

Abstract
The steady and dramatic increase in the incidence of Alzheimer's disease (AD) and the lack of effective treatments have stimulated the search for strategies to prevent or delay its onset and/or progression. Since the diagnosis of dementia requires a number of established features that are present when the disease is fully developed, but not always in the early stages, the need for a biological marker has proven to be urgent, in terms of both diagnosis and monitoring of AD. AD has been shown to affect peripheral blood mononuclear cells (PBMCs) that are a critical component of the immune system which provide defence against infection. Although studies are continuously supplying additional data that emphasize the central role of inflammation in AD, PBMCs have not been sufficiently investigated in this context. Delineating biochemical alterations in AD blood constituents may prove valuable in identifying accessible footprints that reflect degenerative processes within the Central Nervous System (CNS). In this review, we address the role of biomarkers in AD with a focus on the notion that PBMCs may serve as a peripheral laboratory to find molecular signatures that could aid in differential diagnosis with other forms of dementia and in monitoring of disease progression.
1. Introduction
The prevalence of dementia has increased globally, most noticeably in the ageing populations of the developed world. Alzheimer's disease (AD) is the most common type of dementia (60% of cases). Individuals affected by AD are 5.4 million in the United States and more than 33.9 million worldwide [1]. Moreover, AD prevalence is estimated to triple over the next 40 years and this will place a heavy burden on society and its health-care systems in terms of both economic costs and human impacts. The steady and dramatic increase in the incidence of AD and the lack of effective treatments have stimulated the search for strategies to prevent or delay its onset and/or progression.
There is general agreement that the epidemiological impact of dementia can be reduced by detecting and treating classical vascular risk factors since different studies provide evidence in favour of a coexistence of vascular and degenerative components in its pathogenesis [2].
In western countries vascular dementia (VD) is the second most common cause of dementia after AD among the elderly. A meta-analysis of the European studies on the incidence of dementia showed that VD constitutes 17.6% of all dementias [3]. In Europe and North America, AD is more common than VD in a 2 : 1 ratio; in contrast, in Japan and China VD accounts for almost 50% of all dementias. Also, the possibility of concomitant AD often confounds the relationship between cerebrovascular disease and VD.
AD is characterized by neurofibrillary tangles (NFT) and extracellular amyloid deposits. The former are composed of intraneuronal aggregates of hyperphosphorylated tau proteins and the latter are made of amyloid-beta (Aβ) peptides stemming from the sequential cleavage of a transmembrane precursor named amyloid precursor protein (APP).
Vascular pathology, namely arteriosclerosis, endothelial proliferation, and neovascularization, have been often found to be associated with NTF and amyloid plaques [4].
A number of autopsy studies have confirmed that among cases of dementia, AD-related pathology was associated with vascular lesions in nearly one-third of cases [5]. In addition, many epidemiological reports have demonstrated that the presence of vascular factors increases the risk of developing AD.
However, it is still a matter of controversy whether neurodegenerative AD-like disease and cerebrovascular lesions are coexisting but unrelated pathologies or whether they represent different results of synergistic pathogenic mechanisms.
It is hypothesized that an alteration of the neurovascular unit, which is the functional unit encompassing vascular cells, astrocytes, and perivascular neurons, is an early event in the pathogenesis of AD [6]. Dysfunction of the neurovascular unit results in impaired blood brain barrier (BBB) functions, dysregulation of cerebral blood flow, and impairment of Aβ clearance leading to an increase of oligomers and soluble Aβ forms [7]. Vascular oxidative stress and inflammation underlie many of these deleterious effects and are potential therapeutic targets even if, at present, there is no cure for AD and only a few medications aimed at slowing down memory deficits and clinical symptoms are available, with limited benefits.
Consequently, there is a pressing need for the identification of biomarkers that will aid in the differential diagnosis between AD and other forms of dementia and that will allow the detection of AD at early stages. Within the scenario of dementia, biomarker research may thus play an important role in paving the way towards novel diagnostic or therapeutic strategies.
2. Biomarkers
A biomarker is a characteristic that can be objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes, or pharmacological responses to a therapeutic intervention [8, 9]. Many tests commonly used in clinical practice are biomarkers; biochemical tests provide soluble biomarkers, whereas physiological assessment and imaging measures provide anatomical and functional biomarkers. The majority have been identified on the basis of biological insight or underlying physiology. With increasing knowledge and practical experience, many of these tests have evolved into measurable end points in clinical research, applied as indicators of change, be it for the better or for the worse [10].
Biomarkers have also gained an important role in the field of clinical management and have established a close link with bedside medicine, by providing metrics of quality in medical care alongside meaningful costing. With effective translation into many clinical guidelines, biomarkers can facilitate the delivery of evidence-based medical care [11].
The evaluation of biomarkers may aid in the identification of diseases and may also allow correlations to be made with the progression or the susceptibility to a disease or a given treatment.
Yet, single biomarkers are unlikely to capture the complex process of human pathophysiology. Thus research may need to be geared towards sets of biomarkers, reflecting different, but intercalated, processes, which may enable a better assessment of disease states.
Biomarkers can be measured, for instance, in saliva, sweat, breath, blood/serum, urine, and cerebrospinal fluid (CSF). The fact that the collection of these biological fluids is significantly less invasive than biopsies is an important practical issue when studying neurodegenerative disorders like AD [12].
It has been reported that the sensitivity (definitely positive/(definitely positive + false negative)) of an “ideal” biomarker to detect AD should be at least 85%. Similarly, the specificity (definitely negative/(definitely negative + false positive)) in differentiating AD patients from controls and from patients with other forms of dementia should be at least 75% [10].
2.1. Biomarkers and Alzheimer
Despite the enormous advances in modern medicine, the diagnosis of AD remains largely clinical, based on patient history/examination, neuropsychological testing, and imaging techniques. Unfortunately the clinical diagnosis of AD suffers from limitations in that it only allows us to speak of probable or possible AD [13] with a 93% sensitivity and a 55% specificity. Furthermore, the diagnosis becomes far more difficult in the case of early or unusual presentations of the disease.
With the expansion of current knowledge on AD and the increasing availability of technical tools, there is an emerging need for the development of accurate biochemical and imaging tests that support the diagnosis [14, 15]. In this context the diagnostic criteria for AD proposed in 2007 [14] highlight the usefulness of genetic studies since they would enable a definite diagnosis to be made based on the demonstration of mutations in any of the three genes responsible for autosomal dominant disease: the gene for APP on chromosome 21, for presenilin 1 (on chromosome 14), and for presenilin 2 (on chromosome 1).
As to the more prevalent sporadic cases of AD, the need for a biological marker has proven to be urgent, for both the diagnosis and monitoring of the disease [16, 17].
Indeed, an ideal biomarker for AD would assist in the identification of preclinical disease, early disease diagnosis, staging of disease progression, and response to treatment [18]. Early diagnosis and identification of preclinical AD are particularly important issues considering the development of underlying neuropathology in those yet to display clinical symptoms. In particular, Mild Cognitive Impairment (MCI) is a well-described prodromal state of cognitive decline preceding dementia, with an accelerated conversion to AD estimated at 10–12% per year [19].
Over the past decade, biomarker discovery has become a rapidly advancing area of AD research.
With the development of structural, functional, and molecular techniques, neuroimaging is increasingly being employed as a diagnostic and prognostic tool in AD. Quantitative magnetic resonance imaging (MRI) is used to assess neurodegenerative changes in AD, which include reducing whole brain volume and cortical thickness associated with ventricular enlargement [20]. Early degeneration is also apparent in the hippocampus, entorhinal cortex, and medial temporal lobe of AD patients relative to controls [21]. In fact, MRI-determined hippocampal atrophy is currently the most established structural biomarker for AD and has been shown to predict the conversion from MCI to AD in about 80% of cases [22]. Additional neuroimaging techniques include functional MRI (fMRI), positron emission tomography (PET), and single-photon emission computed tomography (SPECT) which reveal abnormalities in brain synaptic activity, metabolism, and perfusion, respectively. Recent advances include the development of a number of amyloid-binding compounds, the most extensively studied being 11C-PIB (Pittsburgh Compound B, PIB). Several PET studies have detected an increased uptake of PIB in AD patients, which was found to correlate with the extent of cerebral atrophy and memory impairment [23]. Notably, longitudinal studies suggest that PIB imaging is able to predict the progression from normal cognition and MCI to symptomatic AD [24].
In view of the close relationship of the CSF with the brain and spinal cord, it is believed that the composition of this fluid may reflect biochemical changes in the CNS and thus provide information on the pathological changes occurring in neurodegenerative disorders [18]. Multiple studies have examined CSF for potential AD biomarkers. It is generally recognized that AD subjects compared to age-matched controls exhibit decreased CSF levels of soluble Aβ42 and increased CSF levels of total tau and phosphotau [25]. Importantly, diagnostic accuracy is improved by using the tau/Aβ42 ratio instead of either single biomarker, and this is reflected in an increase in sensitivity and specificity to 86% and 97%, respectively [26]. Moreover, this combination also appears to predict the subsequent development of AD in both cognitively normal and MCI patients [27, 28]. These findings have thus established CSF Aβ42 and tau as the most sensitive and specific diagnostic and predictive biomarkers for AD.
It should however be remarked that although neuroimaging and CSF biomarkers seem to be the most promising, they also carry some limitations. They are generally expensive to perform routinely and lumbar puncture is invasive and often unpleasant. Moreover a large variability exists in the literature as to CSF biomarker diagnostic accuracies and cut-offs, hampering or delaying their everyday application in the clinical setting [29, 30] and their potential use as indicators of prodromal AD.
Also, it is worth noting that the process of biomarker discovery involves many critical steps including study design, sample preparation, protein and peptide separation and identification, and bioinformatics and data integration issues that must be carefully controlled before achieving independent confirmation and validation.
Lastly, patient age is an important confounding factor in these biomarker studies and could explain some of the variability in published diagnostic accuracies and cut-offs [30]. Indeed a consistent number of subjects affected by Lewy Body Dementia (LBD), Frontotemporal Lobar Degeneration (FTLD), VD, and Corticobasal Degeneration (CBD) display an AD-like CSF biomarker profile [31].
3. Inflammation and Alzheimer
In the human brain several cell types are responsible for initiating and amplifying a specific inflammatory response. In AD signs of an inflammatory activation of microglia and astroglia are present both inside and outside amyloid deposits. Cell cultures and animal models suggest an interactive relationship between inflammatory response activation, reduced neuronal functioning, and amyloid deposition. Furthermore cells associated with extracellular plaques within AD brains can produce a variety of cytokines, chemokines, and other related proteins that influence plaque and tangle formation [32].
There is strong evidence that inflammation exacerbates neuronal loss [33, 34]. In fact, local inflammatory processes can exert a direct neurotoxicity, interfere with Aβ expression and metabolism, and maintain a chronic intracerebral acute phase protein secretion, which in turn favours formation of Aβ oligomers [35].
On the other hand, microglial activation leads to an increased brain expression of major histocompatibility complex type II and an increased secretion of proinflammatory cytokines and chemokines such as interleukin-1 (IL-1), interleukin-6 (IL-6), tumour necrosis factor-α (TNF-α), and interleukin-8 (IL-8), as well as complement components and acute phase proteins [36].
A “cytokine cycle” has been proposed where [37] the anti-inflammatory cytokines (IL-4, IL-10, and IL-13) regulate Aβ-induced microglial/macrophage inflammatory responses and modify the microglial activity surrounding amyloid neuritic plaques [38]. Such cytokines can inhibit the induction of IL-1, TNF-α, and MCP-1 in differentiated human monocytes and, above all, IL-10 causes dose-dependent inhibition of the IL-6 secretion induced by Aβ in these cells and in murine microglia [37].
Accordingly, several reports make it appears that the risk of AD is substantially influenced by polymorphisms in the promoter region and other untranslated regions, of genes encoding inflammatory mediators. Alleles that favour an increased or decreased expression of inflammatory mediators are more frequent in patients with AD than in controls [39].
Aβ has also been shown to induce a phagocytic response in microglia, suggesting a neuroprotective defense mechanism [40]. This is, however, coupled to an increased release of signalling molecules and reactive oxygen and nitrogen species, which may further promote neuronal damage [41].
Despite these findings, clinical trials of nonsteroidal anti-inflammatory drugs (NSAIDs) in AD patients have been disappointing [42].
3.1. Peripheral Blood Mononuclear Cells
Nowadays it remains the need for a reliable, minimally invasive, and inexpensive biomarker for dementia, leading many to investigate peripheral blood. Blood collection is simple, inexpensive, and less invasive than lumbar puncture, allowing for repeated sampling. Approximately 500 mL of CSF is absorbed into the blood daily [43] and there is also evidence for blood-brain barrier (BBB) dysfunction in AD and other neurodegenerative disorders, which may enhance protein exchange between both fluids [44]. Consequently, the leakage of CNS metabolites into the peripheral system may reflect neurodegenerative disease status and could offer a suitable source of disease biomarkers.
AD also affects PBMCs that are defined as any blood cell with a round nucleus (i.e., lymphocytes, monocytes, or macrophages). These blood cells are a critical component of the immune system which provide defence against infection and respond to intruders. The lymphocyte population consists of CD4+ and CD8+ T cells, B cells and natural killer cells, CD14+ monocytes, basophils, neutrophils, eosinophils, and dendritic cells.
Although studies are continuously providing additional data that emphasize the central role of inflammation in AD, PBMCs have not been sufficiently investigated in this context. Indeed, only scant studies have used PBMCs to measure cytokine release, showing a significantly different production of these inflammatory components in AD and MCI subjects compared to controls [39, 45] (Figure 1) as well as a greater IL-1 and TNF-α production, associated with an increased risk of AD, in elderly controls [46].
Figure 1.
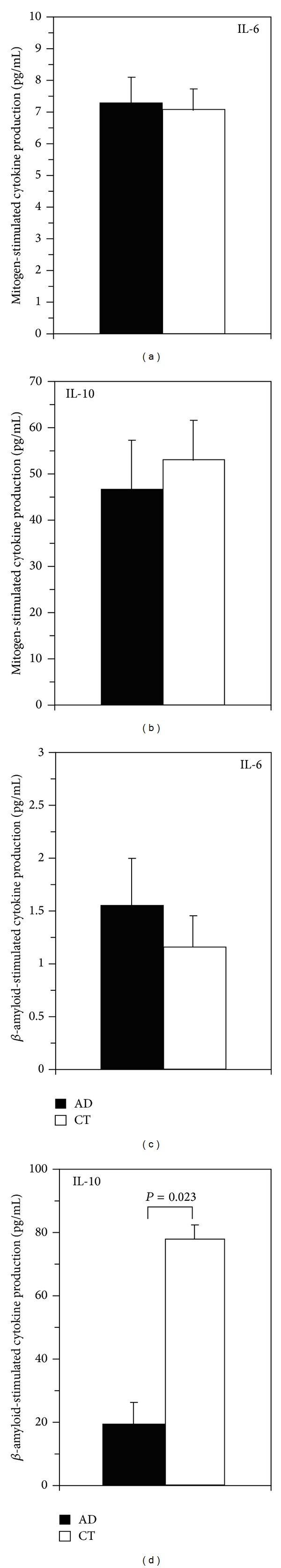
PBMCs of AD patients and age- and sex-matched controls (CT) were stimulated with a mitogen (LPS) and with a pool of three Aβ peptides (Aβ fragment 25–35; Aβ fragment 1–40; Aβ fragment 1–16). The production of IL-10 and IL-6 was measured by means of ELISA. There were no differences in mitogen-stimulated IL-6 and IL-10 production in AD and controls. In contrast, when Aβ-stimulated production of IL-6 and IL-10 was analysed, a marginally increased IL-6 production and a significantly decreased IL-10 generation were observed in AD patients compared to controls, suggesting an antigen-specific impairment in the production of these cytokines.
Delineating biochemical alterations in AD blood constituents may enable the identification of accessible footprints that mirror degenerative processes within the CNS.
Moreover PBMCs could reflect inflammatory mechanisms in a more specific way compared with the serum/plasma, and PBMC-associated biomarkers could thus provide novel insight into the pathogenesis of AD.
In the following paragraphs we discuss the potential of PBMCs to serve as a peripheral laboratory to find molecular signatures in AD that could aid both in the differential diagnosis with other forms of dementia and in the monitoring of disease progression.
4. Peptidyl-prolyl cis-/trans-Isomerase Pin1 in PBMCs
The peptidyl-prolyl cis-/trans-isomerase Pin1 is a cytosolic protein that isomerizes the peptide bond of a phosphorylated serine or a phosphorylated threonine followed by a proline (pSer-/pThr-Pro). Pin1 catalyzes the cis-/trans-isomerization of its substrates, consequently potentiating the accessibility of the phosphate residue for further dephosphorylation by protein phosphatases such as the protein phosphatase PP2A. Alternatively, the binding of Pin1 to other highly phosphorylated substrates can repress their dephosphorylation by calcineurin. Therefore, through isomerization of pSer-/pThr-Pro, Pin1 regulates the function or degradation of a growing number of proteins including transcription factors and cytoskeletal, mitotic, or proapoptotic proteins [47].
Pin1 consists of 2 functional domains. The binding domain corresponds to the amino-terminal region consisting of a group IV WW domain (Trp-Trp domain) that specifically binds to pSer-/pThr-Pro motifs. The carboxyl-terminal region is the catalytic domain [48]. Pin1 substrate-binding and isomerase activity are regulated by phosphorylation. Indeed, 3 phosphorylation sites of Pin1 have been characterized. In particular, serine 16 is located in the WW domain and is phosphorylated by protein kinase A. Phosphorylation of Pin1 at serine 16 represses substrate recognition [49]. Pin1 has several additional putative phosphorylation sites (e.g., human Pin1 has 29 residues of serine or threonine and 3 tyrosines).
Phosphorylation of proteins is a key signalling mechanism in diverse of physiological and pathological processes. Pin1-catalysed conformational changes can have profound effects on phosphorylation signalling by regulating a spectrum of target activities. Interestingly, Pin1 deregulation is implicated in a number of conditions, notably ageing and age-related diseases, including cancer and AD. Pin1 is overexpressed in most human cancers; it activates numerous oncogenes or growth enhancers and also inactivates a large number of tumour suppressors or growth inhibitors. By contrast, ablation of Pin1 prevents cancer but eventually leads to premature ageing and neurodegeneration. Recent studies have demonstrated the reemergence within the brain of cell cycle proteins as patients progress from MCI into AD. Pin1 plays an important role in regulating the activity of key proteins, such as CDK5, GSK3-β, and PP2A, that are involved not only in the cell cycle but also in the phosphorylation state of Tau [50].
Indeed, Pin1 facilitates tau dephosphorylation [51] and regulates APP metabolism, thus providing additional support to the hypothesis that it has a neuroprotective function against AD [52–56].
It has been reported that Pin1 activity is repressed by oxidation in AD [52–58] and that Pin1 is localized to granular vesicles in AD and FTD but not to tau aggregates [55, 59–63].
It should be remarked that the expression and activity of Pin1 are tightly regulated at a transcriptional level and that a Pin1 gene polymorphism (−842G/C) has been found to be associated with reduced levels of Pin1 in blood cells and with an increased risk for AD in an Italian cohort [64].
Interestingly, a depletion of the soluble form of Pin1 has been described in neurons from AD subjects [57, 65] and differences in Pin 1 molecular and biochemical parameters have been reported in PBMCs from late-onset AD (LOAD) compared with control subjects [66].
In particular, in PBMCs from LOAD we observed a significant increase in Pin1 gene expression together with a significant decrease in gene promoter methylation [66].
This latter finding holds particular relevance, since so far little is known about epigenetic patterns in AD. Moreover, epigenetic mechanisms have already been proposed as markers of AD in PBMC-derived DNA [67] and it has been claimed that DNA methylation in peripheral cells could be taken as a model of epigenetic gene regulation in the brain [68].
We have also shown that in LOAD subjects Ser16 phosphorylation levels of Pin1 were lower than in controls (Figure 2).
Figure 2.
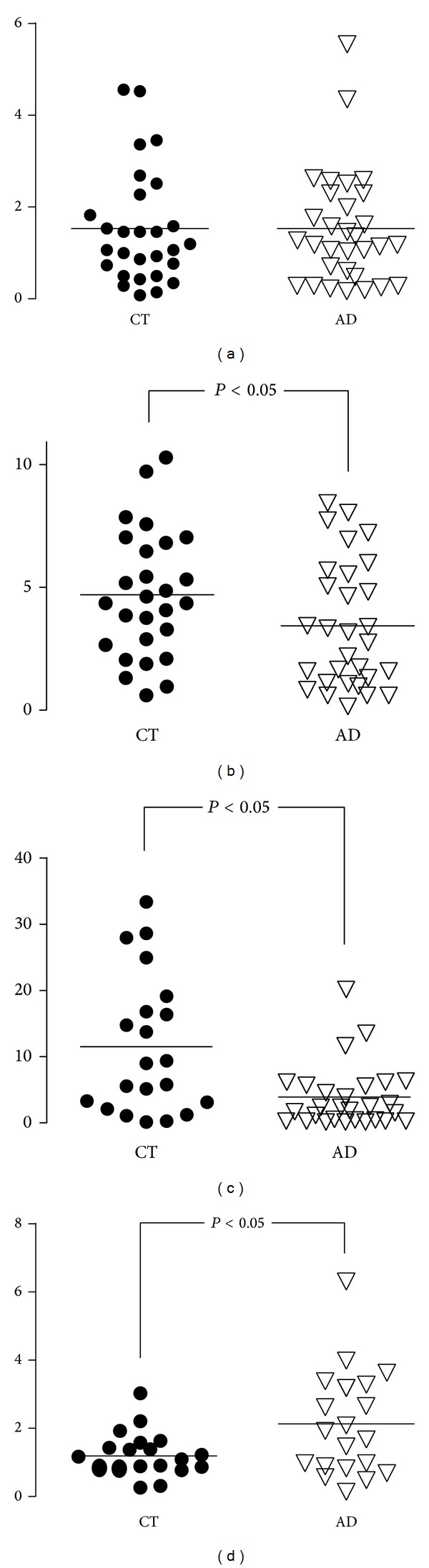
Scatter dot plots showing the distributions of molecular and biochemical parameters of PBMCs from controls (CT) and LOAD: activity (a), Ser16 phosphorylation (b), methylation (c), and gene expression (d). The lines across the boxes indicate median values.
Phosphorylation of Pin1 must therefore be a key factor in regulating its localization, function, and metabolism and tau seems to be involved in controlling the balance between the phosphorylation/dephosphorylation of Pin1 in brain cellular lysate [69].
Moreover, Wang et al. [70] suggested that reduced Pin1 activity in the frontal cortex of patients with MCI contributes to the initial accumulation of hyperphosphorylated tau and is then followed, in a more advanced stage of the disease, by a compensatory upregulation of the Pin1 gene that counteracts Aβ plaque formation.
In particular, with regard to our finding of lower Ser16 phosphorylation levels of Pin1 in LOAD subjects relative to controls, different interpretations can be put forward: the presence in LOAD patients of rare gene variants of Pin1 that could influence its phosphorylation state [71] and the effects on Pin1 of a higher blood concentration of Aβ42 [72]. In keeping with the latter hypothesis, in rat hippocampal cells, treatment with Aβ42 oligomers has been shown to promote a transient Pin1 dephosphorylation on Ser16 associated with a decrease in phosphorylated TauThr231 [73]. Whatever the specific explanation is, the modifications of Pin1 observed in LOAD subjects make it reasonable to suppose that Pin1 is involved in AD [74] and that epigenetic mechanisms (i.e., Pin1 promoter methylation) play a role in the disease. Therefore, alterations in easily accessible peripheral cells may prove to be valuable biomarkers in the diagnosis and follow-up of AD and, potentially, also of some tauopathies.
5. Epigenetics
Literally meaning “above the genome” the epigenome comprises the heritable changes in gene expression that occur in the absence of changes to the DNA sequence itself. Epigenetic mechanisms include chromatin folding and attachment to the nuclear matrix, packaging of DNA around nucleosomes, covalent modifications of histone tails, and DNA methylation in the whole genome and/or in specific gene promoters [75].
DNA methylation, in particular, consists of the transfer of a methyl group to position 5 of the cytosine pyrimidine ring of a cytosine guanine dinucleotide (CpG), which ultimately blocks the binding of transcription factors causing chromatin compaction and gene silencing [76].
The influence of regulatory small RNAs and microRNAs on gene transcription is also increasingly recognized as a key mechanism of epigenetic gene regulation [77].
Indeed, microRNAs (miRNAs), small regulatory RNAs in cells, probably constitute one of the most investigated extracellular RNAs in body fluidsand the levels of certain miRNAs in the circulation correlate well with different pathological conditions (i.e., miR-499 and miR-1 are associated with cardiovascular conditions) [78–81].
Epigenetic mechanisms are important in cell growth and differentiation [82]. Epigenetic change can be stochastic [83] or internally orchestrated as part of ageing [84]. Longitudinal changes in global and gene-specific DNA methylation clusters within families suggest there is a genetic control to methylation status [85].
Epigenetics is destined to change across the lifespan. In fact a loss of global DNA methylation and promoter hypermethylation of several specific genes occurs during ageing.
In particular ageing-associated DNA hypermethylation occurs predominantly in genes involved in the development of anatomical structures, organs, and multicellular organisms and in the regulation of transcription.
This phenomenon may be considered a new aspect of the age remodeling process, a continuous adaptation of the body to the deteriorative changes occurring over time. However, it is not clear how relevant these epigenetic changes are in the context of functional changes in gene expression [86].
Inappropriate epigenetic changes are associated with many diseases including cancer [87], Rett syndrome [88], Beckwith-Wiedemann syndrome [89], and other imprinting disorders.
Environmental signals can trigger epigenetic responses and may be an important mechanism by which environmental exposures are associated with disease [90]. Furthermore, epigenetic mechanisms may play an important role in the developmental origins of adult health and disease by providing a mechanism underlying the latent effects of adverse fetal, infant, and childhood environments on late-life chronic disease [91–93].
5.1. Epigenetic Epidemiology and Alzheimer's Disease
Epigenetic epidemiology is the study of the effects of heritable epigenetic changes on the occurrence and distribution of diseases in populations [94]. This research includes both transgenerational and intraindividual cellular epigenetic inheritance systems. Epigenetic changes are associated not only with ageing [95, 96], but also with psychiatric outcomes [97, 98] and neurodegeneration [99].
Evidence for the role of epigenetics in AD pathogenesis can be found in human studies of various tissues, in animal models, and in cell cultures [100–102]. Global changes associated with AD have been observed in DNA methylation, miRNAs, and histone modifications.
Discordant data have been reported on specific epigenetic modifications of tau- and amyloid-processing genes. On the one hand an altered regulation was reported across multiple brain regions [103–105], and on the other hand no differences were seen in DNA methylation in regions associated with MAPT, PSEN1, and APP [103].
Human postmortem case-control studies have demonstrated global hypomethylation in the entorhinal cortex of AD subjects [106] and in the temporal neocortex of an AD monozygotic twin relative to the cognitively normal twin [107].
An AD case-control study in the postmortem human parietal lobe cortex has revealed a differential regulation of several miRNAs, including miR-204, miR-211, and miR-44691 [108].
Age-matched AD cases have been found to exhibit an increased neuronal global phosphorylation of histone 3 relative to controls, as determined by immunolabeling in the hippocampus, and such histone modification suggests mitotic activation [109].
In experiments where neuroblastoma cells were cultured under low folate and vitamin B12 conditions, PSEN1 and BACE1 were hypomethylated, mRNA expression of BACE1 and PSEN1 was significantly induced, and Aβ production was increased [110].
An additional study using human neuroblastoma cells and male rat brain tissue reports that APP mRNA expression is repressed by thyroid hormone (T3) sensitive histone modifications [111].
5.2. Epigenetics in PBMCs
The study of gene regulation in blood cells from living patients offers the possibility to go through the whole history of the disorder (including the response to pharmacological, metabolic, and environmental events) in a more comprehensive perspective, compared to postmortem studies which allow only pinpoint assessment.
It is important to note that PBMCs may also be a useful model of epigenetic gene regulation in the brain [68]. In fact, it has been shown that PBMCs share much of the nonsynaptic biochemical environment of neurons and contain the full complement of epigenetic enzymes found in most tissues, including neurons and peripheral nucleated cells [112, 113].
For instance, our group has investigated the role of DNA methylation in the PBMCS from LOAD subjects compared to controls and has demonstrated an altered Pin1 gene expression and promoter methylation [66], as detailed above, along with changes in fatty acid amide hydrolase (FAAH) and 5-lipoxygenases (5-LOX) genes (Faah EC 3.5.1.99 and Alox5 EC 1.13.11.34), proteins, and activity [114].
Also, by comparing DNA methylation of Faah and Alox5 promoters we found a direct correlation between these two genes [114, 115].
It has been shown that oxygenation of the FAAH substrates by lipoxygenase activity modulates recognition of these molecules by their protein targets [116], with potential implications for their biological activity [117].
These results might suggest that a parallel increase of FAAH and 5-LOX expression in AD patients could evoke a sustained inflammatory condition, thus reinforcing neurodegeneration [114, 115].
This finding in peripheral cells is in agreement with previous results in postmortem AD brains [118], where FAAH protein upregulation within plaques was suggested to lead to an increase in metabolites from endocannabinoid anandamide (AEA) degradation (such as arachidonic acid). Such metabolites could contribute to the inflammatory process occurring in AD.
Recently, there has been considerable interest in exploring the therapeutic potential of anti-inflammatory agents to prevent, treat, or slow down the progression of AD [119]. However, nonsteroidal anti-inflammatory drugs were found to be ineffective in AD patients with mild to moderate cognitive impairment [120], emphasizing the importance of an early diagnosis and therapy. Furthermore, pharmacological interventions based on chronic treatment with COX inhibitors, or treatment with anticytokine therapies, are not ideal for a long-term use, due to their gastrointestinal (COX1-selective inhibitors), cardiovascular (COX2-selective inhibitors), and immunosuppressive (anticytokine therapies) side effects [121].
Taken together, these lines of research converge towards the notion that novel anti-inflammatory targets may provide a safer strategy for the prevention and the treatment of AD. In such scenario PBMCs stand out as potential peripheral markers of disease within the CNS.
6. Adenosine A2A Receptors in PBMCs
Nutritional alterations have been linked to the epigenetic modulation of some AD-related genes and seem to play a role in AD pathology. There is also evidence in favour of the epigenetic modulation of genes involved in the pathways activated by some dietary factors, both in ageing and disease, further supporting the involvement of epigenetic mechanisms in AD. A number of dietary elements have been reported to be either risk or protective factors for the development of AD. These include fat, fatty acids, antioxidants, fish, vitamins, alcohol, and, more recently, caffeine [122].
The neuroprotective effect of caffeine consumption on AD pathology is currently emerging from both basic and epidemiological studies [123]. In vitro and animal studies have provided convincing data on caffeine's neuroprotective effects against and in the presence of AD pathology [124–126]. Human studies have begun to demonstrate the presence of a similar neuroprotective role in the ageing and demented population.
However, due to the conflicting results from some longitudinal studies, there is no consensus about the role of caffeine in the onset of AD [124–128].
Caffeine is one of the most consumed psychoactive drugs and acts mostly by blocking adenosine receptors [129]. The purine ribonucleoside adenosine (Ado) is a naturally occurring metabolite that is ubiquitously distributed throughout the body as a metabolic intermediary. Intra- and extracellular Ado levels rise in response to physiological stimuli and with metabolic/energetic perturbations, inflammatory challenges, and tissue injury [130, 131].
The physiological responses to Ado take place as a result of the binding and activation of different transmembrane receptors: the high-affinity A1 and A2A (A2A) receptors, the low-affinity A2B receptor, or the low-abundance A3 receptor [132].
These receptors are G-protein coupled receptors that regulate, in opposite directions, the second messenger cAMP; while A1 is inhibitory Gi-coupled, A2A is excitatory Gs-coupled, thereby decreasing and increasing cAMP levels, respectively [133]. The activation of these receptors is also able to modulate Ca2+ channels and the phospholipase C pathway. Through these actions and by modulating both the release and the uptake of different neurotransmitters, the balance between the activation of adenosine A1 and A2A receptors allows the fine tuning of synaptic transmission and plasticity in the hippocampus [134].
In particular A2A is present in a wide variety of tissues, including the nervous system and the peripheral immune system, where they display different levels of expression: significant levels in neurons and peripheral cells (lymphocytes and neutrophils) and lower levels in glial cells [132]. The different levels of expression of A2A in different tissues are consistent with the sophisticated, multifaceted neurochemical, and molecular effects of the Ado system. On the basis of in vitro [135, 136] and in vivo [137] studies, it has become clear that A2A, through complex mechanisms which are still poorly understood [138–141], plays a critical role in the modulation of inflammatory reactions, influencing functional outcome in a broad spectrum of pathologies including neurodegeneration [142, 143].
Moreover it has been demonstrated that A2A is able to prevent Aβ-induced synaptotoxicity in animal models and cell cultures [144] and it has been shown to control NMDA currents and glutamate outflow in the hippocampus [145, 146].
Contrasting data have been reported so far on the beneficial/detrimental effects of A2A on brain cells [147]. The blockade of A2A alleviates the long-term burden of brain disorders such as ischaemia, epilepsy, Parkinson's disease, or AD [138, 145, 148, 149]. On the other hand, agonists of A2A can protect the CNS against several insults, including ischemia and excitotoxins [143, 150].
In the periphery A2A contributes to coronary endothelial dilatation in mice [151], can inhibit endothelial apoptosis [152], and preserves vascular reactivity following hemorrhagic shock in rats [153].
We recently investigated A2A gene expression and density in the PBMCs of patients with amnestic MCI (a-MCI), multiple cognitive domain MCI (mcd-MCI), outright AD, VD, and controls. We found that A2A expression is upregulated in the peripheral cells of a-MCI but not AD subjects, supporting an involvement of the Ado system in the early stages of AD [154]. We also showed that A2A expression is lower in the PBMCs of subjects with VD than AD, highlighting its possible relevance as a biomarker that may help differentiate two forms of dementia that are often closely associated (Figure 3).
Figure 3.
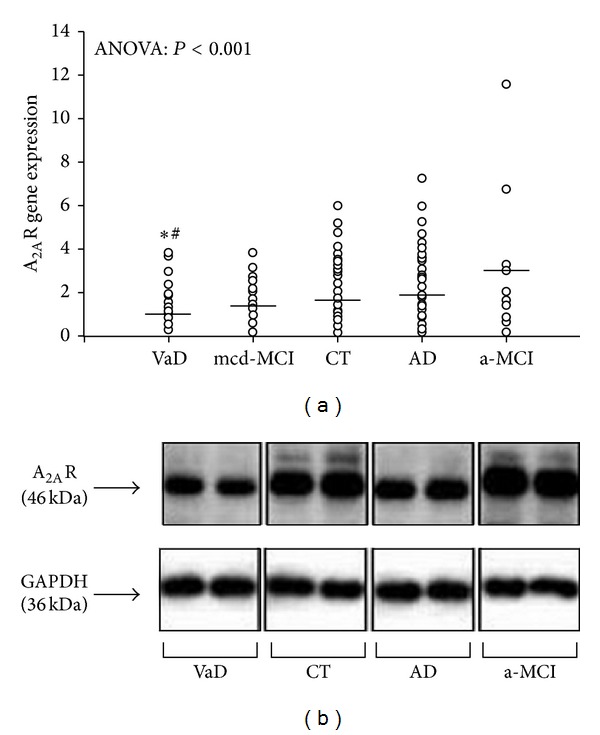
(a) Scatter plot of A2A gene expression in PBMCs from VD, mcd-MCI, controls (CT), AD, and a-MCI subjects (the lines represent the mean value for each group). *P < 0.001 versus AD; # P < 0.05 versus a-MCI. (b) Representative picture of the western blot analysis of the A2A densities in PBMCs extracts, running in duplicate, from one subject from the VD, CT, AD, and a-MCI groups, respectively.
Indeed, ROC analysis data showed that A2A possesses a moderate degree of sensitivity and specificity for identifying VD patients from a heterogeneous group composed of VD and AD patients. The lower A2A expression in VD compared to AD subjects seems to suggest a differential role of the Ado system in these dementias [155].
The methylation of the ADORA2A promoter gene, which codes for A2A, may explain its different expression in these pathological conditions as well as in the ageing process, as already mentioned [156].
Moreover, A2A represents the main Ado receptor involved in inflammation and it is interesting to note that also other inflammatory biomarkers are differently expressed in VD and AD subjects, such as alpha1-globulin and alpha2-globulin in the serum [157] and C3a and C4a in the CSF [158].
On the other hand the decreased A2A levels in VD could be a defence mechanism since it has been demonstrated that pharmacologic inactivation or genetic deletion of A2AR reduces neuronal injury after global and focal cerebral ischemia in many animal models [149, 159, 160]. From our results it can be concluded that A2A may play an important but differential role in both types of dementia: its upregulation in the preclinical stages of AD could counterbalance the existing inflammatory state and its downregulation in VD could reflect the effects of A2A on the brain vasculature [161]. It can therefore be suggested that A2A could serve as a biomarker in the differential diagnosis between VD and AD.
7. Conclusions
Peripheral cells and in particular PBMCs seem to directly participate to neurodegenerative processes. They play critical roles in immune response, metabolism, and communication with other cells as already pointed out many years ago [162]. Moreover, PBMCs have been shown to share much of the nonsynaptic biochemical environment of neurons and contain the full complement of epigenetic enzymes and machinery, which are found in both neurons and peripheral nucleated cells, as in most other tissues.
The substantial evidence in favour of the notion that PBMCs provide a window into the CNS holds particular relevance in neurodegenerative disorders in which, unlike most other diseases, the affected tissue is not directly accessible to evaluation. On a final note, it should be mentioned that the value of biochemical dysfunctions in PBMCs as mirrors of CNS defects appears to extend well beyond dementia.For instance, FAAH and other elements of the endocannabinoid system show alterations in the blood that resemble those within the CNS in a broad spectrum of clinical conditions including Parkinson's and Huntington's disease, multiple sclerosis, schizophrenia, minor depression, and headache [163].
Nowadays we do not know if PBMCs biomarkers are better or worse than the CSF biomarkers. Our study is only a preliminary study, instead multiple studies have examined CSF to establish sensitivity and specificity of CSF biomarkers. Moreover, despite these many studies, a large variability exists in the literature as to CSF biomarker diagnostic accuracies and cut-offs. As biomarker discovery in PBMCs is an ongoing process and PBMCs biomarkers are still immature, we need further analysis to enlarge design population.
It will be also of relevance the possibility to utilize intracellular biomarkers in specific blood cell subpopulations. In fact the differences in between subjects could also be due to different composition of their PBMCs pools, even if separating PBMCs into subpopulations would not permit the cell-cell interactions required for activation of lymphocytes.
Finally, we assume that the combination of peripheral and CSF markers may be utilized to categorize patients since early stages of dementia and to understand mechanisms underlying dementia.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. The Lancet Neurology. 2011;10(9):819–828. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorelick PB, Scuteri A, Black SE, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42(9):2672–2713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fratiglioni L, Launer LJ, Andersen K, et al. Incidence of dementia and major subtypes in Europe: a collaborative study of population-based cohorts. Neurology. 2000;54(supplement 11):S10–S15. [PubMed] [Google Scholar]
- 4.Battistin L, Cagnin A. Vascular cognitive disorder. A biological and clinical overview. Neurochemical Research. 2010;35(12):1933–1938. doi: 10.1007/s11064-010-0346-5. [DOI] [PubMed] [Google Scholar]
- 5.Jellinger KA. The enigma of vascular cognitive disorder and vascular dementia. Acta Neuropathologica. 2007;113(4):349–388. doi: 10.1007/s00401-006-0185-2. [DOI] [PubMed] [Google Scholar]
- 6.Iadecola C. Neurovascular regulation in the normal brain and in Alzheimer’s disease. Nature Reviews Neuroscience. 2004;5(5):347–360. doi: 10.1038/nrn1387. [DOI] [PubMed] [Google Scholar]
- 7.Zlokovic BV. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurotherapeutics. 2008;5(3):409–414. doi: 10.1016/j.nurt.2008.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Atkinson J, Colburn WA, DeGruttola VG, et al. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clinical Pharmacology and Therapeutics. 2001;69(3):89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 9.Tardif J-C, Heinonen T, Orloff D, Libby P. Vascular biomarkers and surrogates in cardiovascular disease. Circulation. 2006;113(25):2936–2942. doi: 10.1161/CIRCULATIONAHA.105.598987. [DOI] [PubMed] [Google Scholar]
- 10.Puntmann VO. How-to guide on biomarkers: biomarker definitions, validation and applications with examples from cardiovascular disease. Postgraduate Medical Journal. 2009;85(1008):538–545. doi: 10.1136/pgmj.2008.073759. [DOI] [PubMed] [Google Scholar]
- 11.Wade A. Derivation versus validation. Archives of Disease in Childhood. 2000;83(6):459–460. doi: 10.1136/adc.83.6.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lausted C, Lee I, Zhou Y, et al. Systems approach to neurodegenerative disease biomarker discovery. Annual Review of Pharmacology and Toxicology. 2014;54:457–481. doi: 10.1146/annurev-pharmtox-011613-135928. [DOI] [PubMed] [Google Scholar]
- 13.McKhann G, Drachman D, Folstein M. Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s disease. Neurology. 1984;34(7):939–944. doi: 10.1212/wnl.34.7.939. [DOI] [PubMed] [Google Scholar]
- 14.Dubois B, Feldman HH, Jacova C, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS-ADRDA criteria. The Lancet Neurology. 2007;6(8):734–746. doi: 10.1016/S1474-4422(07)70178-3. [DOI] [PubMed] [Google Scholar]
- 15.Wiltfang J, Esselmann H, Bibl M, et al. Amyloid β peptide ratio 42/40 but not Aβ42 correlates with phospho-Tau in patients with low- and high-CSF Aβ40 load. Journal of Neurochemistry. 2007;101(4):1053–1059. doi: 10.1111/j.1471-4159.2006.04404.x. [DOI] [PubMed] [Google Scholar]
- 16.Farias G, Perez P, Slachevsky A, Maccioni RB. Platelet tau pattern correlates with cognitive status in Alzheimer's disease. Journal of Alzheimer's Disease. 2012;31(1):65–69. doi: 10.3233/JAD-2012-120304. [DOI] [PubMed] [Google Scholar]
- 17.Neumann K, Farías G, Slachevsky A, Perez P, MacCioni RB. Human platelets tau: a potential peripheral marker for Alzheimer’s disease. Journal of Alzheimer’s Disease. 2011;25(1):103–109. doi: 10.3233/JAD-2011-101641. [DOI] [PubMed] [Google Scholar]
- 18.Hooper C, Lovestone S, Sainz-Fuertes R. Alzheimer’s disease, diagnosis and the need for biomarkers. Biomarker Insights. 2008;2008(3):317–323. doi: 10.4137/bmi.s682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Petersen RC, Smith GE, Waring SC, Ivnik RJ, Tangalos EG, Kokmen E. Mild cognitive impairment: clinical characterization and outcome. Archives of Neurology. 1999;56(3):303–308. doi: 10.1001/archneur.56.3.303. [DOI] [PubMed] [Google Scholar]
- 20.Simmons A, Westman E, Muehlboeck S, et al. The AddNeuroMed framework for multi-centre MRI assessment of Alzheimer’s disease: experience from the first 24 months. International Journal of Geriatric Psychiatry. 2011;26(1):75–82. doi: 10.1002/gps.2491. [DOI] [PubMed] [Google Scholar]
- 21.de Leon MJ, Desanti S, Zinkowski R, et al. MRI and CSF studies in the early diagnosis of Alzheimer’s disease. Journal of Internal Medicine. 2004;256(3):205–223. doi: 10.1111/j.1365-2796.2004.01381.x. [DOI] [PubMed] [Google Scholar]
- 22.Hampel H, Bürger K, Teipel SJ, Bokde ALW, Zetterberg H, Blennow K. Core candidate neurochemical and imaging biomarkers of Alzheimer’s disease. Alzheimer’s and Dementia. 2008;4(1):38–48. doi: 10.1016/j.jalz.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 23.Craig-Schapiro R, Fagan AM, Holtzman DM. Biomarkers of Alzheimer’s disease. Neurobiology of Disease. 2009;35(2):128–140. doi: 10.1016/j.nbd.2008.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris JC, Roe CM, Grant EA, et al. Pittsburgh compound B imaging and prediction of progression from cognitive normality to symptomatic Alzheimer disease. Archives of Neurology. 2009;66(12):1469–1475. doi: 10.1001/archneurol.2009.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Perrin RJ, Fagan AM, Holtzman DM. Multimodal techniques for diagnosis and prognosis of Alzheimer’s disease. Nature. 2009;461(7266):916–922. doi: 10.1038/nature08538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maddalena A, Papassotiropoulos A, Müller-Tillmanns B, et al. Biochemical diagnosis of Alzheimer disease by measuring the cerebrospinal fluid ratio of phosphorylated tau protein to β-amyloid peptide42. Archives of Neurology. 2003;60(9):1202–1206. doi: 10.1001/archneur.60.9.1202. [DOI] [PubMed] [Google Scholar]
- 27.de Meyer G, Shapiro F, Vanderstichele H, et al. Diagnosis-independent Alzheimer disease biomarker signature in cognitively normal elderly people. Archives of Neurology. 2010;67(8):949–956. doi: 10.1001/archneurol.2010.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Visser PJ, Verhey F, Knol DL, et al. Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. The Lancet Neurology. 2009;8(7):619–627. doi: 10.1016/S1474-4422(09)70139-5. [DOI] [PubMed] [Google Scholar]
- 29.Ingelson M, Blomberg M, Benedikz E, et al. Tau immunoreactivity detected in human plasma, but no obvious increase in dementia. Dementia and Geriatric Cognitive Disorders. 1999;10(6):442–445. doi: 10.1159/000017187. [DOI] [PubMed] [Google Scholar]
- 30.Mattsson N, Roseń E, Hansson O, et al. Age and diagnostic performance of Alzheimer disease CSF biomarkers. Neurology. 2012;78(7):468–476. doi: 10.1212/WNL.0b013e3182477eed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoonenboom NSM, Reesink FE, Verwey NA, et al. Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology. 2012;78(1):47–54. doi: 10.1212/WNL.0b013e31823ed0f0. [DOI] [PubMed] [Google Scholar]
- 32.Hüll M, Lieb K, Fiebich BL. Pathways of inflammatory activation in Alzheimer’s disease: potential targets for disease modifying drugs. Current Medicinal Chemistry. 2002;9(1):83–88. doi: 10.2174/0929867023371292. [DOI] [PubMed] [Google Scholar]
- 33.McGeer EG, McGeer PL. Innate immunity in Alzheimer’s disease: a model for local inflammatory reactions. Molecular Interventions. 2001;1(1):22–29. [PubMed] [Google Scholar]
- 34.McGeer EG, McGeer PL. Chronic inflammation in Alzheimer’s disease offers therapeutic opportunities. Expert Review of Neurotherapeutics. 2001;1(1):53–60. doi: 10.1586/14737175.1.1.53. [DOI] [PubMed] [Google Scholar]
- 35.Licastro F, Chiappelli M. Brain immune responses cognitive decline and dementia: relationship with phenotype expression and genetic background. Mechanisms of Ageing and Development. 2003;124(4):539–548. doi: 10.1016/s0047-6374(03)00034-4. [DOI] [PubMed] [Google Scholar]
- 36.Rogers J, Lue L-F. Microglial chemotaxis, activation, and phagocytosis of amyloid β-peptide as linked phenomena in Alzheimer’s disease. Neurochemistry International. 2001;39(5-6):333–340. doi: 10.1016/s0197-0186(01)00040-7. [DOI] [PubMed] [Google Scholar]
- 37.Szczepanik AM, Funes S, Petko W, Ringheim GE. IL-4, IL-10 and IL-13 modulate Aβ(1-42)-induced cytokine and chemokine production in primary murine microglia and a human monocyte cell line. Journal of Neuroimmunology. 2001;113(1):49–62. doi: 10.1016/s0165-5728(00)00404-5. [DOI] [PubMed] [Google Scholar]
- 38.Chao CC, Molitor TW, Hu S. Neuroprotective role of IL-4 against activated microglia. Journal of Immunology. 1993;151(3):1473–1481. [PubMed] [Google Scholar]
- 39.Arosio B, Trabattoni D, Galimberti L, et al. Interleukin-10 and interleukin-6 gene polymorphisms as risk factors for Alzheimer’s disease. Neurobiology of Aging. 2004;25(8):1009–1015. doi: 10.1016/j.neurobiolaging.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 40.Tuppo EE, Arias HR. The role of inflammation in Alzheimer’s disease. International Journal of Biochemistry and Cell Biology. 2005;37(2):289–305. doi: 10.1016/j.biocel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 41.Holmes C, Butchart J. Systemic inflammation and Alzheimer’s disease. Biochemical Society Transactions. 2011;39(4):898–901. doi: 10.1042/BST0390898. [DOI] [PubMed] [Google Scholar]
- 42.McGeer PL, McGeer EG. NSAIDs and Alzheimer disease: epidemiological, animal model and clinical studies. Neurobiology of Aging. 2007;28(5):639–647. doi: 10.1016/j.neurobiolaging.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 43.Hye A, Lynham S, Thambisetty M, et al. Proteome-based plasma biomarkers for Alzheimer’s disease. Brain. 2006;129, part 11:3042–3050. doi: 10.1093/brain/awl279. [DOI] [PubMed] [Google Scholar]
- 44.Begley DJ. Delivery of therapeutic agents to the central nervous system: the problems and the possibilities. Pharmacology and Therapeutics. 2004;104(1):29–45. doi: 10.1016/j.pharmthera.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 45.Guerreiro RJ, Santana I, Brás JM, Santiago B, Paiva A, Oliveira C. Peripheral inflammatory cytokines as biomarkers in Alzheimer’s disease and mild cognitive impairment. Neurodegenerative Diseases. 2007;4(6):406–412. doi: 10.1159/000107700. [DOI] [PubMed] [Google Scholar]
- 46.Tan ZS, Beiser AS, Vasan RS, et al. Inflammatory markers and the risk of Alzheimer disease: the Framingham study. Neurology. 2007;68(22):1902–1908. doi: 10.1212/01.wnl.0000263217.36439.da. [DOI] [PubMed] [Google Scholar]
- 47.Rudrabhatla P, Pant HC. Phosphorylation-specific peptidyl-prolyl isomerization of neuronal cytoskeletal proteins by pin1: implications for therapeutics in neurodegeneration. Journal of Alzheimer’s Disease. 2010;19(2):389–403. doi: 10.3233/JAD-2010-1243. [DOI] [PubMed] [Google Scholar]
- 48.Wulf G, Finn G, Suizu F, Lu KP. Phosphorylation-specific prolyl isomerization: is there an underlying theme? Nature Cell Biology. 2005;7(5):435–441. doi: 10.1038/ncb0505-435. [DOI] [PubMed] [Google Scholar]
- 49.Lu P-J, Zhou XZ, Liou Y-C, Noel JP, Lu KP. Critical role of WW domain phosphorylation in regulating phosphoserine binding activity and Pin1 function. Journal of Biological Chemistry. 2002;277(4):2381–2384. doi: 10.1074/jbc.C100228200. [DOI] [PubMed] [Google Scholar]
- 50.Pastorino L, Ma SL, Balastik M, et al. Alzheimer's disease-related loss of Pin1 function influences the intracellular localization and the processing of AbetaPP . Journal of Alzheimer's Disease. 2012;30(2):277–297. doi: 10.3233/JAD-2012-111259. [DOI] [PubMed] [Google Scholar]
- 51.Zhou XZ, Kops O, Werner A, et al. Pin1-dependent prolyl isomerization regulates dephosphorylation of Cdc25C and Tau proteins. Molecular Cell. 2000;6(4):873–883. doi: 10.1016/s1097-2765(05)00083-3. [DOI] [PubMed] [Google Scholar]
- 52.Galas M-C, Dourlen P, Bégard S, et al. The peptidylprolyl cis/trans-isomerase Pin1 modulates stress-induced dephosphorylation of Tau in neurons: implication in a pathological mechanism related to Alzheimer disease. Journal of Biological Chemistry. 2006;281(28):19296–19304. doi: 10.1074/jbc.M601849200. [DOI] [PubMed] [Google Scholar]
- 53.Hamdane M, Dourlen P, Bretteville A, et al. Pin1 allows for differential Tau dephosphorylation in neuronal cells. Molecular and Cellular Neuroscience. 2006;32(1-2):155–160. doi: 10.1016/j.mcn.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 54.Liou Y-C, Sun A, Ryo A, et al. Role of the prolyl isomerase Pin1 in protecting against age-dependent neurodegeneration. Nature. 2003;424(6948):556–561. doi: 10.1038/nature01832. [DOI] [PubMed] [Google Scholar]
- 55.Lu P-J, Wulf G, Zhou XZ, Davies P, Lu KP. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature. 1999;399(6738):784–788. doi: 10.1038/21650. [DOI] [PubMed] [Google Scholar]
- 56.Pastorino L, Sun A, Lu PJ, et al. The prolyl isomerase Pin1 regulates amyloid precursor protein processing and amyloid-beta production. Nature. 2006;440(7083):528–534. doi: 10.1038/nature04543. [DOI] [PubMed] [Google Scholar]
- 57.Butterfield DA, Abdul HM, Opii W, et al. Pin1 in Alzheimer’s disease. Journal of Neurochemistry. 2006;98(6):1697–1706. doi: 10.1111/j.1471-4159.2006.03995.x. [DOI] [PubMed] [Google Scholar]
- 58.Sultana R, Boyd-Kimball D, Poon HF, et al. Oxidative modification and down-regulation of Pin1 in Alzheimer’s disease hippocampus: a redox proteomics analysis. Neurobiology of Aging. 2006;27(7):918–925. doi: 10.1016/j.neurobiolaging.2005.05.005. [DOI] [PubMed] [Google Scholar]
- 59.Dakson A, Yokota O, Esiri M, et al. Granular expression of prolyl-peptidyl isomerase PIN1 is a constant and specific feature of Alzheimer’s disease pathology and is independent of tau, Aβ and TDP-43 pathology. Acta Neuropathologica. 2011;121(5):635–649. doi: 10.1007/s00401-011-0798-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Holzer M, Gärtner U, Stöbe A, et al. Inverse association of Pin1 and tau accumulation in Alzheimer’s disease hippocampus. Acta Neuropathologica. 2002;104(5):471–481. doi: 10.1007/s00401-002-0581-1. [DOI] [PubMed] [Google Scholar]
- 61.Ramakrishnan P, Dickson DW, Davies P. Pin1 colocalization with phosphorylated tau in Alzheimer’s disease and other tauopathies. Neurobiology of Disease. 2003;14(2):251–564. doi: 10.1016/s0969-9961(03)00109-8. [DOI] [PubMed] [Google Scholar]
- 62.Thorpe JR, Morley SJ, Rulten SL. Utilizing the peptidyl-prolyl cis-trans isomerase Pin1 as a probe of its phosphorylated target proteins: examples of binding to nuclear proteins in a human kidney cell line and to tau in Alzheimer’s diseased brain. Journal of Histochemistry and Cytochemistry. 2001;49(1):97–108. doi: 10.1177/002215540104900110. [DOI] [PubMed] [Google Scholar]
- 63.Thorpe JR, Mosaheb S, Hashemzadeh-Bonehi L, et al. Shortfalls in the peptidyl-prolyl cis-trans isomerase protein Pin1 in neurons are associated with frontotemporal dementias. Neurobiology of Disease. 2004;17(2):237–249. doi: 10.1016/j.nbd.2004.07.008. [DOI] [PubMed] [Google Scholar]
- 64.Segat L, Pontillo A, Annoni G, et al. PIN1 promoter polymorphisms are associated with Alzheimer’s disease. Neurobiology of Aging. 2007;28(1):69–74. doi: 10.1016/j.neurobiolaging.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 65.Butterfield DA, Poon HF, St. Clair D, et al. Redox proteomics identification of oxidatively modified hippocampal proteins in mild cognitive impairment: insights into the development of Alzheimer’s disease. Neurobiology of Disease. 2006;22(2):223–232. doi: 10.1016/j.nbd.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 66.Arosio B, Bulbarelli A, Bastias Candia S, et al. Pin1 contribution to Alzheimer’s disease: transcriptional and epigenetic mechanisms in patients with late-onset Alzheimer’s disease. Neurodegenerative Diseases. 2012;10(1–4):207–211. doi: 10.1159/000333799. [DOI] [PubMed] [Google Scholar]
- 67.Ray S, Britschgi M, Herbert C, et al. Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nature Medicine. 2007;13(11):1359–1362. doi: 10.1038/nm1653. [DOI] [PubMed] [Google Scholar]
- 68.Gavin DP, Sharma RP. Histone modifications, DNA methylation, and Schizophrenia. Neuroscience and Biobehavioral Reviews. 2010;34(6):882–888. doi: 10.1016/j.neubiorev.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ando K, Dourlen P, Sambo AV, et al. Tau pathology modulates Pin1 post-translational modifications and may be relevant as biomarker. Neurobiology of Aging. 2013;34(3):757–769. doi: 10.1016/j.neurobiolaging.2012.08.004. [DOI] [PubMed] [Google Scholar]
- 70.Wang S, Simon BP, Bennett DA, Schneider JA, Malter JS, Wang D-S. The significance of Pin1 in the development of Alzheimer’s disease. Journal of Alzheimer’s Disease. 2007;11(1):13–23. doi: 10.3233/jad-2007-11105. [DOI] [PubMed] [Google Scholar]
- 71.Maruszak A, Safranow K, Gustaw K, et al. PIN1 gene variants in Alzheimer’s disease. BMC Medical Genetics. 2009;10, article 115 doi: 10.1186/1471-2350-10-115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Storace D, Cammarata S, Borghi R, et al. Elevation of β-amyloid 1-42 autoantibodies in the blood of amnestic patients with mild cognitive impairment. Archives of Neurology. 2010;67(7):867–872. doi: 10.1001/archneurol.2010.137. [DOI] [PubMed] [Google Scholar]
- 73.Bulbarelli A, Lonati E, Cazzaniga E, Gregori M, Masserini M. Pin1 affects Tau phosphorylation in response to Aβ oligomers. Molecular and Cellular Neuroscience. 2009;42(1):75–80. doi: 10.1016/j.mcn.2009.06.001. [DOI] [PubMed] [Google Scholar]
- 74.Lonati E, Masserini M, Bulbarelli A. Pin1: a new outlook in alzheimer’s disease. Current Alzheimer Research. 2011;8(6):615–622. doi: 10.2174/156720511796717140. [DOI] [PubMed] [Google Scholar]
- 75.Bakulski KM, Rozek LS, Dolinoy DC, Paulson HL, Hu H. Alzheimer's disease and environmental exposure to lead: the epidemiologic evidence and potential role of epigenetics. Current Alzheimer Research. 2012;9(5):563–573. doi: 10.2174/156720512800617991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pidsley R, Mill J. Epigenetic studies of psychosis: current findings, methodological approaches, and implications for postmortem research. Biological Psychiatry. 2011;69(2):146–156. doi: 10.1016/j.biopsych.2010.03.029. [DOI] [PubMed] [Google Scholar]
- 77.Morris KV. The emerging role of RNA in the regulation of gene transcription in human cells. Seminars in Cell and Developmental Biology. 2011;22(4):351–358. doi: 10.1016/j.semcdb.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gidlöf O, Andersson P, van der Pals J, Götberg M, Erlinge D. Cardiospecific microRNA plasma levels correlate with troponin and cardiac function in patients with ST elevation myocardial infarction, are selectively dependent on renal elimination, and can be detected in urine samples. Cardiology. 2011;118(4):217–226. doi: 10.1159/000328869. [DOI] [PubMed] [Google Scholar]
- 79.Kuwabara Y, Ono K, Horie T, et al. Increased microRNA-1 and microRNA-133a levels in serum of patients with cardiovascular disease indicate myocardial damage. Circulation: Cardiovascular Genetics. 2011;4(4):446–454. doi: 10.1161/CIRCGENETICS.110.958975. [DOI] [PubMed] [Google Scholar]
- 80.Mitchell PS, Parkin RK, Kroh EM, et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(30):10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang K, Zhang S, Marzolf B, et al. Circulating microRNAs, potential biomarkers for drug-induced liver injury. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(11):4402–4407. doi: 10.1073/pnas.0813371106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Jones PA, Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20(1):85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 83.Feinberg AP, Irizarry RA. Stochastic epigenetic variation as a driving force of development, evolutionary adaptation, and disease. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(supplement 1):1757–1764. doi: 10.1073/pnas.0906183107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fraga MF, Esteller M. Epigenetics and aging: the targets and the marks. Trends in Genetics. 2007;23(8):413–418. doi: 10.1016/j.tig.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 85.Bjornsson HT, Sigurdsson MI, Fallin MD, et al. Intra-individual change over time in DNA methylation with familial clustering. Journal of the American Medical Association. 2008;299(24):2877–2883. doi: 10.1001/jama.299.24.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gentilini D, Mari D, Castaldi D, et al. Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenarians' offspring. Age. 2013;35(5):1961–1973. doi: 10.1007/s11357-012-9463-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Esteller M. Molecular origins of cancer: epigenetics in cancer. The New England Journal of Medicine. 2008;358(11):1148–1059. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- 88.Horike S-I, Cai S, Miyano M, Cheng J-F, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nature Genetics. 2005;37(1):31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 89.DeBaun MR, Niemitz EL, McNeil DE, Brandenburg SA, Lee MP, Feinberg AP. Epigenetic alterations of H19 and LIT1 distinguish patients with Beckwith-Wiedemann syndrome with cancer and birth defects. The American Journal of Human Genetics. 2002;70(3):604–611. doi: 10.1086/338934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Faulk C, Dolinoy DC. Timing is everything: the when and how of environmentally induced changes in the epigenome of animals. Epigenetics. 2011;6(7):791–797. doi: 10.4161/epi.6.7.16209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Barker DJP. The developmental origins of chronic adult disease. Acta Paediatrica, Supplement. 2004;93(446):26–33. doi: 10.1111/j.1651-2227.2004.tb00236.x. [DOI] [PubMed] [Google Scholar]
- 92.Hanson M, Godfrey KM, Lillycrop KA, Burdge GC, Gluckman PD. Developmental plasticity and developmental origins of non-communicable disease: theoretical considerations and epigenetic mechanisms. Progress in Biophysics and Molecular Biology. 2011;106(1):272–280. doi: 10.1016/j.pbiomolbio.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 93.Wadhwa PD, Buss C, Entringer S, Swanson JM. Developmental origins of health and disease: brief history of the approach and current focus on epigenetic mechanisms. Seminars in Reproductive Medicine. 2009;27(5):358–368. doi: 10.1055/s-0029-1237424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jablonka E. Epigenetic epidemiology. International Journal of Epidemiology. 2004;33(5):929–935. doi: 10.1093/ije/dyh231. [DOI] [PubMed] [Google Scholar]
- 95.Calvanese V, Lara E, Kahn A, Fraga MF. The role of epigenetics in aging and age-related diseases. Ageing Research Reviews. 2009;8(4):268–276. doi: 10.1016/j.arr.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 96.Fraga MF. Genetic and epigenetic regulation of aging. Current Opinion in Immunology. 2009;21(4):446–453. doi: 10.1016/j.coi.2009.04.003. [DOI] [PubMed] [Google Scholar]
- 97.D’Addario C, Dell’Osso B, Palazzo MC, et al. Selective DNA methylation of BDNF promoter in bipolar disorder: differences among patients with BDI and BDII. Neuropsychopharmacology. 2012;37(7):1647–1655. doi: 10.1038/npp.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sananbenesi F, Fischer A. The epigenetic bottleneck of neurodegenerative and psychiatric diseases. Biological Chemistry. 2009;390(11):1145–1153. doi: 10.1515/BC.2009.131. [DOI] [PubMed] [Google Scholar]
- 99.Urdinguio RG, Sanchez-Mut JV, Esteller M. Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. The Lancet Neurology. 2009;8(11):1056–1072. doi: 10.1016/S1474-4422(09)70262-5. [DOI] [PubMed] [Google Scholar]
- 100.Chouliaras L, Rutten BPF, Kenis G, et al. Epigenetic regulation in the pathophysiology of Alzheimer’s disease. Progress in Neurobiology. 2010;90(4):498–510. doi: 10.1016/j.pneurobio.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 101.Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic mechanisms in Alzheimer’s disease. Neurobiology of Aging. 2011;32(7):1161–1180. doi: 10.1016/j.neurobiolaging.2010.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Mill J. Toward an integrated genetic and epigenetic approach to Alzheimer’s disease. Neurobiology of Aging. 2011;32(7):1188–1191. doi: 10.1016/j.neurobiolaging.2010.10.021. [DOI] [PubMed] [Google Scholar]
- 103.Barrachina M, Ferrer I. DNA methylation of Alzheimer disease and tauopathy-related genes in postmortem brain. Journal of Neuropathology and Experimental Neurology. 2009;68(8):880–891. doi: 10.1097/NEN.0b013e3181af2e46. [DOI] [PubMed] [Google Scholar]
- 104.Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Genda Y, Ukitsu M. Reduction with age in methylcytosine in the promoter region -224~-101 of the amyloid precursor protein gene in autopsy human cortex. Molecular Brain Research. 1999;70(2):288–292. doi: 10.1016/s0169-328x(99)00163-1. [DOI] [PubMed] [Google Scholar]
- 105.Tohgi H, Utsugisawa K, Nagane Y, Yoshimura M, Ukitsu M, Genda Y. The methylation status of cytosines in a τ gene promoter region alters with age to downregulate transcriptional activity in human cerebral cortex. Neuroscience Letters. 1999;275(2):89–92. doi: 10.1016/s0304-3940(99)00731-4. [DOI] [PubMed] [Google Scholar]
- 106.Mastroeni D, Grover A, Delvaux E, Whiteside C, Coleman PD, Rogers J. Epigenetic changes in Alzheimer’s disease: decrements in DNA methylation. Neurobiology of Aging. 2010;31(12):2025–2037. doi: 10.1016/j.neurobiolaging.2008.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mastroeni D, McKee A, Grover A, Rogers J, Coleman PD. Epigenetic differences in cortical neurons from a pair of monozygotic twins discordant for Alzheimer’s disease. PLoS ONE. 2009;4(8) doi: 10.1371/journal.pone.0006617.e6617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nunez-Iglesias J, Liu C-C, Morgan TE, Finch CE, Zhou XJ. Joint genome-wide profiling of miRNA and mRNA expression in Alzheimer’s disease cortex reveals altered miRNA regulation. PloS ONE. 2010;5(2) doi: 10.1371/journal.pone.0008898.e8898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ogawa O, Zhu X, Lee H-G, et al. Ectopic localization of phosphorylated histone H3 in Alzheimer’s disease: a mitotic catastrophe? Acta Neuropathologica. 2003;105(5):524–528. doi: 10.1007/s00401-003-0684-3. [DOI] [PubMed] [Google Scholar]
- 110.Fuso A, Seminara L, Cavallaro RA, D’Anselmi F, Scarpa S. S-adenosylmethionine/homocysteine cycle alterations modify DNA methylation status with consequent deregulation of PS1 and BACE and beta-amyloid production. Molecular and Cellular Neuroscience. 2005;28(1):195–204. doi: 10.1016/j.mcn.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 111.Belakavadi M, Dell J, Grover GJ, Fondell JD. Thyroid hormone suppression of β-amyloid precursor protein gene expression in the brain involves multiple epigenetic regulatory events. Molecular and Cellular Endocrinology. 2011;339(1-2):72–80. doi: 10.1016/j.mce.2011.03.016. [DOI] [PubMed] [Google Scholar]
- 112.Dangond F, Gullans SR. Differential expression of human histone deacetylase mRNAs in response to immune cell apoptosis induction by Trichostatin A and butyrate. Biochemical and Biophysical Research Communications. 1998;247(3):833–837. doi: 10.1006/bbrc.1998.8891. [DOI] [PubMed] [Google Scholar]
- 113.de Ruijter AJM, van Gennip AH, Caron HN, Kemp S, van Kuilenburg ABP. Histone deacetylases (HDACs): characterization of the classical HDAC family. Biochemical Journal. 2003;370, part 3:737–749. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.di Francesco A, Arosio B, Gussago C, et al. Involvement of 5-lipoxygenase in Alzheimer's disease: a role for DNA methylation. Journal of Alzheimer's Disease. 2013;37(1):3–8. doi: 10.3233/JAD-130506. [DOI] [PubMed] [Google Scholar]
- 115.D'Addario C, Di Francesco A, Arosio B, et al. Epigenetic regulation of fatty acid amide hydrolase in Alzheimer disease. PLoS ONE. 2012;7(6) doi: 10.1371/journal.pone.0039186.e39186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.van der Stelt M, van Kuik JA, Bari M, et al. Oxygenated metabolites of anandamide and 2-arachidonoylglycerol: conformational analysis and interaction with cannabinoid receptors, membrane transporter, and fatty acid amide hydrolase. Journal of Medicinal Chemistry. 2002;45(17):3709–3720. doi: 10.1021/jm020818q. [DOI] [PubMed] [Google Scholar]
- 117.Rouzer CA, Marnett LJ. Endocannabinoid oxygenation by cyclooxygenases, lipoxygenases, and cytochromes P450: cross-talk between the eicosanoid and endocannabinoid signaling pathways. Chemical Reviews. 2011;111(10):5899–5921. doi: 10.1021/cr2002799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Benito C, Núñez E, Tolón RM, et al. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. Journal of Neuroscience. 2003;23(35):11136–11141. doi: 10.1523/JNEUROSCI.23-35-11136.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cunningham C, Skelly DT. Non-steroidal anti-inflammatory drugs and cognitive function: are prostaglandins at the heart of cognitive impairment in dementia and delirium? Journal of NeuroImmune Pharmacology. 2012;7(1):60–73. doi: 10.1007/s11481-011-9312-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Imbimbo BP, Solfrizzi V, Panza F. Are NSAIDs useful to treat Alzheimer's disease or mild cognitive impairment? Frontiers in Aging Neuroscience. 2010 doi: 10.3389/fnagi.2010.00019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Ng SC, Chan FK. NSAID-induced gastrointestinal and cardiovascular injury. Current Opinion in Gastroenterology. 2010;26(6):611–617. doi: 10.1097/MOG.0b013e32833e91eb. [DOI] [PubMed] [Google Scholar]
- 122.Petot GJ, Friedland RP. Lipids, diet and Alzheimer disease: an extended summary. Journal of the Neurological Sciences. 2004;226(1-2):31–33. doi: 10.1016/j.jns.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 123.Mendonça A, Cunha RA. Therapeutic opportunities for caffeine in Alzheimer’s disease and other neurodegenerative disorders. Journal of Alzheimer’s Disease. 2010;20(supplement 1):S1–S2. doi: 10.3233/JAD-2010-01420. [DOI] [PubMed] [Google Scholar]
- 124.Arendash GW, Schleif W, Rezai-Zadeh K, et al. Caffeine protects Alzheimer’s mice against cognitive impairment and reduces brain β-amyloid production. Neuroscience. 2006;142(4):941–952. doi: 10.1016/j.neuroscience.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 125.Carter AJ, O’Connor WT, Carter MJ, Ungerstedt U. Caffeine enhances acetylcholine release in the hippocampus in vivo by a selective interaction with adenosine A1 receptors. Journal of Pharmacology and Experimental Therapeutics. 1995;273(2):637–642. [PubMed] [Google Scholar]
- 126.Dall’Igna OP, Fett P, Gomes MW, Souza DO, Cunha RA, Lara DR. Caffeine and adenosine A2A receptor antagonists prevent β-amyloid (25–35)-induced cognitive deficits in mice. Experimental Neurology. 2007;203(1):241–245. doi: 10.1016/j.expneurol.2006.08.008. [DOI] [PubMed] [Google Scholar]
- 127.Johnson-Kozlow M, Kritz-Silverstein D, Barrett-Connor E, Morton D. Coffee consumption and cognitive function among older adults. The American Journal of Epidemiology. 2002;156(9):842–850. doi: 10.1093/aje/kwf119. [DOI] [PubMed] [Google Scholar]
- 128.van Boxtel MPJ, Schmitt JAJ, Bosma H, Jolles J. The effects of habitual caffeine use on cognitive change: a longitudinal perspective. Pharmacology Biochemistry and Behavior. 2003;75(4):921–927. doi: 10.1016/s0091-3057(03)00171-0. [DOI] [PubMed] [Google Scholar]
- 129.Fredholm BB, Bättig K, Holmén J, Nehlig A, Zvartau EE. Actions of caffeine in the brain with special reference to factors that contribute to its widespread use. Pharmacological Reviews. 1999;51(1):83–133. [PubMed] [Google Scholar]
- 130.Haskó G, Linden J, Cronstein B, Pacher P. Adenosine receptors: therapeutic aspects for inflammatory and immune diseases. Nature Reviews Drug Discovery. 2008;7(9):759–770. doi: 10.1038/nrd2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Headrick JP, Ashton KJ, Rose'meyer RB, Peart JN. Cardiovascular adenosine receptors: expression, actions and interactions. Pharmacology & Therapeutics. 2013;140(1):92–111. doi: 10.1016/j.pharmthera.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 132.Fredholm BB, Chern Y, Franco R, Sitkovsky M. Aspects of the general biology of adenosine A2A signaling. Progress in Neurobiology. 2007;83(5):263–276. doi: 10.1016/j.pneurobio.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 133.Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annual Review of Pharmacology and Toxicology. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- 134.Sebastião AM, Ribeiro JA. Tuning and fine-tuning of synapses with adenosine. Current Neuropharmacology. 2009;7(3):180–194. doi: 10.2174/157015909789152128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cronstein BN, Kramer SB, Rosenstein ED, Weissmann G, Hirschhorn R. Adenosine modulates the generation of superoxide anion by stimulated human neutrophils via interaction with a specific cell surface receptor. Annals of the New York Academy of Sciences. 1985;451:291–301. doi: 10.1111/j.1749-6632.1985.tb27120.x. [DOI] [PubMed] [Google Scholar]
- 136.Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV. Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. Journal of Biological Chemistry. 1997;272(41):25881–25889. doi: 10.1074/jbc.272.41.25881. [DOI] [PubMed] [Google Scholar]
- 137.Sitkovsky M, Lukashev D. Regulation of immune cells by local-tissue oxygen tension: HIF1α and adenosine receptors. Nature Reviews Immunology. 2005;5(9):712–721. doi: 10.1038/nri1685. [DOI] [PubMed] [Google Scholar]
- 138.Chen J-F, Sonsalla PK, Pedata F, et al. Adenosine A2A receptors and brain injury: broad spectrum of neuroprotection, multifaceted actions and “fine tuning” modulation. Progress in Neurobiology. 2007;83(5):310–331. doi: 10.1016/j.pneurobio.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 139.Rodrigues RJ, Canas PM, Lopes LV, Oliveira CR, Cunha RA. Modification of adenosine modulation of acetylcholine release in the hippocampus of aged rats. Neurobiology of Aging. 2008;29(10):1597–1601. doi: 10.1016/j.neurobiolaging.2007.03.025. [DOI] [PubMed] [Google Scholar]
- 140.Sitkovsky MV. Use of the A2A adenosine receptor as a physiological immunosuppressor and to engineer inflammation in vivo. Biochemical Pharmacology. 2003;65(4):493–501. doi: 10.1016/s0006-2952(02)01548-4. [DOI] [PubMed] [Google Scholar]
- 141.Sitkovsky MV, Lukashev D, Apasov S, et al. Physiological control of immune response and inflammatory tissue damage by hypoxia-inducible factors and adenosine A2A receptors. Annual Review of Immunology. 2004;22:657–682. doi: 10.1146/annurev.immunol.22.012703.104731. [DOI] [PubMed] [Google Scholar]
- 142.Sebastião AM, Ribeiro JA. Adenosine receptors and the central nervous system. Handbook of Experimental Pharmacology. 2009;193:471–534. doi: 10.1007/978-3-540-89615-9_16. [DOI] [PubMed] [Google Scholar]
- 143.Stone TW, Ceruti S, Abbracchio MP. Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. Handbook of Experimental Pharmacology. 2009;193:535–587. doi: 10.1007/978-3-540-89615-9_17. [DOI] [PubMed] [Google Scholar]
- 144.Canas PM, Porciúncula LO, Cunha GMA, et al. Adenosine A2A receptor blockade prevents synaptotoxicity and memory dysfunction caused by β-amyloid peptides via p38 mitogen-activated protein kinase pathway. Journal of Neuroscience. 2009;29(47):14741–14751. doi: 10.1523/JNEUROSCI.3728-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Cunha RA. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signalling. 2005;1(2):111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A2A receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57(1):121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- 147.Fredholm BB, Chen J-F, Cunha RA, Svenningsson P, Vaugeois J-M. Adenosine and brain function. International Review of Neurobiology. 2005;63:191–270. doi: 10.1016/S0074-7742(05)63007-3. [DOI] [PubMed] [Google Scholar]
- 148.Gomes CV, Kaster MP, Tomé AR, Agostinho PM, Cunha RA. Adenosine receptors and brain diseases: neuroprotection and neurodegeneration. Biochimica et Biophysica Acta. 2011;1808(5):1380–1399. doi: 10.1016/j.bbamem.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 149.Phillis JW. The effects of selective A1 and A2A adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Research. 1995;705(1-2):79–84. doi: 10.1016/0006-8993(95)01153-6. [DOI] [PubMed] [Google Scholar]
- 150.Jones PA, Smith RA, Stone TW. Protection against kainate-induced excitotoxicity by adenosine A2A receptor agonists and antagonists. Neuroscience. 1998;85(1):229–237. doi: 10.1016/s0306-4522(97)00613-1. [DOI] [PubMed] [Google Scholar]
- 151.Talukder MAH, Morrison RR, Ledent C, Mustafa SJ. Endogenous adenosine increases coronary flow by activation of both A2A and A2B receptors in mice. Journal of Cardiovascular Pharmacology. 2003;41(4):562–570. doi: 10.1097/00005344-200304000-00008. [DOI] [PubMed] [Google Scholar]
- 152.Delikouras A, Fairbanks LD, Simmonds AH, Lechler RI, Dorling A. Endothelial cell cytoprotection induced in vitro by allo- or xenoreactive antibodies is mediated by signaling through adenosine A2 receptors. European Journal of Immunology. 2003;33(11):3127–3135. doi: 10.1002/eji.200323566. [DOI] [PubMed] [Google Scholar]
- 153.Zhu Y, Liu L, Peng X, Ding X, Yang G, Li T. Role of adenosine A2A receptor in organ-specific vascular reactivity following hemorrhagic shock in rats. Journal of Surgical Research. 2013;184(2):951–958. doi: 10.1016/j.jss.2013.03.039. [DOI] [PubMed] [Google Scholar]
- 154.Arosio B, Viazzoli C, Mastronardi L, Bilotta C, Vergani C, Bergamaschini L. Adenosine A {2A} receptor expression in peripheral blood mononuclear cells of patients with mild cognitive impairment. Journal of Alzheimer’s Disease. 2010;20(4):991–996. doi: 10.3233/JAD-2010-090814. [DOI] [PubMed] [Google Scholar]
- 155.Gussago C, Arosio B, Casati M, et al. Different adenosine A2A receptor expression in peripheral cells from elderly patients with vascular dementia and alzheimer's disease. Journal of Alzheimer's Disease. 2014;40(1):45–49. doi: 10.3233/JAD-131652. [DOI] [PubMed] [Google Scholar]
- 156.Marques S, Batalha VL, Lopes LV, Outeiro TF. Modulating alzheimer’s disease through caffeine: a putative link to epigenetics. Journal of Alzheimer’s Disease. 2011;24(supplement 2):161–171. doi: 10.3233/JAD-2011-110032. [DOI] [PubMed] [Google Scholar]
- 157.Helmy AA, Abdel Naseer MM, El Shafie S, Nada MAF. Role of interleukin 6 and alpha-globulins in differentiating Alzheimer and vascular dementias. Neurodegenerative Diseases. 2012;9(2):81–86. doi: 10.1159/000329568. [DOI] [PubMed] [Google Scholar]
- 158.Simonsen AH, Hagnelius NO, Waldemar G, Nilsson TK, McGuire J. Protein markers for the differential diagnosis of vascular dementia and Alzheimer's disease . International Journal of Proteomics. 2012;2012:8 pages. doi: 10.1155/2012/824024.824024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Chen J-F, Huang Z, Ma J, et al. A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. Journal of Neuroscience. 1999;19(21):9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Yang ZJ, Wang B, Kwansa H, et al. Adenosine A2A receptor contributes to ischemic brain damage in newborn piglet. Journal of Cerebral Blood Flow & Metabolism. 2013;33(10):1612–1620. doi: 10.1038/jcbfm.2013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pelligrino DA, Xu H-L, Vetri F. Caffeine and the control of cerebral hemodynamics. Journal of Alzheimer’s Disease. 2010;20(supplement 1):S51–S62. doi: 10.3233/JAD-2010-091261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Govoni S, Gasparini L, Racchi M, Trabucchi M. Peripheral cells as an investigational tool for Alzheimer’s disease. Life Sciences. 1996;59(5-6):461–468. doi: 10.1016/0024-3205(96)00325-6. [DOI] [PubMed] [Google Scholar]
- 163.Centonze D, Battistini L, Maccarrone M. The endocannabinoid system in peripheral lymphocytes as a mirror of neuroinflammatory diseases. Current Pharmaceutical Design. 2008;14(23):2370–2342. doi: 10.2174/138161208785740018. [DOI] [PubMed] [Google Scholar]