–6479. doi: 10.1128/MCB.22.18.6471-6479.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacCollin M, Chiocca EA, Evans DG, Friedman JM, Horvitz R, Jaramillo D, Lev M, Mautner VF, Niimura M, Plotkin SR, Sang CN, Stemmer-Rachamimov A, Roach ES. Diagnostic criteria for schwannomatosis. Neurology. 2005;64:1838–1845. doi: 10.1212/01.WNL.0000163982.78900.AD. [DOI] [PubMed] [Google Scholar]
- MacCollin M, Willett C, Heinrich B, Jacoby LB, Acierno JS, Jr, Perry A, Louis DN. Familial schwannomatosis: Exclusion of the NF2 locus as the germline event. Neurology. 2003;60:1968–1974. doi: 10.1212/01.wnl.0000070184.08740.e0. [DOI] [PubMed] [Google Scholar]
- MacCollin M, Woodfin W, Kronn D, Short MP. Schwannomatosis: A clinical and pathologic study. Neurology. 1996;46:1072–1079. doi: 10.1212/wnl.46.4.1072. [DOI] [PubMed] [Google Scholar]
- Mahmoudi T, Parra M, Vries RG, Kauder SE, Verrijzer CP, Ott M, Verdin E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J Bio Chem. 2006;281:19960–19968. doi: 10.1074/jbc.M603336200. [DOI] [PubMed] [Google Scholar]
- Martens JA, Winston F. Recent advances in understanding chromatin remodeling by Swi/Snf complexes. Curr Opin Genet Dev. 2003;13:136–142. doi: 10.1016/s0959-437x(03)00022-4. [DOI] [PubMed] [Google Scholar]
- Matsumoto S, Banine F, Struve J, Xing R, Adams C, Liu Y, Metzger D, Chambon P, Rao MS, Sherman LS. Brg1 is required for murine neural stem cell maintenance and gliogenesis. Dev Biol. 2006;289:372–383. doi: 10.1016/j.ydbio.2005.10.044. [DOI] [PubMed] [Google Scholar]
- Medjkane S, Novikov E, Versteege I, Delattre O. The tumor suppressor hSNF5/INI1 modulates cell growth and actin cytoskeleton organization. Cancer Res. 2004;64:3406–3413. doi: 10.1158/0008-5472.CAN-03-3004. [DOI] [PubMed] [Google Scholar]
- Merker VL, Esparza S, Smith MJ, Stemmer-Rachamimov A, Plotkin SR. Clinical features of schwannomatosis: a retrospective analysis of 87 patients. Oncologist. 2012;17:1317–1322. doi: 10.1634/theoncologist.2012-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messing A, Behringer RR, Hammang JP, Palmiter RD, Brinster RL, Lemke G. P0 promoter directs expression of reporter and toxin genes to Schwann cells of transgenic mice. Neuron. 1992;8:507–520. doi: 10.1016/0896-6273(92)90279-m. [DOI] [PubMed] [Google Scholar]
- Messing A, Behringer RR, Wrabetz L, Hammang JP, Lemke G, Palmiter RD, Brinster RL. Hypomyelinating peripheral neuropathies and schwannomas in transgenic mice expressing SV40 T-antigen. J Neurosci. 1994;14:3533–3539. doi: 10.1523/JNEUROSCI.14-06-03533.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morozov A, Lee SJ, Zhang ZK, Cimica V, Zagzag D, Kalpana GV. INI1 induces interferon signaling and spindle checkpoint in rhabdoid tumors. Clin Cancer Res. 2007;13:4721–4730. doi: 10.1158/1078-0432.CCR-07-0054. [DOI] [PubMed] [Google Scholar]
- Morozov A, Yung E, Kalpana G. Structure-function analysis of integrase interactor 1/hSNF5L1 reveals differential properties of two repeat motifs present in the highly conserved region. Proc Natl Acad Sci USA. 1998;95:1120–1125. doi: 10.1073/pnas.95.3.1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A, Hughes TAT, Neal JW, Howard E, Evans DGR, Harper PS. A case of multiple cutaneous schwannomas; schwannomatosis or neurofibromatosis type 2? J Neurol Neurosurg Psychiatry. 2006;77:269–271. doi: 10.1136/jnnp.2005.067017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadler Y, Camp RL, Schwartz C, Rimm DL, Kluger HM, Kluger Y. Expression of Aurora A (but not Aurora B) is predictive of survival in breast cancer. Clin Cancer Res. 2008;14:4455–4462. doi: 10.1158/1078-0432.CCR-07-5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narlikar GJ, Fan HY, Kingston RE. Cooperation between complexes that regulate chromatin structure and transcription. Cell. 2002;108:475–487. doi: 10.1016/s0092-8674(02)00654-2. [DOI] [PubMed] [Google Scholar]
- Niranjan A, Mathieu D, Flickinger JC, Kondziolka D, Lunsford LD. Hearing preservation after intracanalicular vestibular schwannoma radiosurgery. Neurosurgery. 2008;63:1054–1062. doi: 10.1227/01.NEU.0000335783.70079.85. [DOI] [PubMed] [Google Scholar]
- Nowak SJ, Aihara H, Gonzalez K, Nibu Y, Baylies MK. Akirin links twist-regulated transcription with the Brahma chromatin remodeling complex during embryogenesis. PLoS Genet. 2012;8:e1002547. doi: 10.1371/journal.pgen.1002547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osley MA, Tsukuda T, Nickoloff JA. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat Res. 2007;618:65–80. doi: 10.1016/j.mrfmmm.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil S, Perry A, MacCollin M, Dong S, Betensky RA, Yeh TH, Gutmann DH, Stemmer-Rachamimov AO. Immunohistochemical analysis supports a role for INI1/SMARCB1 in hereditary forms of schwannomas, but not in solitary, sporadic schwannomas. Brain Pathol. 2008;18:517–519. doi: 10.1111/j.1750-3639.2008.00155.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattenden SG, Klose R, Karaskov E, Bremner R. Interferon-gamma-induced chromatin remodeling at the CIITA locus is BRG1 dependent. EMBO J. 2002;21:1978–1986. doi: 10.1093/emboj/21.8.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin SR, Bredella MA, Cai W, Kassarjian A, Harris GJ, Esparza S, Merker VL, Munn LL, Muzikansky A, Askenazi M, Nguyen R, Wenzel R, Mautner VF. Quantitative assessment of whole-body tumor burden in adult patients with neurofibromatosis. PLoS One. 2012;7:e35711. doi: 10.1371/journal.pone.0035711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard JR, Mortimore M. Discovery and development of aurora kinase inhibitors as anticancer agents. J Med Chem. 2009;52:2629–2651. doi: 10.1021/jm8012129. [DOI] [PubMed] [Google Scholar]
- Racki LR, Narlikar GJ. ATP-dependent chromatin remodeling enzymes: Two heads are not better, just different. Curr Opin Genet Dev. 2008;18:137–144. doi: 10.1016/j.gde.2008.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reyes JC, Barra J, Muchardt C, Camus A, Babinet C, Yaniv M. Altered control of cellular proliferation in the absence of mammalian brahma (SNF2alpha) EMBO J. 1998;17:6979–6991. doi: 10.1093/emboj/17.23.6979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieske P, Zakrzewska M, Piaskowski S, Jaskolski D, Sikorska B, Papierz W, Zakrzewski K, Liberski PP. Molecular heterogeneity of meningioma with INI1 mutation. Mol Pathol. 2003;56:299–301. doi: 10.1136/mp.56.5.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Galusha SA, McMenamin ME, Fletcher CD, Orkin SH. Haploinsufficiency of Snf5 (integrase interactor 1) predisposes to malignant rhabdoid tumors in mice. Proc Natl Acad Sci USA. 2000;97:13796–13800. doi: 10.1073/pnas.250492697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CW, Leroux MM, Fleming MD, Orkin SH. Highly penetrant, rapid tumorigenesis through conditional inversion of the tumor suppressor gene Snf5. Cancer Cell. 2002;2:415–425. doi: 10.1016/s1535-6108(02)00185-x. [DOI] [PubMed] [Google Scholar]
- Rousseau G, Noguchi T, Bourdon V, Sobol H, Olschwang S. SMARCB1/INI1 germline mutations contribute to 10% of sporadic schwannomatosis. BMC Neurol. 2011;11:9–9. doi: 10.1186/1471-2377-11-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblatt-Rosen O, Rozovskaia T, Burakov D, Sedkov Y, Tillib S, Blechman J, Nakamura T, Croce CM, Mazo A, Canaani E. The C-terminal SET domains of ALL-1 and TRITHORAX interact with the INI1 and SNR1 proteins, components of the SWI/SNF complex. Proc Natl Acad Sci U S A. 1998;95:4152–4157. doi: 10.1073/pnas.95.8.4152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz U, Mueller W, Weber M, Sevenet N, Delattre O, von Deimling A. INI1 mutations in meningiomas at a potential hotspot in exon 9. Br J Cancer. 2001;84:199–201. doi: 10.1054/bjoc.2000.1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo S, Richardson GA, Kroll KL. The SWI/SNF chromatin remodeling protein Brg1 is required for vertebrate neurogenesis and mediates transactivation of Ngn and NeuroD. Development. 2005;132:105–115. doi: 10.1242/dev.01548. [DOI] [PubMed] [Google Scholar]
- Sestini R, Bacci C, Provenzano A, Genuardi M, Papi L. Evidence of a four-hit mechanism involving SMARCB1 and NF2 in schwannomatosis-associated schwannomas. Hum Mutat. 2008;29:227–231. doi: 10.1002/humu.20679. [DOI] [PubMed] [Google Scholar]
- Smith ME, Cimica V, Chinni S, Challagulla K, Mani S, Kalpana GV. Rhabdoid tumor growth is inhibited by flavopiridol. Clin Cancer Res. 2008;14:523–532. doi: 10.1158/1078-0432.CCR-07-1347. [DOI] [PubMed] [Google Scholar]
- Smith ME, Cimica V, Chinni S, Jana S, Koba W, Yang Z, Fine E, Zagzag D, Montagna C, Kalpana GV. Therapeutically targeting cyclin D1 in primary tumors arising from loss of Ini1. Proc Natl Acad Sci USA. 2011;108:319–324. doi: 10.1073/pnas.0913297108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MJ, Wallace AJ, Bowers NL, Rustad CF, Woods CG, Leschziner GD, Ferner RE, Evans DG. Frequency of SMARCB1 mutations in familial and sporadic schwannomatosis. Neurogenetics. 2012;13:141–145. doi: 10.1007/s10048-012-0319-8. [DOI] [PubMed] [Google Scholar]
- Sorin M, Kalpana GV. Dynamics of virus-host interplay in HIV-1 replication. Curr HIV Res. 2006;4:117–130. doi: 10.2174/157016206776055048. [DOI] [PubMed] [Google Scholar]
- Swensen JJ, Keyser J, Coffin CM, Biegel JA, Viskochil DH, Williams MS. Familial occurrence of schwannomas and malignant rhabdoid tumour associated with a duplication in SMARCB1. J Med Genet. 2009;46:68–72. doi: 10.1136/jmg.2008.060152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsikitis M, Zhang Z, Edelman W, Zagzag D, Kalpana GV. Genetic ablation of Cyclin D1 abrogates genesis of rhabdoid tumors resulting from Ini1 loss. Proc Natl Acad Sci USA. 2005;102:12129–12134. doi: 10.1073/pnas.0505300102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Munckhof P, Christiaans I, Kenter SB, Baas F, Hulsebos TJ. Germline SMARCB1 mutation predisposes to multiple meningiomas and schwannomas with preferential location of cranial meningiomas at the falx cerebri. Neurogenetics. 2012;13:1–7. doi: 10.1007/s10048-011-0300-y. [DOI] [PubMed] [Google Scholar]
- van Vugt JJ, Ranes M, Campsteijn C, Logie C. The ins and outs of ATP-dependent chromatin remodeling in budding yeast: Biophysical and proteomic perspectives. Biochim Biophys Acta. 2007;1769:153–171. doi: 10.1016/j.bbaexp.2007.01.013. [DOI] [PubMed] [Google Scholar]
- Versteege I, Sevenet N, Lange J, Rousseau-Merck MF, Ambros P, Handgretinger R, Aurias A, Delattre O. Truncating mutations of hSNF5/INI1 in aggressive paediatric cancer. Nature. 1998;394:203–206. doi: 10.1038/28212. [DOI] [PubMed] [Google Scholar]
- Vries RG, Bezrookove V, Zuiderduijn LM, Kia SK, Houweling A, Oruetxebarria I, Raap AK, Verrijzer CP. Cancer-associated mutations in chromatin remodeler hSNF5 promote chromosomal instability by compromising the mitotic checkpoint. Genes Dev. 2005;19:665–670. doi: 10.1101/gad.335805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GG, Allis CD, Chi P. Chromatin remodeling and cancer, Part II: ATP-dependent chromatin remodeling. Trends Mol Med. 2007;13:373–380. doi: 10.1016/j.molmed.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Cote J, Xue Y, Zhou S, Khavari PA, Biggar SR, Muchardt C, Kalpana GV, Goff SP, Yaniv M, Workman JL, Crabtree GR. Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 1996a;15:5370–5382. [PMC free article] [PubMed] [Google Scholar]
- Wang W, Xue Y, Zhou S, Kuo A, Cairns BR, Crabtree GR. Diversity and specialization of mammalian SWI/SNF complexes. Genes Dev. 1996b;10:2117–2130. doi: 10.1101/gad.10.17.2117. [DOI] [PubMed] [Google Scholar]
- Wang X, Dong L, Xie J, Tong T, Zhan Q. Stable knockdown of Aurora-A by vector-based RNA interference in human esophageal squamous cell carcinoma cell line inhibits tumor cell proliferation, invasion and enhances apoptosis. Cancer Biol Ther. 2009;8:1852–1859. doi: 10.4161/cbt.8.19.9550. [DOI] [PubMed] [Google Scholar]
- Wen X, Casey Klockow L, Nekorchuk M, Sharifi HJ, de Noronha CM. The HIV1 protein Vpr acts to enhance constitutive DCAF1-dependent UNG2 turnover. PLoS One. 2012;7:e30939. doi: 10.1371/journal.pone.0030939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson BG, Roberts CW. SWI/SNF nucleosome remodellers and cancer. Nat Rev Cancer. 2011;11:481–492. doi: 10.1038/nrc3068. [DOI] [PubMed] [Google Scholar]
- Wilson BG, Wang X, Shen X, McKenna ES, Lemieux ME, Cho YJ, Koellhoffer EC, Pomeroy SL, Orkin SH, Roberts CW. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18:316–328. doi: 10.1016/j.ccr.2010.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DY, Kalpana GV, Goff SP, Schubach WH. Epstein-Barr virus nuclear protein 2 (EBNA2) binds to a component of the human SNF-SWI complex, hSNF5/Ini1. J Virol. 1996;70:6020–6028. doi: 10.1128/jvi.70.9.6020-6028.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DY, Krumm A, Schubach WH. Promoter-specific targeting of human SWI-SNF complex by Epstein-Barr virus nuclear protein 2. J Virol. 2000;74:8893–8903. doi: 10.1128/jvi.74.19.8893-8903.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu DY, Tkachuck DC, Roberson RS, Schubach WH. The human SNF5/INI1 protein facilitates the function of the growth arrest and DNA damage-inducible protein (GADD34) and modulates GADD34-bound protein phosphatase-1 activity. J Biol Chem. 2002;277:27706–27715. doi: 10.1074/jbc.M200955200. [DOI] [PubMed] [Google Scholar]
- Wurzenberger C, Gerlich DW. Phosphatases: Providing safe passage through mitotic exit. Nat Rev Mol Cell Biol. 2011;12:469–482. doi: 10.1038/nrm3149. [DOI] [PubMed] [Google Scholar]
- Xiao GH, Gallagher R, Shetler J, Skele K, Altomare DA, Pestell RG, Jhanwar S, Testa JR. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Cell Biol. 2005;25:2384–2394. doi: 10.1128/MCB.25.6.2384-2394.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi Y, Abe K, Mantani A, Hitoshi Y, Suzuki M, Osuzu F, Kuratani S, Yamamura K. A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev Biol. 1999;212:191–203. doi: 10.1006/dbio.1999.9323. [DOI] [PubMed] [Google Scholar]
- Yan A, Wang L, Xu S, Xu J. Aurora-A kinase inhibitor scaffolds and binding modes. Drug Discov Today. 2011;16:260–269. doi: 10.1016/j.drudis.2010.12.003. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Crabtree GR. ATP-dependent chromatin remodeling in neural development. Curr Opin Neurobiol. 2009;19:120–126. doi: 10.1016/j.conb.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young DW, Pratap J, Javed A, Weiner B, Ohkawa Y, van Wijnen A, Montecino M, Stein GS, Stein JL, Imbalzano AN, Lian JB. SWI/SNF chromatin remodeling complex is obligatory for BMP2-induced, Runx2-dependent skeletal gene expression that controls osteoblast differentiation. J Cell Biochem. 2005;94:720–730. doi: 10.1002/jcb.20332. [DOI] [PubMed] [Google Scholar]
- Yung E, Sorin M, Pal A, Craig E, Morozov A, Delattre O, Kappes J, Ott D, Kalpana GV. Inhibition of HIV-1 virion production by a trans-dominant mutant of Integrase interactor 1. Nat Med. 2001;7:920–926. doi: 10.1038/90959. [DOI] [PubMed] [Google Scholar]
- Yung E, Sorin M, Wang EJ, Perumal S, Ott D, Kalpana GV. Specificity of interaction of INI1/hSNF5 with retroviral integrases and its functional significance. J Virol. 2004;78:2222–2231. doi: 10.1128/JVI.78.5.2222-2231.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Wang J, Liu SJ, Hua W, Xin XY. Correlation between Aurora-A expression and the prognosis of cervical carcinoma patients. Acta Obstet Gynecol Scand. 2009;88:521–527. doi: 10.1080/00016340902835927. [DOI] [PubMed] [Google Scholar]
- Zhang ZK, Davies KP, Allen J, Zhu L, Pestell RG, Zagzag D, Kalpana GV. Cell cycle arrest and repression of cyclin D1 transcription by INI1/hSNF5. Mol Cell Biol. 2002;22:5975–5988. doi: 10.1128/MCB.22.16.5975-5988.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]