

Simvastatin modulates rhinovirus-induced cell signaling pathways in human monocytic cells, resulting in attenuation of IFN-κ and CXCL10.
Keywords: macrophages, virus, inflammation, chemokines, statins
Abstract
RV infections frequently trigger exacerbations of respiratory diseases, such as asthma, yet treatment and intervention options remain limited. Statin drugs are the treatment of choice for dyslipidemia and can also modulate immune cell function. To determine whether statin drugs modify antiviral responses of human monocytic cells, we obtained blood monocytes from donors with allergies and/or asthma and treated the cells with sim prior to challenge with RV. RV-induced secretion of CXCL10 was attenuated significantly, irrespective of RV type (RV-16, -14, or -1A), which corresponded with decreases in IFN-α secretion and pSTAT1. Sim pretreatment also reduced RV-induced CXCL10 secretion from human alveolar macrophages. The addition of mev and GGPP—two intermediates of the cholesterol biosynthesis pathway—was able to rescue CXCL10 release fully, demonstrating that effects of sim were related to inhibition of cholesterol biosynthesis and not to an off-target effect. In addition, sim pretreatment attenuated IFN-α-induced pSTAT1 and CXCL10 secretion, providing evidence that sim additionally can affect type I IFNR signaling. SOCS1 and 3 mRNA are both induced with RV stimulation, but sim did not elevate SOCS1 or SOCS3 mRNA expression basally or in the presence of RV. Our findings suggest that sim inhibition of the cholesterol biosynthesis pathway leads to decreased RV-induced chemokine secretion in monocytes and macrophages. These findings suggest that statin drugs have the potential to curb the inflammatory response to RV infection.
Introduction
Viral infections are an established risk factor for exacerbations of chronic lower-airway diseases, such as asthma. In adults, up to 50% of asthma exacerbations are a result of viral infections, with two-thirds of those infections attributed to RV, the principal cause of the common cold [1]. Virus-induced asthma exacerbations lead to increased doctor and hospital visits, loss of control of asthma symptoms, and overall decreased quality of life [2]. Treatment options remain limited, despite the prevalence of viral-induced exacerbations.
The primary target of RV infection and site of replication are the bronchial epithelium in the upper and lower airway [3]. Recently, RV has been shown to colocalize with airway monocytes/macrophages [4]. Additionally, we have shown previously that coculture of epithelial and monocytic cells results in a synergistic release of proinflammatory mediators, such as CXCL10 [5]. Monocytic cells represent up to 90% of immune cells in the resting lung and alone contribute to the immune response to RV through the synthesis of chemokines, cytokines, and IFNs, independent of viral replication [6, 7].
RV binding and cellular internalization promote monocytic cells to produce IFN-α, which acts via the type I IFNR to activate STAT1 and promote CXCL10 secretion [7]. CXCL10 is a chemoattractant for a variety of immune cells, including T cells, NK cells, and eosinophils, all of which play a role in asthma pathophysiology. During RV illnesses and exacerbations of asthma, CXCL10 is increased in airway secretions and serum and may contribute to virus-induced inflammation [8]. As such, understanding the RV-induced sensing and signaling pathways that may modulate CXCL10 production and secretion may provide novel therapeutic targets.
HMG-CoA reductase inhibitors, also known as statin drugs, are ubiquitous and effective therapeutics for dyslipidemia, by reducing LDL levels. In addition to their cholesterol-lowering effects, statin drugs are immunomodulators [9]. The therapeutic dosage of statins for potential anti-inflammatory effects has not yet been identified and may be different from doses required for cholesterol-lowering effects. Many of the in vitro anti-inflammatory effects in immune cell lines require doses much higher than physiological statin blood levels [10]. However, there is evidence that administration of statins via inhalation increases anti-inflammatory effects by 20- to 100-fold compared with i.p. injection or gavage in a mouse model of allergic asthma [11], and local concentrations after inhalation would be quite high. The anti-inflammatory effects of statins are mainly attributed to the inhibition of cholesterol biosynthesis intermediates FPP and GGPP. Depletion of FPP and GGPP reduces intracellular concentrations of isoprenylated, small GTPase signaling molecules, important in a number of cell-signaling pathways, many of which are involved in immune responses. As such, statin drugs have been proposed for the treatment of asthma and other inflammatory diseases [12–14].
Clinical studies to test anti-inflammatory properties of statins in asthmatic patients have yielded conflicting results. In some reports, statin use reduces asthma-associated hospitalization and enhances anti-inflammatory effects of inhaled corticosteroids [15, 16]. Conversely, other findings suggest a lack of steroid-sparing effects or improvement in lung function associated with statin treatment [17, 18]. However, these studies did not consider effects of statin drugs on seasonal and environmental modulators of asthma exacerbations, such as viral infections.
With the increasing appreciation of statins as immunomodulators, there are no data in human cell models testing the role of statins in modulating the immune response to common viral pathogens known to worsen asthma symptoms in monocytic cells. To test the hypothesis that statins modulate the viral immune response to RV, we examined the effects of sim, a commonly prescribed statin drug, on monocytic cells isolated from subjects with respiratory allergies or persistent asthma. We found that sim inhibits monocyte activation in response to RV, as measured by IFN-α and CXCL10 release and pSTAT1. These data suggest that statin drugs dampen the inflammatory response to RV infections.
MATERIALS AND METHODS
Reagents
Activated InSolution sim sodium salt (veh H2O, 0.1%) was purchased from Calbiochem (Darmstadt, Germany). Mev (veh H2O, 0.03%), FPP (veh MeOH, 0.2%), GGPP (veh MeOH, 0.2%), and atorvastatin (veh DMSO, 0.06%) were purchased from Sigma (St. Louis, MO, USA). Human rIFN-α2b (Intron A) was purchased from Schering (Kenilworth, NJ, USA). Immunoblotting antibodies anti-STAT1, anti-rabbit IgG, and anti-mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA); anti-actin from BD Transduction Laboratories (Franklin Lakes, NJ, USA); and anti-pSTAT1 pY701 from Cell Signaling Technologies (Danvers, MA, USA). Coating/biotinylated antibodies and the recombinant protein for CXCL10 ELISAs were purchased from R&D Systems (Minneapolis, MN, USA). Flow cytometry antibodies V450-conjugated anti-CD14 (Clone MΦPg) was purchased from BD Horizon (San Jose, CA, USA), and PE-conjugated anti-CD54 (ICAM-1, Clone HA58) was from BD PharMingen (San Jose, CA, USA).
Purification of peripheral blood monocytes and isolation of alveolar macrophages
PBMCs and BAL macrophages were received from volunteer donors under informed consent at the University of Wisconsin Hospital, in accordance with a protocol approved by the Human Subjects Committee. Donors were men and women between the ages of 18 and 50 with confirmed respiratory allergies and/or mild, persistent asthma, according to American Thoracic Society criteria [19]. Exclusion criteria included smoking, pregnant/lactating females, albuterol use within 2 weeks or corticosteroid use within the last month of the screen, unstable asthma based on self-report, and presence of other major health problems (heart disease, diabetes, other lung diseases besides asthma). Peripheral blood monocytes and BAL macrophages were purified and isolated as described previously [7]. Blood monocytes were maintained in MCM containing RPMI 1640 (Mediatech, Herndon, VA, USA), supplemented with 10% FBS (Hyclone, Logan, UT, USA), 100 U/ml penicillin/streptomycin, 2 mM sodium pyruvate, and 2 mM L-glutamine (all from Mediatech). In addition, 0.25 μg/mL amphotericin B was added to BAL macrophage media (Mediatech).
RV production and purification
RV-16, -14, and -1A were grown in HeLa cells and sucrose-purified, and infectivity (plaque-forming units) was determined by plaque assay in HeLa cell monolayers, as described previously [20]. Viruses were suspended in HBSS plus 0.1% HSA.
Cell viability
Blood monocytes were plated at 1 × 105 cells/0.1 mL media in 96-well plates (Sarstedt, Newton, NC, USA) and incubated overnight (16 h) at 37°C. The cells were treated with 0.1–10 μM sim for 24 h. The cells were then treated with MTS and the electron-coupling reagent phenazine methosulfate, according to the manufacturer's protocol (Promega, Madison, WI, USA). The cells were incubated at 37°C for 2 h, and the amount of formazan was measured by absorbance at 490 nm.
Detection of IFN-α and CXCL10 secretion
Blood monocytes were cultured at 1 × 106 cells/well and BAL macrophages at 5 × 105 cells/well in a 24-well Costar plate in 0.5 mL MCM (Corning, Corning, NY, USA). Blood monocytes were pretreated with 0.1–10 μM sim and BAL macrophages with 10 μm sim for 24 h at 37°C. For cholesterol intermediate rescue experiments, sim (10 μM)-treated cells were also cosupplemented with mev (100 μM), FPP (5 μM), or GGPP (5 μM). Cells were subsequently stimulated with control veh, RV-16, at a MOI of 10 or IFN-α (10 ng/ml) for 24 h at 34.5°C. Supernatants were collected, centrifuged at 15,800 g to remove nonadherent cells and debris, and stored at −40°C until analysis. IFN-α was detected using by ELISA (PBI Interferon Source, Piscataway, NJ, USA). CXCL10 concentration was determined using a two-step, sandwich-type ELISA, as described previously [21].
Flow cytometric analysis of cell-surface expression of ICAM-1
Blood monocytes were cultured at 2 × 106 cells/well in a six-well plate in 2 mL MCM. Cells were treated with veh or 10 μM sim for 24 h. Cells were lifted by pipetting and centrifuged at 200 g. Cells were suspended in PBS, supplemented with 1% FBS, and 5 × 105 cells were aliquoted per flow tube (BD Falcon; Becton Dickinson, Franklin Lakes, NJ, USA). With the use of fluorescence-minus one controls [22], cells were immunostained with 5 μl V450-conjugated anti-CD14 and 5 μl PE-conjugated anti-ICAM-1 for 45 min at 4°C. Cells were washed, centrifuged at 200 g, and suspended in 200 μl PBS + 1% FBS. Dead cells were excluded with 0.3 μg/mL PI. Ten thousand events were collected on a LSRII flow cytometer (Becton Dickinson, Bedford, MA, USA). The data were analyzed on FlowJo analysis software (TreeStar, Ashland, OR, USA). Cells positive for CD14 were gated, and the geometric mean fluorescence intensity was assessed for ICAM-1.
Immunoblotting
Blood monocytes were pretreated with 0.1–10 μM sim for 24 h at 37°C and stimulated with control veh or RV-16 (MOI=10) for 2 h or IFN-α for 30 min at 34.5°C. Supernatants were collected and centrifuged at 15,800 g for 1 min to collect nonadherent cells. Cells were lysed in 2× sample buffer (20 mM Tris, pH 6.8, 2 mM EDTA, 1 mM Na3VO4, 2 mM DTT, 2% SDS, and 20% glycerol) and added to the cell pellet. Samples were sonicated, boiled, and centrifuged at 15,800 g for 5 min to pellet cellular debris. Equal volume of cell lysate was separated by electrophoresis on 10% SDS-polyacrylamide gels, transferred to PVDF membranes, and blocked in 5% BSA/TBST (for pSTAT1) or 5% nonfat dry milk in TBST (STAT1, actin). Membranes were incubated with the primary antibodies to pSTAT1 or actin, followed by secondary antibodies conjugated to HRP. For α-STAT1 blots, pSTAT1 blots were stripped with One-Minute Western Blot Stripping Buffer (GM Biosciences, Rockville, MD, USA) and incubated with α-STAT1, or the lysate from the same sample was run on a separate gel for reference but not for protein-loading control. All blots were normalized to the actin-loading control. The immunoreactive bands were imaged using Epichemi II Darkroom (UVP, Upland CA, USA), and band densitometry was quantified using ImageJ (U.S. NIH, Bethesda, MD, USA).
Real-time qPCR
Blood monocytes were cultured at 1 × 106 cells/well in a 12-well Costar plate (Corning). Cells were treated with veh or 10 μM sim for 24 h and stimulated with RV-16 for 1, 3, 6, or 12 h. The 0-h time-point represents unstimulated cells. Total RNA was extracted using the RNeasy Mini Kit (Qiagen, Valencia, CA, USA). cDNA was obtained using the Superscript III system (Invitrogen/Life Technologies, Grand Island, NY, USA). Expression of mRNA was determined by qPCR using SYBR Green Master Mix (SABiosciences, Frederick, MD, USA). SOCS1 (forward-GCTGGCCCCTTCTGTAGGAT, reverse-TGCTGTGGAGACTGCATTGTC)- and SOCS3 (forward-GAGACTTCGATTCGGGACCAG, reverse-GAAACTTGCTGTGGGTGACCA)-specific primers were designed using Primer Express 3.0 (Applied Biosystems, Carlsbad, CA, USA), and sequences were compared with the human genome to determine specificity using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast; National Center for Biotechnology Information, Bethesda, MD, USA). The reference gene, β-glucuronidase (forward-CAGGACCTGCGCACAAGAG, reverse-TCGCACAGCTGGGGTAAG), was used to normalize the samples. Data are expressed as fold change compared with veh (0 h) using the ΔΔ comparative cycle threshold method.
Statistical analysis
Data were analyzed with paired Student's t-tests (see Figs. 1A, 2A, 3A, 5C, and 6A and B), or two-way ANOVA was used to assess differences between control and sim treatment at multiple time-points (see Fig. 6C and D). One-way ANOVA, followed by Bonferroni's corrected t-test for pair-wise comparisons, was used for all other statistical analyses.
Figure 1. Sim reduces RV-induced CXCL10 secretion from human blood monocytes in a dose-dependent manner.

(A) Human blood monocytes pretreated with veh or sim (10 μM) for 24 h, and RV-16 (MOI=10)-induced CXCL10 release at 24 h was measured (n=6). (B) Human blood monocytes were pretreated with increasing doses of sim (0.1, 1, or 10 μM) for 24 h and then stimulated with RV-16 (MOI=10) for 24 h. Data were pooled and normalized to veh-pretreated/RV-16-stimulated CXCL10 release (n=6). (C) Human blood moncytes were pretreated with veh or sim (10 μM) for 24 h, and RV-14 (MOI=10)- and (D) RV-1A-induced CXCL10 release at 24 h was determined. (E) Human blood monocytes were treated for 24 h with veh or increasing doses of sim (0.1, 1, or 10 μM), and metabolic activity was measured by MTS assay. Metabolic activity was normalized to veh (n=3). (F) Human blood monocytes pretreated with veh or sim (10 μM) and supplemented with mev (100 μM), FPP (5 μM), or GGPP (5μM) for 24 h. RV-16 (MOI=10)-induced CXCL10 secretion was normalized to veh-pretreated/RV-16-stimulated monocytes (n=7). Error bars indicate sem. *P < 0.05, ***P < 0.001 compared with RV treatment alone; ###P < 0.001 compared with sim + RV-16.
Figure 2. Sim attenuates RV-induced CXCL10 secretion from BAL macrophages.
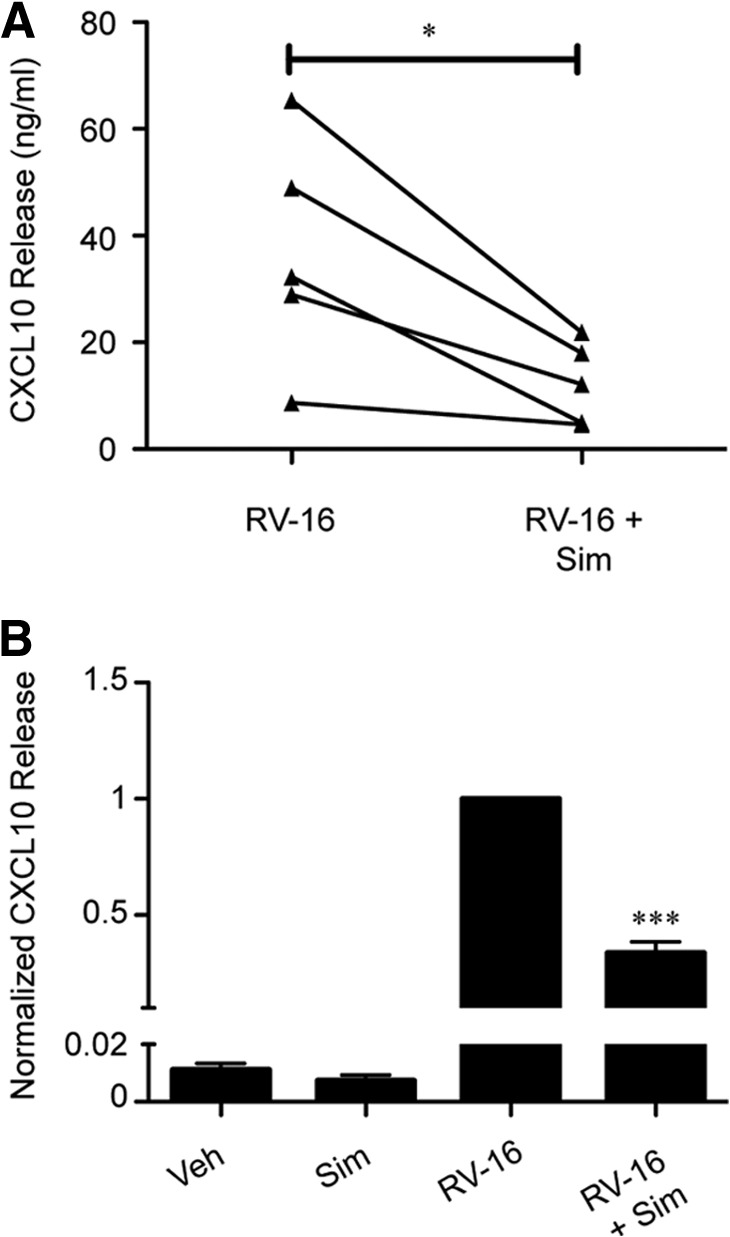
(A) BAL macrophages were pretreated with veh or sim (10 μM) for 24 h, and RV-16 (MOI=10)-induced CXCL10 release at 24 h was determined (n=5). (B) Data were pooled and normalized to veh-pretreated/RV-stimulated CXCL10 release. Error bars indicate sem. *P < 0.05, ***P < 0.001 compared with RV treatment alone.
Figure 3. Sim attenuates RV-induced IFN-α release from human blood monocytes.
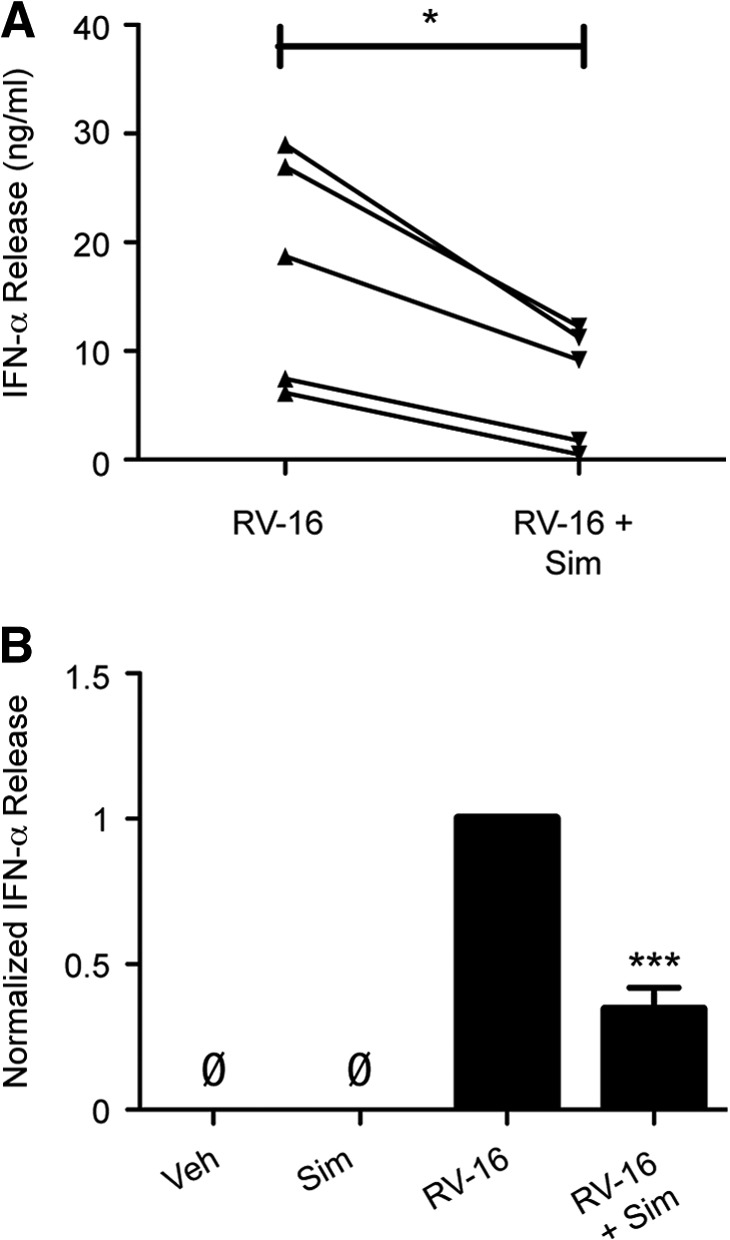
(A) Human blood monocytes, pretreated with veh or sim (10 μM) for 24 h, and RV-16 (MOI=10)-induced IFN-α (pg/mL) release at 24 h were measured (n=5). (B) Data were pooled and normalized to veh-pretreated, RV-16-stimulated IFN-α release (n=5). Error bars indicate sem. *P < 0.05, ***P < 0.001 compared with RV-16 treatment alone; ØBasal level of IFN-α below detection limit of 12.5 pg/ml.
Figure 5. Sim pretreatment reduces IFN-α-induced pSTAT1 and CXCL10 release from human blood monocytes.

(A) Representative immunoblots for IFN-α (10 ng/ml) induced pSTAT1, total STAT1, and actin (loading control) at 30 min from human blood monocytes pretreated with veh or sim (10 μM) for 24 h. (B) Band densitometry averages for pSTAT1 were corrected with actin and normalized to IFN-α treatment (n=4). (C) Human blood monocytes were pretreated with veh or sim (10 μM) for 24 h, and IFN-α (10 ng/ml)-induced CXCL10 release at 24 h was measured (n=5). (D) Data were pooled and normalized to veh-pretreated/IFN-α-stimulated CXCL10 release. (E) Human blood monocytes pretreated with veh or sim (10 μM) and supplemented with mev (100 μM), FPP (5 μM), or GGPP (5μM) for 24 h. IFN-α (10 ng/ml)-induced CXCL10 release was normalized to veh-pretreated/RV-16-stimulated monocytes (n=5). *P < 0.05, ***P < 0.001 compared with IFN-α treatment alone; ###P < 0.001 compared with sim + IFN-α.
Figure 6. Sim does not induce SOCS1 or SOCS3 mRNA.
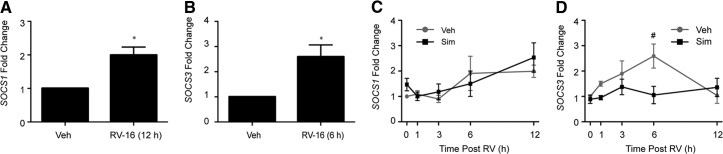
Human blood monocytes stimulated with veh or RV-16, and (A) SOCS1 mRNA at 12 h or (B) SOCS3 mRNA at 6 h were determined (n=4). (C and D) Human blood monocytes pretreated with veh or sim (10 μM) for 24 h and stimulated with RV-16 (0–12 h). SOCS1 mRNA (C) or SOCS3 mRNA (D) is represented as fold change relative to cells pretreated with veh alone for 24 h (n=4). Error bars indicate sem. *P < 0.05 compared with veh; #P < 0.05 compared with sim + RV at 6 h.
RESULTS
Sim attenuates RV-induced CXCL10 release in a dose-dependent manner from human blood monocytes
Monocytic cells and bronchial epithelial cells produce CXCL10 in response to RV-16 stimulation [7, 23]. Thus, we tested the hypothesis that sim pretreatment modulates RV-induced CXCL10 secretion, which is a potential biomarker for RV-induced viral exacerbation [8]. Blood monocytes were pretreated with increasing doses of sim for 24 h and then stimulated with RV-16. Sim significantly attenuated RV-induced CXCL10 release in a dose-dependent manner (Fig. 1A and B). At 0.1 μM sim, CXCL10 levels trend 24% lower than RV treatment alone. At 1 μM and 10 μM, sim significantly inhibited RV-induced CXCL10 release by 41% and 54%, respectively. Accordingly, pretreatment with 10 μM atorvastatin, another commonly prescribed statin medication, significantly reduced RV-induced CXCL10 secretion by 57% (Supplemental Fig. 1).
Experiments using an additional major group virus, RV-14, and a minor group virus, RV-1A, yielded results similar to those obtained with RV-16. (Fig. 1C and D). Major group RVs (i.e., RV-16 and RV-14) use ICAM-1, and minor group viruses (i.e., RV-1A) use the LDLR family members as receptors to enter cells [24]. Sim has been reported to reduce ICAM-1 surface expression on circulating monocytes [25] but has no effects on surface expression of LDLR [26]. To test if sim effects on CXCL10 production were secondary to down-regulation of ICAM-1, monocytes were treated for 24 h with veh or sim, and ICAM-1 surface expression was measured on CD14-positive monocytes. Sim had no significant effects on monocyte ICAM-1 expression at 24 h (Supplemental Fig. 2). These data suggest that at the 24-h time-point, sim effects on RV-induced CXCL10 secretion are independent of RV entry receptor expression.
To confirm that the doses of sim did not affect viability and thus, chemokine secretion, metabolic activity of blood monocytes was assessed 24 h after sim treatment (Fig. 1E). There was no statistical difference in metabolic activity at any of the sim concentrations tested.
Mev and GGPP rescue RV-induced CXCL10 release from human blood monocytes
In the presence of sim, monocytes obtain all cholesterol from media, supplemented with 10% FBS. However, the cholesterol biosynthesis pathway is not reversible; thus, products dependent on mev production, such as the precursors for isoprenylated proteins FPP and GGPP, are diminished. Also, statin drugs can have off-target effects in cells, in addition to the inhibition of HMG-CoA reductase [27]. To test whether the effects of sim on chemokine responses are a result of the direct inhibition of the cholesterol biosynthesis pathway, we supplemented sim-treated monocytes with mev, the product of HMG-CoA reductase. The supplementation of sim-pretreated blood monocytes with mev rescued RV-induced CXCL10 release completely compared with cells treated with RV in the absence of sim (Fig. 1F). In addition to mev, sim-treated monocytes were supplemented with FPP and GGPP, two intermediates of the cholesterol biosynthesis pathway that are crucial for the isoprenylation of small G-protein families, such as Ras and Rho family members, respectively. GGPP supplementation rescued RV-induced CXCL10 release completely compared with veh-pretreated/RV-stimulated monocytes (Fig. 1F). FPP had similar but less striking effects. Together, these data support the idea that sim-induced inhibition of the cholesterol biosynthesis pathway is responsible for differences in RV-induced CXCL10 release.
Sim attenuates RV-induced CXCL10 secretion from BAL macrophages
There are intrinsic differences between blood monocytes and terminally differentiated tissue macrophages [28]. To determine whether sim exerted similar effects on airway cells, BAL macrophages were pretreated with veh or 10 μM sim for 24 h and then stimulated with RV-16 for 24 h. Similar to observations in blood monocytes, sim pretreatment reduced RV-induced CXCL10 release by 60% (Fig. 2A and B). These observations confirm that sim has similar effects on virus-induced responses of human blood monocytes and airway macrophages. Taken together, these data indicate that sim pretreatment blunts a monocytic, proinflammatory response to RV.
Sim attenuates RV-induced IFN-α release from human blood monocytes
Secretion of type I IFNs is a hallmark of the innate immune response to RV [23, 29]. Additionally, IFN-α secretion is required for the production of CXCL10 in monocytic cells. To test whether sim additionally attenuates a monocytic cell type I IFN response, human blood monocytes were pretreated with veh or sim for 24 h and then stimulated with RV-16 for 24 h. Similarly to CXCL10, sim pretreatment reduced RV-induced IFN-α release by 65% (Fig. 3A and B).
Sim reduces RV-induced pSTAT1 in human blood monocytes
IFN-α binds to the type I IFNR and promotes pSTAT1, and this process contributes to RV-induced CXCL10 release [7]. Based on the observations that sim pretreatment decreased RV-induced IFN-α and CXCL10 secretion, we hypothesized that sim would also reduce RV-induced pSTAT1. Human blood monocytes were treated with increasing doses of sim for 24 h and then stimulated with RV-16 for 2 h. sim significantly attenuated RV-induced pSTAT1 (52% and 68% reduction at 1 μM and 10 μM sim, respectively) but did not affect total STAT1 (Fig. 4).
Figure 4. Sim pretreatment decreases RV-induced pSTAT1 in human blood monocytes in a dose-dependent manner.
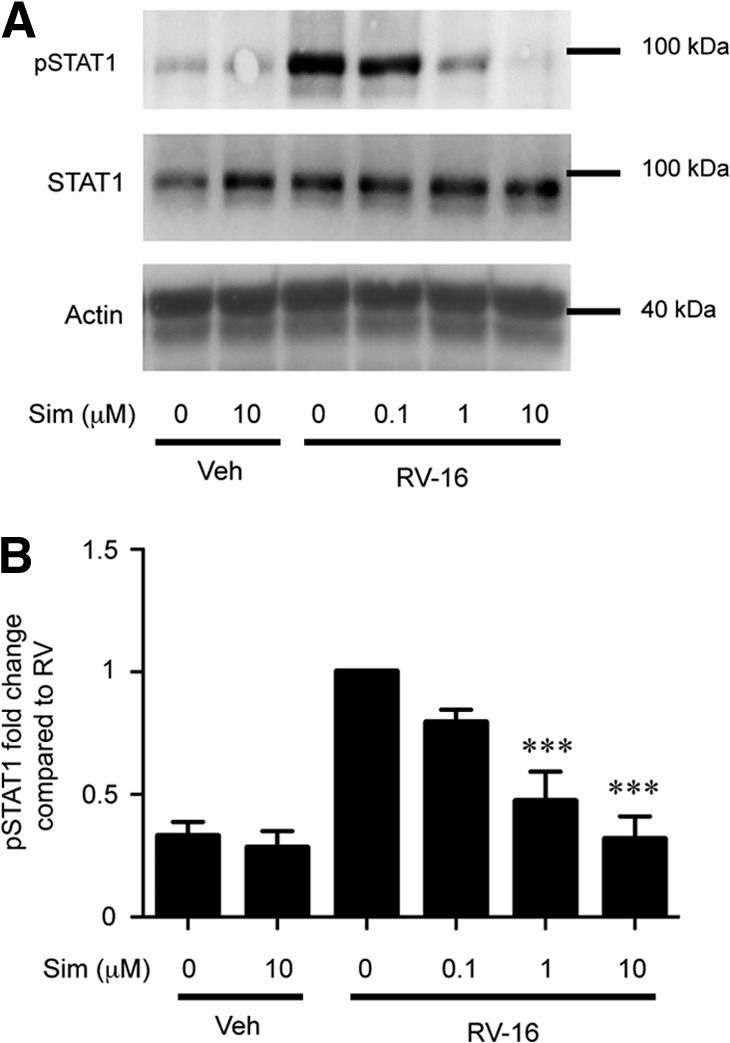
(A) Representative immunoblots for RV-16 (MOI=10) induced pSTAT1, total STAT1, and actin (loading control) at 2 h from human blood monocytes pretreated with veh or sim (0.1, 1, or 10 μM) for 24 h. (B) Band densitometry averages for pSTAT1 corrected with actin and normalized to RV treatment (n=5). Error bars indicate sem. ***P < 0.001 compared with RV treatment alone.
Sim reduces IFN-α-induced pSTAT1 and CXCL10 release
As RV-induced IFN-α contributes to CXCL10 production, we tested whether sim pretreatment additionally blunts IFN-α (10 ng/ml)-stimulated pSTAT1 and CXCL10 release. Blood monocytes were pretreated with veh or sim for 24 h and then stimulated with IFN-α for 30 min to detect pSTAT1 activation or 24 h to assess CXCL10 release. Compared with veh-pretreated/IFN-α-stimulated monocytes, sim pretreatment attenuated IFN-α-induced pSTAT1 levels by 48% (Fig. 5A and B) and CXCL10 release by 40% (Fig. 5C and D). These data suggest that sim can affect cell-signaling pathways upstream and downstream of the type I IFNR.
Mev, GGPP, and FPP rescue sim-attenuated, IFN-α-induced CXCL10 release
To test whether attenuation in IFN-α-induced effects were also a result of inhibition of the cholesterol biosynthesis pathway, sim-pretreated blood monocytes were supplemented with mev (100 μM), FPP (5 μM), or GGPP (5μM) and then stimulated with IFN-α (10 ng/ml). All three cholesterol-biosynthesis intermediates were able to rescue sim-pretreated/IFN-α-induced CXCL10 release to levels of veh-pretreated/IFN-α-stimulated monocytes (Fig. 5E).
SOCS1 and SOCS3 are not involved in the sim-induced blockade of type I IFN response to RV
SOCS1 and -3 regulate type I IFNR signaling by preventing pSTAT transcription factors [30]. Previous work in murine RAW264.7 macrophages and human THP-1 premonocytic cell lines found that sim induced expression of SOCS1 and SOCS3 mRNA [31]. Therefore, we investigated whether sim pretreatment increased SOCS1 and SOCS3 mRNA expression levels in blood monocytes, with or without RV stimulation. Cells were pretreated for 24 h with veh or sim and then stimulated with RV for 0–12 h, and SOCS1 and SOCS3 mRNA levels were determined. RV induced SOCS1 (Fig. 6A) and SOCS3 mRNA (Fig. 6B) at 12 h and 6 h, respectively, but sim did not augment either mRNA in the presence or absence of RV (Fig. 6C and D). These data suggest that sim attenuates RV-induced chemokine release independently of two known regulators of pSTAT1.
DISCUSSION
Herein, we demonstrate that sim can modulate the immune response to RV in primary human monocytic cells. Sim pretreatment attenuates RV-induced CXCL10 secretion in BAL macrophages and blood monocytes. This corresponds with decreases in IFN-α release and pSTAT1 in blood monocytes. Sim did not alter expression of ICAM-1, indicating that this effect was not a result of reduced binding capacity of the virus. Additionally, sim blocks IFN-α-stimulated pSTAT1 and CXCL10 release, suggesting that sim affects upstream and downstream of the type I IFNR. However, SOCS1 and SOCS3 do not appear to be involved in sim inhibition of RV- or IFN-α-induced STAT signaling and CXCL10 release. Overall, these data provide evidence that statin drugs may attenuate RV-induced inflammatory responses.
Significant immunomodulatory effects of sim were seen at levels higher than those achieved with oral administration [32, 33]. However, inhaled sim in a murine model of allergic asthma was well-tolerated and reduced inflammatory responses, such as Th2 cytokines and chemokines, and physiologic outcomes, such as airway remodeling and overall lung function [11]. Additionally, in the dsRNA-induced mouse pneumonia model, intranasal administration of sim decreased STAT3 activation, RANTES release, and neutrophilia [34]. Inhaled sim would be associated with high drug concentrations in the lung; thus, the investigation of the effects of higher levels of sim is of interest.
Our findings also suggest sim as a specific tool to better understand the effects of cholesterol pathways on antiviral responses. We provide evidence that sim modulates the sensing and response pathways of monocytic cells to RV, both upstream of IFN-α production and downstream of the type I IFNR, leading to attenuation of CXCL10 release. Notably, CXCL10 is a potential therapeutic target for viral-induced asthma exacerbation, and the corticosteroid budesonide can decrease RV-induced CXCL10 secretion [8, 35]. The effects of sim on RV-induced CXCL10 are reversed by the cholesterol biosynthesis intermediates mev and GGPP but not FPP. This suggests that Rho small GTPase family members, which are isoprenylated by GGPP, may play a role in RV-induced CXCL10 production. Rho family members are involved in inflammatory cell activation via MAPK and NF-κB signaling [36]. In plasmacytoid DCs, sim decreased TLR9 ligand stimulation of IFN regulatory factory 7 nuclear translocation and IFN-α production in a GGPP-dependent mechanism [37]. Our model suggests that sim may inhibit RV-induced IFN-α and CXCL10 release by affecting transcription factor activation in response to RV, leading to downstream decreases in chemokine secretion. Further understanding the mechanism by which sim attenuates RV-induced CXCL10 will give insight into the regulatory components of this pathway in monocytic cells.
RV infection stimulates IFN production in epithelial and monocytic cells, and IFN secretion secondarily enhances chemokine production (such as CXCL10) and immune cell recruitment to clear the virus. In asthmatics, an excessive proinflammatory response to stimulants, such as RV, can trigger an asthmatic episode [38]. However, whether IFN production is increased or decreased during virus-induced asthma exacerbation remains controversial [39]. In our study, sim pretreatment reduced RV-induced IFN-α release, on average, by 60% (Fig. 4). Previous findings indicate that IFN-α and IFN-λ-1 levels in respiratory secretions tended to be higher in children with wheezing RV infection compared with nonwheezing RV infection [40]. In contrast to these results, infants predisposed to lower antiviral responses, such as lower IFN responses, are more likely to be affected by more severe respiratory infections early in life, which can lead to wheezing, atopy, and higher risk for future asthma development [1]. Furthermore, BAL macrophages isolated from adult asthmatics stimulated with RV-16 have significantly lower IFN-α and IFN-β responses compared with normal patients [41]. Additional studies are warranted to determine whether reduction of IFN-α and CXCL10 impacts the antiviral responses of other cell types to delineate the balance between excessive inflammatory responses to RV seen in asthmatic patients versus a dampened host-defense response to a viral infection.
Our group and others have shown that statins can inhibit the Jak/STAT signaling cascade, which is downstream of IFN release and the type I IFNR [42, 43]. Dysregulation of this pathway has been implicated in the pathogenesis of inflammatory diseases, such as asthma [44]. Even with high-dose stimulation with IFN-α, pSTAT1 expression was still reduced (Fig. 6), suggesting that this pathway can be targeted independently of RV-induced IFN-α secretion. To our knowledge, this is the first study to show that RV induces SOCS1 and SOCS3 mRNA, two negative regulators of the Jak/STAT pathway, in primary human monocytic cells. However, we found no evidence that sim induced SOCS1 or SOCS3 mRNA, contrary to data from transformed cell lines. There are reports that SOCS family members are subject to post-transcriptional regulation [45], and it is possible that sim affects the activity of the SOCS protein through this process. Alternatively, there are a number of tyrosine phosphatases involved in the negative regulation of the Jak/STAT pathway that could be affected directly or indirectly by sim treatment [44].
To our knowledge, this is the first study investigating the immunomodulatory properties of statins to the common respiratory virus, RV, in monocytic cells. There are limited data testing effects of sim on outcomes of other viral infections. For example, cotreatment with sim and antiviral treatments, including supplemental IFN-α, synergistically promoted clearance of hepatitis C from human-derived cell lines [46]. Conversely, in mouse models of influenza, researchers found no difference in viral replication and clearance or overall survival with statin treatment [47]. However, there is clinical evidence that statins reduce hospitalization and mortality rates secondary to influenza [47, 48].
This study has a number of strengths and some limitations that should be considered in interpreting the data. Two commonly prescribed statin drugs—sim and atorvastatin—both reduced RV-induced CXCL10 release significantly. Furthermore, the monocytic immune response to three strains of RV representing major (RV-16, RV-14) and minor (RV-1A) groups, as well as type A (RV-16, RV-1A) and type B (RV-14) viruses, was similarly affected by sim. All experiments used primary human cells (blood monocytes and BAL macrophages), and effects were consistent in blood and airway cells. However, all cells were obtained from patients with allergies or mild asthma, and responses in normal individuals could be distinct. Additionally, the duration of sim pretreatment in all of the presented experiments was 24 h, and the long-term effects of statin use could result in differences in the monocytic immune response to RV stimulation. Finally, additional experiments may be warranted to evaluate effects of statins on metabolic activity of alveolar macrophages, as this was not tested in our study.
In sum, we provide evidence that sim modulates monocytic cell responses to RV infection and IFN-α stimulation. If these effects also occur in vivo, then it is possible that sim may modify the clinical course of RV respiratory infections. The understanding of the clinical effects of a commonly prescribed drug, sim, on viral infections may be especially important for patients with chronic respiratory diseases, such as asthma.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the U.S. National Institutes of Health Grants NIH-NHLBI P01HL088594 (to P.J.B.) and 5T32DK007665 (to L.E.W.).
The authors thank Drs. Yury Bochkov and Wai-Ming Lee for preparation of virus stocks; Dr. Sameer Mathur, Dr. Lei Shi, Elizabeth Schwantes, and Paul Fichtinger for providing PBMCs and macrophages; the University of Wisconsin Carbone Cancer Center (UWCCC) flow cytometry laboratory; Dr. Colleen Curran for assistance with flow experiments; Larissa Delain and Rose Vrtis for technical support; and Mandy Burnham for editorial comments.
The online version of this paper, found at www.jleukbio.org, includes supplemental information.
- BAL
- bronchoalveolar lavage
- FPP
- farnesyl pyrophospate
- GGPP
- geranylgeranyl pyrophosphate
- HMG-CoA
- 3-hydroxy-3-methylglutaryl-CoA
- MCM
- monocyte complete media
- mev
- mevalonate
- MTS
- 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium
- pSTAT
- phosphorylated STAT
- qPCR
- quantitative PCR
- RV
- rhinovirus
- sim
- simvastatin
- SOCS
- suppressor of cytokine signaling molecules
- veh
- vehicle
AUTHORSHIP
L.E.W. designed, performed, and analyzed experiments and wrote the manuscript. M.R.K. assisted with experiments and helped edit the manuscript. A.A. and J.E.G. assisted with the design of experiments and editing the manuscript. P.J.B. assisted with the conception of the project and design of experiments.
DISCLOSURES
L.E.W., M.R.K., A.A., and P.J.B. have no conflicts of interest to disclose. J.E.G. is a consultant to Merck, Centocor, and GlaxoSmithKline and was awarded research funding to the University of Wisconsin-Madison from Merck, AstraZeneca, and GlaxoSmithKline.
REFERENCES
- 1. Gavala M. L., Bertics P. J., Gern J. E. (2011) Rhinoviruses, allergic inflammation, and asthma. Immunol. Rev. 242, 69–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Jackson D. J., Johnston S. L. (2010) The role of viruses in acute exacerbations of asthma. J. Allergy Clin. Immunol. 125, 1178–1187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Proud D., Sanders S. P., Wiehler S. (2004) Human rhinovirus infection induces airway epithelial cell production of human β-defensin 2 both in vitro and in vivo. J. Immunol. 172, 4637–4645 [DOI] [PubMed] [Google Scholar]
- 4. Bentley J. K., Sajjan U. S., Dzaman M. B., Jarjour N. N., Lee W. M., Gern J. E., Hershenson M. B. (2013) Rhinovirus colocalizes with CD68- and CD11b-positive macrophages following experimental infection in humans. J. Allergy Clin. Immunol. 132, 758.e3–761.e3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Korpi-Steiner N. L., Valkenaar S. M., Bates M. E., Evans M. D., Gern J. E., Bertics P. J. (2010) Human monocytic cells direct the robust release of CXCL10 by bronchial epithelial cells during rhinovirus infection. Clin. Exp. Allergy 40, 1203–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Smith D. L., deShazo R. D. (1992) Integrins, macrophages, and sarcoidosis. Chest 102, 659–660 [DOI] [PubMed] [Google Scholar]
- 7. Korpi-Steiner N. L., Bates M. E., Lee W. M., Hall D. J., Bertics P. J. (2006) Human rhinovirus induces robust IP-10 release by monocytic cells, which is independent of viral replication but linked to type I interferon receptor ligation and STAT1 activation. J. Leukoc. Biol. 80, 1364–1374 [DOI] [PubMed] [Google Scholar]
- 8. Wark P. A., Bucchieri F., Johnston S. L., Gibson P. G., Hamilton L., Mimica J., Zummo G., Holgate S. T., Attia J., Thakkinstian A., Davies D. E. (2007) IFN-γ-induced protein 10 is a novel biomarker of rhinovirus-induced asthma exacerbations. J. Allergy Clin. Immunol. 120, 586–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bu D. X., Griffin G., Lichtman A. H. (2011) Mechanisms for the anti-inflammatory effects of statins. Curr. Opin. Lipidol. 22, 165–170 [DOI] [PubMed] [Google Scholar]
- 10. Bjorkhem-Bergman L., Lindh J. D., Bergman P. (2011) What is a relevant statin concentration in cell experiments claiming pleiotropic effects? Br. J. Clin. Pharmacol. 72, 164–165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xu L., Dong X. W., Shen L. L., Li F. F., Jiang J. X., Cao R., Yao H. Y., Shen H. J., Sun Y., Xie Q. M. (2012) Simvastatin delivery via inhalation attenuates airway inflammation in a murine model of asthma. Int. Immunopharmacol. 12, 556–564 [DOI] [PubMed] [Google Scholar]
- 12. Zeki A. A., Franzi L., Last J., Kenyon N. J. (2009) Simvastatin inhibits airway hyperreactivity: implications for the mevalonate pathway and beyond. Am. J. Respir. Crit. Care Med. 180, 731–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rosch P. J., McCully K. (2013) Statin use and reduced cancer-related mortality. N. Engl. J. Med. 368, 576. [DOI] [PubMed] [Google Scholar]
- 14. Janda S., Park K., FitzGerald J. M., Etminan M., Swiston J. (2009) Statins in COPD: a systematic review. Chest 136, 734–743 [DOI] [PubMed] [Google Scholar]
- 15. Hothersall E. J., Chaudhuri R., McSharry C., Donnelly I., Lafferty J., McMahon A. D., Weir C. J., Meiklejohn J., Sattar N., McInnes I., Wood S., Thomson N. C. (2008) Effects of atorvastatin added to inhaled corticosteroids on lung function and sputum cell counts in atopic asthma. Thorax 63, 1070–1075 [DOI] [PubMed] [Google Scholar]
- 16. Huang C-C., Chan W-L., Chen Y-C., Chen T-J., Chou K-T., Lin S-J., Chen J-W., Leu H-B. (2011) Statin use in patients with asthma—a nationwide population-based study. Eur. J. Clin. Invest. 41, 507–512 [DOI] [PubMed] [Google Scholar]
- 17. Menzies D., Nair A., Meldrum K. T., Fleming D., Barnes M., Lipworth B. J. (2007) Simvastatin does not exhibit therapeutic anti-inflammatory effects in asthma. J. Allergy Clin. Immunol. 119, 328–335 [DOI] [PubMed] [Google Scholar]
- 18. Cowan D. C., Cowan J. O., Palmay R., Williamson A., Taylor D. R. (2010) Simvastatin in the treatment of asthma: lack of steroid-sparing effect. Thorax 65, 891–896 [DOI] [PubMed] [Google Scholar]
- 19. (2000) Proceedings of the ATS workshop on refractory asthma: current understanding, recommendations, and unanswered questions. American Thoracic Society. Am. J. Respir. Crit. Care Med. 162, 2341–2351 [DOI] [PubMed] [Google Scholar]
- 20. Sherry B., Rueckert R. (1985) Evidence for at least two dominant neutralization antigens on human rhinovirus 14. J. Virol. 53, 137–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kelly E. A., Rodriguez R. R., Busse W. W., Jarjour N. N. (1997) The effect of segmental bronchoprovocation with allergen on airway lymphocyte function. Am. J. Respir. Crit. Care Med. 156, 1421–1428 [DOI] [PubMed] [Google Scholar]
- 22. Maecker H. T., Trotter J. (2006) Flow cytometry controls, instrument setup, and the determination of positivity. Cytometry A 69, 1037–1042 [DOI] [PubMed] [Google Scholar]
- 23. Spurrell J. C., Wiehler S., Zaheer R. S., Sanders S. P., Proud D. (2005) Human airway epithelial cells produce IP-10 (CXCL10) in vitro and in vivo upon rhinovirus infection. Am. J. Physiol. Lung Cell. Mol. Physiol. 289, L85–L95 [DOI] [PubMed] [Google Scholar]
- 24. Palmenberg A. C., Rathe J. A., Liggett S. B. (2010) Analysis of the complete genome sequences of human rhinovirus. J. Allergy Clin. Immunol. 125, 1190–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rezaie-Majd A., Prager G. W., Bucek R. A., Schernthaner G. H., Maca T., Kress H. G., Valent P., Binder B. R., Minar E., Baghestanian M. (2003) Simvastatin reduces the expression of adhesion molecules in circulating monocytes from hypercholesterolemic patients. Arterioscler. Thromb. Vasc. Biol. 23, 397–403 [DOI] [PubMed] [Google Scholar]
- 26. Gouni-Berthold I., Berthold H. K., Gylling H., Hallikainen M., Giannakidou E., Stier S., Ko Y., Patel D., Soutar A. K., Seedorf U., Mantzoros C. S., Plat J., Krone W. (2008) Effects of ezetimibe and/or simvastatin on LDL receptor protein expression and on LDL receptor and HMG-CoA reductase gene expression: a randomized trial in healthy men. Atherosclerosis 198, 198–207 [DOI] [PubMed] [Google Scholar]
- 27. Planaguma A., Pfeffer M. A., Rubin G., Croze R., Uddin M., Serhan C. N., Levy B. D. (2010) Lovastatin decreases acute mucosal inflammation via 15-epi-lipoxin A4. Mucosal Immunol. 3, 270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gordon S., Taylor P. R. (2005) Monocyte and macrophage heterogeneity. Nat. Rev. Immunol. 5, 953–964 [DOI] [PubMed] [Google Scholar]
- 29. Sen G. C. (2001) Viruses and interferons. Annu. Rev. Microbiol. 55, 255–281 [DOI] [PubMed] [Google Scholar]
- 30. Wormald S., Hilton D. J. (2004) Inhibitors of cytokine signal transduction. J. Biol. Chem. 279, 821–824 [DOI] [PubMed] [Google Scholar]
- 31. Lee C. S., Shin Y. J., Won C., Lee Y. S., Park C. G., Ye S. K., Chung M. H. (2009) Simvastatin acts as an inhibitor of interferon γ-induced cycloxygenase-2 expression in human THP-1 cells, but not in murine RAW264.7 cells. Biocell 33, 107–114 [PubMed] [Google Scholar]
- 32. Lilja J. J., Kivisto K. T., Neuvonen P. J. (1998) Grapefruit juice-simvastatin interaction: effect on serum concentrations of simvastatin, simvastatin acid, and HMG-CoA reductase inhibitors. Clin. Pharmacol. Ther. 64, 477–483 [DOI] [PubMed] [Google Scholar]
- 33. Ucar M., Neuvonen M., Luurila H., Dahlqvist R., Neuvonen P. J., Mjorndal T. (2004) Carbamazepine markedly reduces serum concentrations of simvastatin and simvastatin acid. Eur. J. Clin. Pharmacol. 59, 879–882 [DOI] [PubMed] [Google Scholar]
- 34. Lee C. S., Yi E. H., Lee J. K., Won C., Lee Y. J., Shin M. K., Yang Y. M., Chung M. H., Lee J. W., Sung S. H., Ye S. K. (2013) Simvastatin suppresses RANTES-mediated neutrophilia in polyinosinic-polycytidylic acid-induced pneumonia. Eur. Respir. J. 41, 1147–1156 [DOI] [PubMed] [Google Scholar]
- 35. Skevaki C. L., Christodoulou I., Spyridaki I. S., Tiniakou I., Georgiou V., Xepapadaki P., Kafetzis D. A., Papadopoulos N. G. (2009) Budesonide and formoterol inhibit inflammatory mediator production by bronchial epithelial cells infected with rhinovirus. Clin. Exp. Allergy 39, 1700–1710 [DOI] [PubMed] [Google Scholar]
- 36. Greenwood J., Steinman L., Zamvil S. S. (2006) Statin therapy and autoimmune disease: from protein prenylation to immunomodulation. Nat. Rev. Immunol. 6, 358–370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Amuro H., Ito T., Miyamoto R., Sugimoto H., Torii Y., Son Y., Nakamichi N., Yamazaki C., Hoshino K., Kaisho T., Ozaki Y., Inaba M., Amakawa R., Fukuhara S. (2010) Statins, inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A reductase, function as inhibitors of cellular and molecular components involved in type I interferon production. Arthritis Rheum. 62, 2073–2085 [DOI] [PubMed] [Google Scholar]
- 38. Andreakos E. (2012) Asthma exacerbations: a molecular dichotomy between antiviral and pro-inflammatory responses revealed. EMBO Mol. Med. 4, 1231–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gavala M. L., Bashir H., Gern J. E. (2013) Virus/allergen interactions in asthma. Curr. Allergy Asthma Rep. 13, 298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Miller E. K., Hernandez J. Z., Wimmenauer V., Shepherd B. E., Hijano D., Libster R., Serra M. E., Bhat N., Batalle J. P., Mohamed Y., Reynaldi A., Rodriguez A., Otello M., Pisapia N., Bugna J., Bellabarba M., Kraft D., Coviello S., Ferolla F. M., Chen A., London S. J., Siberry G. K., Williams J. V., Polack F. P. (2012) A mechanistic role for type III IFN-λ1 in asthma exacerbations mediated by human rhinoviruses. Am. J. Respir. Crit. Care Med. 185, 508–516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sykes A., Edwards M. R., Macintyre J., del Rosario A., Bakhsoliani E., Trujillo-Torralbo M. B., Kon O. M., Mallia P., McHale M., Johnston S. L. (2012) Rhinovirus 16-induced IFN-α and IFN-β are deficient in bronchoalveolar lavage cells in asthmatic patients. J. Allergy Clin. Immunol. 129, 1506.e6–1514.e6 [DOI] [PubMed] [Google Scholar]
- 42. Feng X., Han D., Kilaru B. K., Franek B. S., Niewold T. B., Reder A. T. (2012) Inhibition of interferon-β responses in multiple sclerosis immune cells associated with high-dose statins. Arch. Neurol. 69, 1303–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Jougasaki M., Ichiki T., Takenoshita Y., Setoguchi M. (2010) Statins suppress interleukin-6-induced monocyte chemo-attractant protein-1 by inhibiting Janus kinase/signal transducers and activators of transcription pathways in human vascular endothelial cells. Br. J. Pharmacol. 159, 1294–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Shuai K., Liu B. (2003) Regulation of JAK-STAT signalling in the immune system. Nat. Rev. Immunol. 3, 900–911 [DOI] [PubMed] [Google Scholar]
- 45. Cacalano N. A., Sanden D., Johnston J. A. (2001) Tyrosine-phosphorylated SOCS-3 inhibits STAT activation but binds to p120 RasGAP and activates Ras. Nat. Cell Biol. 3, 460–465 [DOI] [PubMed] [Google Scholar]
- 46. Delang L., Paeshuyse J., Vliegen I., Leyssen P., Obeid S., Durantel D., Zoulim F., Op de Beeck A., Neyts J. (2009) Statins potentiate the in vitro anti-hepatitis C virus activity of selective hepatitis C virus inhibitors and delay or prevent resistance development. Hepatology 50, 6–16 [DOI] [PubMed] [Google Scholar]
- 47. Radigan K. A., Urich D., Misharin A. V., Chiarella S. E., Soberanes S., Gonzalez A., Perlman H., Wunderink R. G., Budinger G. R., Mutlu G. M. (2012) The effect of rosuvastatin in a murine model of influenza A infection. PLoS One 7, e35788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kumaki Y., Morrey J. D., Barnard D. L. (2012) Effect of statin treatments on highly pathogenic avian influenza H5N1, seasonal and H1N1pdm09 virus infections in BALB/c mice. Future Virol. 7, 801–818 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.