

Abstract
Primary biliary cirrhosis is a multifactor autoimmune disease characterized by hepatic and systemic manifestations, with immune system dysregulation and abnormalities in the hepatic metabolism of bile salts, lipids, and nutrients, as well as destruction of membrane lipids and mitochondrial dysfunction. Both oxidative and nitrosative stress are associated with ongoing manifestations of the disease. In particular, abnormalities in nitric oxide metabolism and thiol oxidation already occur at early stages, thus leading to the hypothesis that these biochemical events play a pathogenic role in primary biliary cirrhosis. Moreover, the association of these metabolic abnormalities with the progression of the disease may indicate some biochemical parameters as early diagnostic markers of disease evolution, and may open up the potential for pharmacological intervention to inhibit intra- and extra-cellular stress events for resuming hepatocellular functions. The following paragraphs summarize the current knowledge by outlining molecular mechanisms of the disease related to these stress events.
Keywords: Aquaporins, Bile salts, Chronic cholestasis, Glutathione, Mitochondria, Nitrosothiols, Nitrotyrosine, Protein sulfhydryls, Thioredoxin
Core tip: Both oxidative and nitrosative stress are associated with ongoing manifestations of chronic cholestasis, and in particular, primary biliary cirrhosis. Abnormalities in nitric oxide metabolism and thiols oxidation already occur at early stages, thus leading to the hypothesis that these biochemical events play a pathogenic role in primary biliary cirrhosis. The association of these metabolic abnormalities with the progression of the disease may indicate some biochemical parameters as early diagnostic markers of disease evolution, and may open up the potential for pharmacological intervention to inhibit intra- and extra-cellular stress events for resuming hepatocellular functions. This article summarizes the current knowledge by outlining molecular mechanisms of the disease related to these stress events.
INTRODUCTION
Primary biliary cirrhosis (PBC) is an organ-specific autoimmune liver disease that predominantly affects women and is characterized by chronic and progressive destruction of small intrahepatic bile ducts with portal inflammation and ultimately fibrosis. It is a multifactor condition, mainly due to an immunological disorder of the liver, resulting in a chronic intrahepatic cholestasis that often occurs in asymptomatic subjects, with the diagnosis suggested by the occasional finding of elevated hepatic biochemical markers as early indexes. Appearance of jaundice, dark urine, and pale and fatty stools occur in patients who develop more advanced forms of liver disease.
Little is known about the etiology of PBC. Current theories on its pathogenesis support the hypothesis that the disease develops as a result of an inappropriate immune response following stimulation by an environmental or infectious agent (Figure 1). In particular, liver injury may follow a defect in immunologic tolerance, resulting in the activation and expansion of self-antigen specific T and B lymphocyte clones and the production of circulating autoantibodies, together with a myriad of cytokines and other inflammatory mediators (Figure 2)[1]. The resulting effect of this cascade of events is the activation of pathways leading to oxidative and nitrosative stress (Figures 3 and 4)[2].
Figure 1.
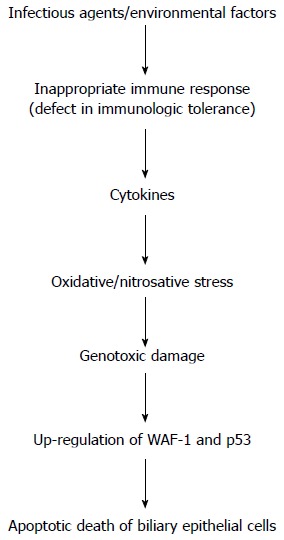
Flow-chart illustrating the cascade of events playing a potential role in the pathogenesis of liver injury in primary biliary cirrhosis. Important steps include the altered immune response linked to increased oxidative/nitrosative stress and genotoxic damage. The final result is the apoptotic death of biliary epithelial cells. WAF1: Cyclin-dependent kinase inhibitor 1; p53: Tumor protein 53.
Figure 2.
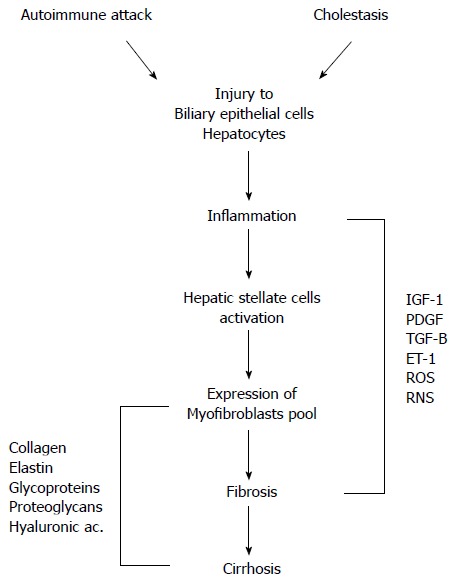
Pathophysiological mechanisms of liver damage in primary biliary cirrhosis. Autoimmune attack and cholestasis are both responsible for the injury to biliary epithelial cells and hepatocytes. Cell damage results in inflammatory processes with activation of hepatic stellate cells and expression of myofibroblasts leading to fibrosis and cirrhosis. A myriad of mediators have a role in these events. IGF-1: Insulin-like growth factor-1; PDGF: Platelet-derived growth factor; TGF-β: Transforming growth factor beta; ET-1: Endothelin-1; ROS: Reactive oxygen species; RNS: Reactive nitrogen species.
Figure 3.
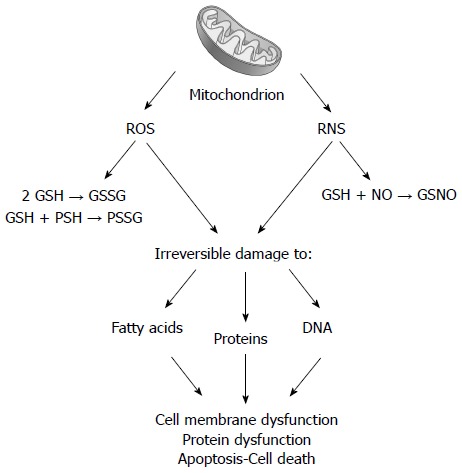
Following chronic cholestasis, liver mitochondrial impairment is associated with increased delivery of reactive oxygen species and reactive nitrogen species. Reactive oxygen species (ROS) promotes the oxidation of glutathione (GSH to GSSG) and of protein sulfhydryls (PSH to PSSG). Reactive nitrogen species (RNS) favors the formation of nitrosothiols (GSNO). Both steps are responsible for irreversible damage to lipids, proteins, and nucleic acids, ultimately leading to membrane and protein alteration.
Figure 4.
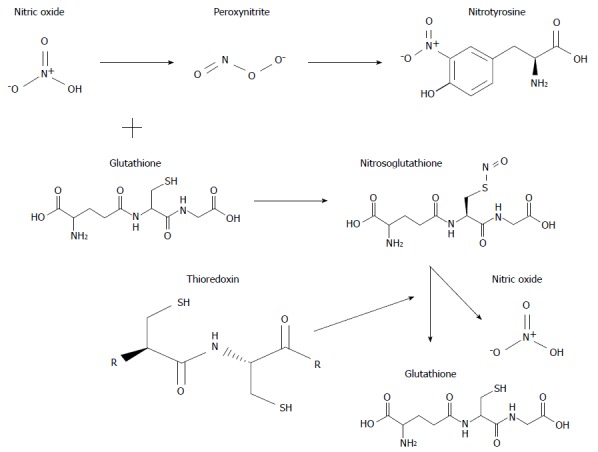
Structural formula and interaction of some important molecules involved in oxidative and nitrosative stress associated with chronic cholestasis.
Although the disease results from a combination of genetic and environmental risk factors, its serological hallmark is the presence of circulating anti-mitochondrial antibodies. This reflects the presence of autoreactive T and B cells to the culprit antigens, the E2 subunits of mitochondrial pyruvate dehydrogenase. The existence of a genetic predisposition is indicated by the higher familial incidence of the disease, particularly among siblings, and the high concordance rate among monozygotic twins[3]. Environmental xenobiotics/chemical compounds triggering events appear to be crucial in disrupting a pre-existing unstable immune tolerance of genetic origin allowing, after a long latency, the emergence of clinical disease[4]. Finally, up-regulation of WAF1 and p53 related to apoptosis of biliary epithelial cells represents the major pathogenic process leading to bile duct loss in PBC as the expression of genotoxic damage (Figure 1)[5].
It remains unknown why the disease evolution displays a faster pace in some PBC patients compared to others in which it is milder. Therefore, one of the major efforts of the current research is to answer this clinical question and, as a consequence, to identify easily assessable biochemical abnormalities serving as prognostic markers, as well as a target for potential therapeutic intervention. Up to now, however, such a search has not been particularly successful, although a number of biochemical and physiological alterations (toxic bile salt accumulation, oxidative mitochondrial dysfunction, and changes in lipid metabolism)[6] appear to be actively implicated in the onset and evolution of the disease.
PBC is mainly characterized by biliary obstruction and impaired canalicular bile secretion[7], with a progressive retention of hydrophobic bile salts in the liver determining detergent activity and cytotoxic damage[8]. Inflammatory changes[9] and parallel metabolic disorders contribute to the reduced detoxification capacity of the liver[8,10,11], and the enhanced generation of reactive oxygen (ROS) and nitrogen species[12] responsible for oxidative and nitrosative stress[13].
The changes in bile secretion involve complex pathways and result in retention of toxic compounds in the liver. This metabolic and functional impairment is associated with alterations of some physiological mechanisms that regulate bile flow and water transport into the bile canaliculus in response to transient osmotic gradients originating from active solute transport[7,14]. Indeed, at least one third of the bile flow is driven osmotically by the amount of hepatic glutathione (GSH) secreted into canalicular spaces. Beyond the importance of this secretory mechanism for bile formation, the excretion of GSH also represents an important way to discharge toxic anionic compounds deriving from hepatic metabolism of exogenous and endogenous substances. Derangement of hepatic and/or biliary GSH status occurs in several experimental animal models of cholestatic liver injury and in patients with cholestasis[15]. Chronic cholestasis, and in particular PBC, are associated with decreased biliary secretion of GSH, which may affect thiol-dependent bile flow and the hepato-biliary transport of toxic organic compounds[16], and can occur before the reduction in biliary bile salt secretion[17].
On the other hand, the canalicular secretion of water is mediated by aquaporin-8 (AQP8)[18], a water channel protein found at multiple compartments of hepatocytes, including the canalicular plasma membrane and related subapical vesicles[19,20], smooth endoplasmic reticulum, and mitochondria[21,22]. The transport of water from the sinusoidal blood into the hepatocyte is conversely facilitated by AQP9, an aquaporin channel with a broad selectivity[23,24]. AQP9 also represents the major pathway by which glycerol is imported by hepatocytes to supply gluconeogenesis during starvation[25].
During choleresis, hepatocytes rapidly increase their canalicular membrane water permeability by modulating the abundance of AQP8. Studies in several experimental models of cholestasis, such as extrahepatic biliary obstruction, estrogen-induced cholestasis, and sepsis-induced cholestasis, have shown that a decrease in canalicular AQP8 expression contributes to the development of cholestasis. Thus, it has been suggested that the combined alterations in hepatic expression of solute transporters and AQP8 hamper the efficient coupling of osmotic gradients and canalicular water flow, indicating that cholestasis may also result from a mutual occurrence of impaired solute transport and decreased water permeability[26,27]. Since glucagon, a hormone stimulating hepatocyte bile formation[28], induces the gene expression of AQP8, but not AQP9 water channels in rodent hepatocytes[29], it is reasonable to hypothesize that the most critical step in coupling solute transport and secretion of water takes place on the canalicular membrane of hepatocytes, and that it may be influenced by stress conditions.
Therefore, the associations between inflammation and oxidative and nitrosative stress-induced structural and functional alterations of hepatocytes, including subcellular organelles, render the interpretation of these relationships extremely intriguing, and open to potential therapeutic challenges. In the following paragraphs, the current knowledge on the molecular mechanisms of the disease related to oxidative and nitrosative stress is concisely summarized.
Oxidative stress
Cholestatic events may evolve towards oxidative changes which may directly affect the nature and function of membrane lipids and embedded proteins[30,31], and occur in both the liver and some extrahepatic tissues, albeit recognizing different pathogenic moments.
The transsulfuration pathway is impaired in rats undergoing bile duct ligation (BDL), an experimental model resembling human chronic cholestatic conditions, as well as in humans with cholestatic liver diseases[6,32,33]. This alteration is associated with changes in thiol disposition and redox status. GSH is the main thiol detoxifying molecule playing a role in bile formation and biliary excretion of toxic compounds, as well as in maintaining protein sulfhydryls (PSH) in their reduced form[34,35]. Among these proteins exerting a number of major cell functions are those involved in mitochondrial respiration and membrane transition pore integrity[36,37]. Indeed, most of the redox changes occurring in proteins involve the redox state of cysteine residues, and lead to the formation of intermolecular or mixed disulfides.
In some studies[6,33], more aggressive forms of cholestasis have been associated with primary changes in hepatic and extrahepatic membrane protein redox status and lipid composition. In particular, abnormalities in hepatocyte membranes have been reported in cirrhotic patients[38] that have been associated with an imbalance between the transmethylation and the transsulfuration pathways[39]. Similar to hypercholesterolemic subjects[40], cirrhotic patients show lipid alterations in erythrocyte (RBC) membranes in relation to changes in the circulating lipoproteins[41,42]. Such alterations are particularly important in patients with PBC who accumulate toxic bile salts and show early alterations in hepatic lipid metabolism with increased synthesis of cholesterol[43]. Indeed, both bile salt excess and lipid changes have been implicated in the early modification of membrane fluidity and transport function[44] at both hepatocyte and RBC levels[45,46]. Common biochemical derangements[47], such as the alteration of Na+/K+ ATPase activity of hepatocyte membranes[48], have also been claimed.
RBC membrane alterations, which parallel those found in hepatocytes[47], include a decreased content of PSH and an increased aliquot of oxidized proteins (carbonyl), especially in patients with stage III-IV PBC[49]. In cell membranes, PSH has structural and functional roles, and confers resistance to damaging insults[50]. Membrane PSH maintenance in the reduced form is therefore a major program for all biological systems to assure functional integrity. Together with changes in ratios of the membrane cholesterol:phospholipids, the described oxidation rate of ghost proteins in RBC likely explains the high susceptibility to in vitro hemolysis observed in PBC patients. Furthermore, with the application of logistic regression analysis, it has been observed that advanced forms of PBC could be identified by the simultaneous presence of significant changes of some RBC membrane parameters[50]: PSH lower than 40 nmol/mg protein (r = -0.817), protein carbonyls higher than 2.7 nmol/mg protein (r = 0.653), cholesterol greater than 550 nmol/mg protein (r = 0.744), and phosphatidylethanolamine lower than 25% ( r = -0.731).
Taken together, these observations explain, at least in part, the changes to membrane fluidity, transport[41], receptor activities[51-53], and the increased hemolysis observed in patients with PBC[54]. Indeed, the possible link between protein oxidation and altered lipid composition in the RBC ghost membrane of PBC patients may be partially explained by metabolic changes occurring in the liver. In particular, the increased hepatic production of phosphatidylcholine, which is formed through the methylation of phosphatidylethanolamine via the transformation of S-adenosyl methionine in S-adenosyl-L-homocysteine, may play a role in these processes[55-57].
Many clinical and experimental observations have found that oxidative stress also participates actively in cholestasis-induced hepatocyte damage, as suggested by the accumulation of lipid and protein oxidative products[9]. However, the molecular regulatory mechanisms by which oxidative stress induces modifications of antioxidant capacities at the same time as maneuvering cellular adaptation by increasing the availability of redox active compounds is still debatable.
Studies performed in BDL rats have shown that both free and protein thiols (GSH and PSH) are lower within the hepatocyte cytosol, and that these alterations occur at early stages. These changes parallel the high concentrations of oxidized glutathione (GSSG) observed with ongoing cholestasis and the increased enzymatic activities of both GSH-peroxidase and GSSG-reductase, thus asserting the participation of oxidative stress in cholestatic hepatic injury[58].
One of the most interesting molecules in this context is thioredoxin, an oxidative stress-induced redox-active protein[59]. Thioredoxin possesses a number of biological activities[60], including the maintenance of protein-SH groups[61], the regulation of redox-sensitive molecules[59], and the protection of membrane permeability[62]. Circulating levels of thioredoxin are increased in patients with a high rate of free radical generation[63], in which it also interferes with the modulation of the immunological response[64]. In PBC patients, circulating thioredoxin is increased in the early disease stage[65] and decreased in patients with a more advanced disease stage, in which it is inversely related with circulating nitrosothiols (r = -0.609), the liver fibrosis marker hyaluronate (r = -0.432), and with histology (r = -0.757)[65]. In RBC, thioredoxin concentrations are higher in PBC stages I to III, but lower in stage IV patients compared to healthy subjects[65].
More detailed changes in thioredoxin disposition have been studied in BDL rats. In these cholestatic animals, serum thioredoxin concentrations were found to be increased at 3 d after surgery and then decreased. Similarly, such changes were observed in the whole liver and in isolated mitochondria, but no variations were documented in RBC. Therefore, by considering the difference in the average time changes from baseline among serum, hepatic, and RBC levels, it emerged that circulating thioredoxin is likely of hepatic origin.
Other important organs having a role in cholestatic liver damage and reparation are oxidatively damaged during chronic cholestasis; this is the case of the intestine. In this regard, significant redox alterations have been early observed in the intestine of BDL rats, where the mucosal concentrations of GSH are low in both the ileum and colon. The hypothesis on oxidative consumption of GSH has been confirmed by early increases of GSSG and protein carbonyls, as well as a decrease of PSH[58]. However, under cholestatic conditions, decreased availability of biliary GSH, which currently represents a considerable source of luminal GSH, may also take part in the depletion of intestinal GSH. At this level, the changes in mucosal proteins are associated with a decreased concentration of total proteins and lower activity of GSH-dependent antioxidant enzymes (peroxidase and reductase). This may depend on the reduced synthesis of proteins linked to a decreased availability of bile salts, which enhance gene expression and regulate the synthesis of proteins at the intestinal level[66,67]. Bile salt deficiency, conversely, promotes mucosal injury[68] and reduces protein concentrations[69], which, at least in part, explains the suppression in protein synthesis in the intestinal mucosa of cholestatic rats. These biochemical changes are associated with profound functional alterations of colonic ionic transport and a significant overall decrease of all electrophysiological parameters.
Considering that the impairment of the GSH-dependent antioxidant system, the changes in mucosal permeability and transport capacity, and the increased susceptibility to toxic injury all occur during the early stage of cholestasis, it is conceivable that these alterations may favor intestinal bacterial translocation and the absorption of toxic molecules[70]. The latter two phenomena are likely to be implicated in the appearance of hepatic injuries, and this would demonstrate the existence of a close interrelation between the liver and the intestine, both in the physiological state and in the case of cholestasis. Finally, cholestatic rats show a progressive reduction in their daily food intake, and as a consequence, show a deficient availability of dietary precursors, which is likely to occur earlier in the intestine and liver than in other organs. This aspect should always be considered when studying cholestatic animals.
Nitrosative stress
It is known that a number of cellular and circulating factors can modulate cell membrane activity and cholestatic disease progression. In cholestatic livers, nitric oxide (NO), a potential free radical intermediate, is mainly released by inducible NO synthase (iNOS)[71,72], which is likely sustained by the enhanced inflow of gut-derived endotoxins[58,72,73]. Indeed, it is ascertained that extrahepatic factors actively participate in the determination of cholestatic liver injury. In this respect, prolonged interruption of enterohepatic circulation of bile salts results in intestinal permeability alteration and favors portal endotoxemia[74] with worsening or even promotion of hepatic damage[75] by enhancing free radicals generation, GSH depletion, and impairing detoxification defense[6,32,33].
The excess generation of NO already present in early disease stages favors the formation of hyperreactive derivatives that ultimately result in nitrotyrosine deposition[76-78]. This compound accumulation reflects the excess formation of the highly reactive molecule peroxynitrite, and supports a major injuring role for NO in chronic cholestasis. This view is also consolidated by the observation that the serum of PBC patients contains significantly higher levels of nitrotyrosine compared with control subjects. Moreover, the trend in nitrotyrosine levels seems to parallel the disease stage.
Conversely, nitrosothiols are formed by conjugation of some reactive NO forms (NO●) with thiols and GSH in particular, and thereby oppose dangerous side reactions such as peroxynitrite formation[60]. Nitrosothiols also act as intracellular messengers, and exert a major regulatory effect on cellular and mitochondrial functions[79] through the nitrosylation of proteins and enzymes[80,81]. Conversely, circulating nitrosothiols mainly act as a reserve for NO, whose release after molecule decomposition serves for vascular tone modulation[81]. Serum nitrosothiols are significantly higher in PBC patients than in healthy controls, with the increase being directly proportional to disease severity and paralleling the serum concentrations of hyaluronate[65] and nitrotyrosine[82]. Indeed, it is known that circulating levels of NO derivatives are particularly high in all cirrhotic conditions[83].
Compared to RBC, in which no variations have been noted, serum and hepatic nitrosothiols significantly and progressively increase with ongoing cholestasis in the BDL model; increasing more than tenfold in value at day 10 after the BDL. Following BDL, the NOx (nitrate plus nitrite) content is significantly increased in the liver.
Overall, the importance of the observations reported for early stages of experimental cholestasis and their presence in patients with milder forms of PBC definitely point to an active participation of these pathophysiological events in the generation of cholestatic liver damage. In fact, the increased hepatic content of NOx and nitrosothiols following BDL point to enhanced hepatocellular NO production, although some studies have shown no increase or even a decreased delivery of hepatic NO by endothelial cells and macrophages in pre-cirrhotic BDL rats[84,85]. On the other hand, others have reported increased hepatic iNOS expression in PBC patients[77] and in the hepatocytes of BDL rats[70]. Indeed, the physiological role of nitrosothiols in biological systems has also not been completely clarified as of yet. Nitrosothiols are unstable thioesters exerting different functions within the cell and in the extracellular compartment[86]. Nitrosothiols are not exclusively formed in cells generating NO, and can be taken up by cells via amino acid transport systems[87]. In a concentration dependent manner, intracellular nitrosothiols seems to be actively involved either in redox signaling or as nitrosative stress mediators[88], whereas, by representing a storage pool of thiols and NO, extracellular nitrosothiols have an importance in the regulation of the protein redox status at the cell surface[89]. In tissues, the formation of nitrosothiols adducts occurs under conditions of nitrosative stress and counteracts excess NO radicals[60]. Therefore, increased levels of tissue nitrosothiols may result from increased formation and/or decreased decomposition[86,90], such as in the presence of reduced thioredoxin availability.
Interaction between oxidative and nitrosative stress
The interplay between antioxidant molecules and oxidative and nitrosative stress derivatives renders these interactions particularly fascinating in the field of chronic cholestasis, where adaptive processes, in particular those at the subcellular level, are ultimately responsible for hepatocyte death or survival. Indeed, mutual and interrelated changes between circulating and hepatic oxidative and nitrosative stress markers have been reported, and point to an active and interrelated participation of these biochemical events in both liver injury and extrahepatic changes during cholestasis[6,33,65]. In fact, chronic cholestasis results in early and significant interactive changes between thioredoxin and nitrosothiols both at the circulating level and in the liver. The close relationship between the levels of these molecules with liver histology in patients with PBC may account for their active implication in the progression of the disease itself.
This is an important observation since thioredoxin represents one major system playing a regulatory role for several subcellular activities. Thioredoxin has an active role in oxidative/nitrosative stress interplay; in fact, it is also involved in the regulation of some NO activities through the cleavage of nitrosothiols[90,91] and the suppression of peroxynitrite formation[60]. Serum thioredoxin levels appear to discriminate between aggressive and benign forms of chronic non-cholestatic liver disease conditions[92]. In the context of PBC, the increased circulating levels of thioredoxin in patients with stage I-II suggest that it is likely induced to counteract increased ongoing oxidative stress or that it is an early adaptive measure to regulate NO metabolism, nitrosothiols decomposition, and to maintain surface PSH in the reduced form. Conversely, the decreased circulating level of thioredoxin in patients with more advanced stages of PBC likely reflects a diminished hepatic synthesis and delivery into the vasculature.
However, with the progression of cholestasis, the decreased production of thioredoxin in the liver, likely due to a down-regulation process associated with excess retention of hydrophobic bile salts and toxic molecules, may indirectly contribute to the progressive appearance of oxidative alterations, and therefore the promotion of fibrosis. In fact, adequate thioredoxin levels effectively protect against stellate cells activation and opposite collagen synthesis and hepatic fibrosis[93]. As a consequence of thioredoxin changes, the serum levels of nitrosothiols and the oxidation of erythrocyte PSH are increased in stage IV patients, indicating that circulating thioredoxin serves to protect surface PSH from oxidation and regulates nitrosothiols levels.
In this regard, it is known that nitrosothiols can be decomposed either by enzymatic homolytic cleavage in a NADPH dependent reaction involving the thioredoxin system, by a redox-sensitive metalloprotein, or via a non-enzymatic one electron reduction[86,91,94]. Notably, one recent study has reported that thioredoxin-deficient cells denitrosate S-nitrosothiols less efficiently[91]. Given the fact that increased nitrosothiols formation is associated with disease progression in PBC patients, they may have a potential use as a prognostic indicator in these patients.
The close link between oxidative and nitrosative stress parameters in PBC is further shown by the inverse relationship existing between serum thioredoxin and nitrotyrosine (r = -0.838), as well as between thioredoxin and cytokeratin 18 (r = -0.838), a major cytoplasmic intermediate filament protein in hepatocytes and cholangiocytes whose serum levels are elevated in chronic liver disease as a consequence of hepatic inflammation, and thus directly reflect the expression of cellular apoptotic processes (Figure 5)[95]. In PBC patients, cytokeratin 18 correlates well with serum nitrotyrosine (r = 0.894)[82].
Figure 5.

Link between nitric oxide derivatives and inflammatory products is linearly evidenced in this graph reporting the relationship between serum levels of nitrotyrosine and keratin-18. Patients with primary biliary cirrhosis (n = 30) were divided in stages according to liver histology: stage I; stage II; stage III; stage IV. Data adapted from Grattagliano and Portincasa (unpublished). Nitrotyrosine and keratin-18 (K-18) concentrations measured as previously reported[82].
Hepatic mitochondrial changes
Prolonged cholestasis is known to be associated with changes in mitochondrial morphology (i.e., shortening of cristae, appearance of inclusions, and matrix darkening) and energy metabolism[8,10,96]. In fact, both the number and volume ratio of mitochondria increase and parallel the duration of biliary obstruction in rats[97]. These findings point to adaptive processes in response to mitochondrial dysfunction[98]. Mitochondria are plastic organelles constantly changing their shape to fulfill their various functional activities[99].
In this regard, the permeability transition pore (PTP), which is formed across the mitochondrial membranes, has been hypothesized to have a major part in the rapid movements of water into and out of the mitochondrial matrix underlying the changes in volume that characterize the organelle[100]. The aqueous pore formed by the AQP8 isoform across the inner mitochondrial membrane is found to be more relevant to mitochondrial processes, such as release of H2O2[101] and import of ammonia by the organelle (urea cycle)[102-104], than underlying mitochondrial volume homeostasis. It has been observed that the mitochondrial changes accompanying the ongoing cholestasis occur early and parallel the progressive decrease of mitochondrial GSH concentrations and the increase of GSSG levels. In our context, therefore, low levels of mitochondrial GSH are associated with increased susceptibility to oxidative damages[105], resulting in protein oxidation responsible for the activation of cell death pathways[106,107]. The amount of mitochondrial PSH lowers with the cholestasis progression and, similarly to the extra-mitochondrial compartment, both GSH-peroxidase and GSSG-reductase activities increase (Table 1). The decrease in PSH parallels the increased content of protein disulfides (PSSG) and decreased expression of AQP8 (Figures 6 and 7), thus testifying that cholestasis is associated with mitochondrial oxidative changes of proteins and sulfhydryls, as well as functional impairment.
Table 1.
Time-related changes of glutathione peroxidase and glutathione reductase activities in liver cytosol and mitochondria of sham-operated and bile duct ligated rats
| Cytosol | Day 0 | Sham Day 3 | BDL Day 3 | Sham Day 10 | BDL Day 10 |
| GSH-Px | 351 ± 32 | 336 ± 35 | 504 ± 541 | 335 ± 20 | 566 ± 3212 |
| GSSG-Rx | 0.31 ± 0.11 | 0.27 ± 0.10 | 0.37 ± 0.081 | 0.28 ± 0.05 | 0.60 ± 0.1012 |
| Mitochondria | |||||
| GSH-Px | 977 ± 27 | 913 ± 44 | 1061 ± 95 | 890 ± 42 | 1568 ± 10512 |
| GSSG-Rx | 1.12 ± 0.17 | 0.93 ± 0.09 | 1.33 ± 0.121 | 0.89 ± 0.07 | 1.98 ± 0.1212 |
Glutathione peroxidase (GSH-Px) and glutathione reductase (GSSG-Rx) are reported as mU/mg protein/minute. Significantly different (0.01 < P < 0.05) compared to
baseline and
sham-operated rats at each time point. Adapted from Grattagliano and Portincasa (unpublished data). Enzyme activities assessed as previously described[58]. BDL: Bile duct ligation.
Figure 6.
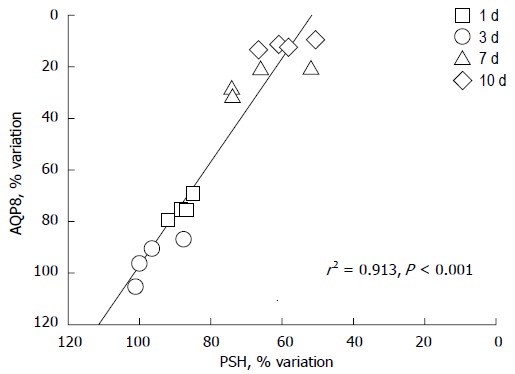
Aquaporin 8 is a specific water channel actively involved in the regulation of mitochondrial volume in the liver. Protein sulfhydryls (PSH) are a diffuse category of proteins playing important roles in mitochondria, including structural proteins and respiratory complexes to channel pores. The graph depicts the correlation between densitometric values of aquaporin 8 (AQP8) immunoreactivity and PSH content in the liver mitochondria of sham-operated and bile duct ligated rats at different time-points. AQP8 expression varies in relation to the changes in PSH content (n = 16, r = +0.96, r2 = 0.913, P < 0.0001). N = 4 experiments per time point with 1 d, 3 d, 7 d and 10 d; BDL. Data adapted from Grattagliano and Portincasa (unpublished). Measurements performed as previously reported[65].
Figure 7.
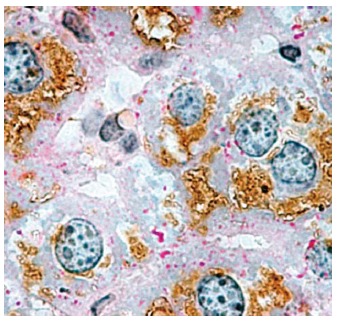
Immunohistochemical localization of aquaporin 8 in mouse hepatocytes. Considerable aquaporin 8 (AQP8) immunoreactivity (brown staining) is seen within the cytoplasmic compartment of most hepatocytes. Periodic acid-Schiff (PAS) reactivity (red staining) is seen over glycogen granules. In rodents, hepatocyte AQP8 is expressed at multiple subcellular levels, including the apical (canalicular) plasma membrane, subapical vesicles, smooth endoplasmic reticulum, and mitochondria. The AQP8 channel features conductance to water, ammonia, and H2O2, and is suggested to be involved in primary bile secretion, ammonia detoxification, ureagenesis, and mitochondrial reactive oxygen species generation. Original magnification, × 1000. (micrograph from Ferri et al[22], reproduced with permission from Wiley Online Library).
These oxidative changes of mitochondria are functionally important for the energetic activities known to be damaged early during cholestasis, as reported previously[10]. With this in mind, the observation that GSH content declines earlier and at a greater extent in mitochondria than in the cytosolic compartment, as observed in rodent models of cholestasis, points to selective mitochondrial damage. This behavior of mitochondrial GSH might imply two different hypotheses: defective ex novo synthesis of cytosolic GSH, which is then imported by mitochondria, or increased mitochondrial oxidative consumption. Both may follow the intrahepatic accumulation of hydrophobic bile salts (i.e., tauro- and glycochenodeoxycholate) and may also coexist. Indeed, it is known that liver mitochondria exposed to hydrophobic bile salts generate a great amount of ROS, and that damaged mitochondria result in a higher release of radicals[108,109]. The former hypothesis is also supported by the increased expression of mRNA for γ-glutamylcysteine synthetase and by the enhanced activity of methionine adenosyl-transferase following administration of ursodeoxycholate (UDCA)[110]. UDCA (molecular formula C24H40O4, molecular weight: 392.572) is the 7β-epimer of the primary dihydroxy bile acid chenodeoxycholic acid (representing the hepatic catabolic product of cholesterol)[111]. UDCA is found in mammalian bile, following bacterial reduction in ileum and colon. As a therapeutic agent, UDCA has hydrophilic and less cytotoxic properties. The latter hypothesis is supported by the association of a marked increase of GSSG and PSSG with the increased activity of GSH-related antioxidant enzymes. Furthermore, mitochondria from BDL rats exhibited an altered lipid composition, with a two- to threefold increase in the cholesterol/phospholipid ratio in the mitochondrial inner membrane[97], thus suggesting the possibility of impaired transport of GSH into mitochondria.
Moreover, it is important to note that mitochondrial protein content is generally decreased in BDL rats and, together with their increased oxidation rate, deserves some consideration. Firstly, mitochondria exhibiting a decrease of functional proteins are less prone to rapidly changing their shape and, therefore, are less able to adapt their function in response to environmental stress. Secondly, the decreased content of proteins in mitochondria may be the consequence of down-regulation processes to which several proteins undergo during cholestasis when the excretory function is severely impaired[112].
Recent studies have shown that mitochondrial concentrations of nitrosothiols are increased after BDL to an even higher extent than that observed in the extra-mitochondrial compartment[65]. Indeed, NO is known to stimulate hepatocyte GMPc dependent signaling pathways and, in mitochondria, it is an important physiological reactant. NO controls mitochondrial ATP synthase, gene expression, and PTP through protein nitrosylation and nitrosation[88,113]. Mitochondria are known to possess their own NO metabolism[114] and a specific NOS that generates the highest rate of intracellular NO●[88,115]. Moreover, NO may provide beneficial effects at this level by supporting cell survival, thus resulting as a determining factor for mitochondriogenesis[116,117].
Among redox active compounds, thioredoxin is a molecule having an important role for the mitochondrial energetic domain. In fact, overexpression of mitochondrial thioredoxin and thioredoxin reductase attenuates damages associated with excess NO[118] by increasing the mitochondrial membrane potential[119]. In particular, mitochondrial thioredoxin is critical for defense against oxidative stress induced cell apoptosis, and thioredoxin reductase is the only known enzyme catalyzing thioredoxin reduction in mitochondria. However, thioredoxin reductase is known to be sensitive to inactivation by electrophiles, such as some lipoperoxidative products. Glutaredoxin, a system helping to keep thioredoxin reduced during stress events, also contributes to anti-apoptotic signaling. This mitochondrial thiol-disulfide oxidoreductase, in fact, is relatively independent from the cytosolic compartment. In particular, thioredoxin and glutaredoxin systems consist of NADPH, thioredoxin reductase[120], and thioredoxin[121], or NADPH, GSH-reductase, GSH, and glutaredoxin[122], respectively. Within mitochondria, thioredoxin and glutaredoxin play critical roles in controlling protein folding and regulating cell growth/apoptosis[123-126]. An experimental-computational study converged on the idea that GSH/thioredoxin scavenging systems in mitochondria are both essential for keeping minimal levels of H2O2 emission[127]. The crosstalk between these two systems may support each other’s functions, but could not substitute for each other, probably due to their respective functions and different substrates. Thioredoxin is more active in catalyzing the reduction of intra- or inter-chain disulfides of protein substrates, is an electron donor for peroxiredoxins, a major player in the elimination of H2O2[128], and regulates the activity/activation of transcription factors or apoptosis signaling factors through modulating the redox-regulatory disulfides of the protein. Mitochondrial thioredoxin in the reduced form is usually an indicator of cell survival, while the predominance of its oxidized form is an indicator of cell death. Glutaredoxin is not inactivated by oxidation[122] and its insensitiveness increases the chance of protecting mitochondrial thioredoxin from oxidation when the reductase is inactivated. It is specifically active in catalyzing the deglutathionylation of proteins at the expense of GSH.
Protein S-glutathionylation is an important reversible post-translational modification of proteins that performs thiol-redox signaling for many cellular events, including apoptosis[129]. Indeed, it is known that nitrosative stress is critically important in promoting S-nitrosylation and S-glutathiolation of various mitochondrial proteins, leading to mitochondrial dysfunction, decreased energy supply[130], and increased hepatic injury[130]. With the progression of cholestasis, however, the decreased availability of thioredoxin, together with excess NO and GSH depletion[131], may result in enhanced protein nitrosation[76] and PSH oxidation. These events have obviously negative consequences for hepatocyte survival and bile duct integrity[78,132]. Of course, whether over-expression of mitochondrial glutaredoxin and thioredoxin systems might have therapeutic potential in diseases such as PBC where mitochondrial oxidation plays a dominant role is an intriguing challenge for future investigations.
CONCLUSION
The data discussed here from integrated and translational studies unequivocally suggests that oxidative and nitrosative events determine disease appearance and progression in patients with PBC. Changes in circulating markers, such as thioredoxin and nitrosothiols, may be used to monitor disease progression and to identify patients at risk of disease evolution. Finally, it may be argued that pharmacological interventions directed to sustain hepatic GSH, thioredoxin, and glutaredoxin levels by activating nuclear factors and up-regulating gene transcription[133-135] may favorably contrast NO deranged metabolism in the early phase of chronic cholestatic conditions and may yield hepatocyte protection by favoring mitochondrial proliferation through the maintenance of protein redox status. In fact, the modulation of the protein oxidation/denitrosation process has been suggested as a mechanism of either cytoprotection or cellular damage. In this connection, recent studies have revealed new therapeutic aspects of some compounds. UDCA, initially introduced in therapy to counteract the cholestatic components of PBC, was subsequently shown to have also anti-inflammatory and immunomodulatory properties (Table 2). The use of farnesoid X receptor agonists in animal models of cholestasis has confirmed that bile acids are not only toxicants and inflammagens, but also repressors of innate and adaptive immunity[136]. In fact, other than a well-established role in the digestion and absorption of dietary lipids, bile acids are currently recognized as signaling molecules in a wide range of metabolic processes in which they contribute to improve hyperglycemia and insulin resistance[137].
Table 2.
Therapeutic doses of ursodeoxycholic acid and its potential effects in the treatment of patients with primary biliary cirrhosis
| Effective dose: 13-15 mg/kg per day indefinitely |
| Mechanisms of action |
| Promotion of endogenous bile acids secretion |
| Replacement of hepatotoxic (endogenous) bile acids |
| Stabilization of biliary epithelial cell membranes |
| Alteration of HLA I-II expression on biliary epithelial cell |
| Inhibition of biliary cell apoptosis |
| Improvement of hepatocyte redox status |
| Signaling for glucose and insulin metabolic pathways |
| Delays disease progression and improves transplant-free survival |
For this reason, understanding the complexity of the inflammatory mechanisms leading to bile duct epithelial injury represents a crucial step for the future development of therapies aimed at inhibiting ongoing biliary tract destruction in PBC. Both oxidative and nitrosative injurious mechanisms are effectors of damage in the liver, and therefore, interventions aimed at rectifying these processes may have therapeutic effects. Recently, UDCA has shown to protect against oxidative and nitrosative stress, at least in the early stages of PBC[65,138].
Footnotes
P- Reviewers: Bubnov RV, Guerrieri F, Kaufmann R, Mekky MAS, Micuda S, Solinas A S- Editor: Qi Y L- Editor: Rutherford A E- Editor: Zhang DN
References
- 1.Reshetnyak VI. Concept on the pathogenesis and treatment of primary biliary cirrhosis. World J Gastroenterol. 2006;12:7250–7262. doi: 10.3748/wjg.v12.i45.7250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu BR, Mack CL. Inflammation and biliary tract injury. Curr Opin Gastroenterol. 2009;25:260–264. doi: 10.1097/mog.0b013e328325aa10. [DOI] [PubMed] [Google Scholar]
- 3.Kar SP, Seldin MF, Chen W, Lu E, Hirschfield GM, Invernizzi P, Heathcote J, Cusi D, Gershwin ME, Siminovitch KA, et al. Pathway-based analysis of primary biliary cirrhosis genome-wide association studies. Genes Immun. 2013;14:179–186. doi: 10.1038/gene.2013.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lleo A, Invernizzi P, Mackay IR, Prince H, Zhong RQ, Gershwin ME. Etiopathogenesis of primary biliary cirrhosis. World J Gastroenterol. 2008;14:3328–3337. doi: 10.3748/wjg.14.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harada K, Nakanuma Y. Molecular mechanisms of cholangiopathy in primary biliary cirrhosis. Med Mol Morphol. 2006;39:55–61. doi: 10.1007/s00795-006-0321-z. [DOI] [PubMed] [Google Scholar]
- 6.Krähenbühl S, Talos C, Lauterburg BH, Reichen J. Reduced antioxidative capacity in liver mitochondria from bile duct ligated rats. Hepatology. 1995;22:607–612. doi: 10.1002/hep.1840220234. [DOI] [PubMed] [Google Scholar]
- 7.Masyuk AI, Marinelli RA, LaRusso NF. Water transport by epithelia of the digestive tract. Gastroenterology. 2002;122:545–562. doi: 10.1053/gast.2002.31035. [DOI] [PubMed] [Google Scholar]
- 8.Krähenbühl S, Talos C, Reichen J. Mechanisms of impaired hepatic fatty acid metabolism in rats with long-term bile duct ligation. Hepatology. 1994;19:1272–1281. doi: 10.1002/hep.1840190528. [DOI] [PubMed] [Google Scholar]
- 9.Shoda J, Kano M, Asano T, Irimura T, Ueda T, Iwasaki R, Furukawa M, Kamiya J, Nimura Y, Todoroki T, et al. Secretory low-molecular-weight phospholipases A2 and their specific receptor in bile ducts of patients with intrahepatic calculi: factors of chronic proliferative cholangitis. Hepatology. 1999;29:1026–1036. doi: 10.1002/hep.510290440. [DOI] [PubMed] [Google Scholar]
- 10.Krähenbühl S, Talos C, Fischer S, Reichen J. Toxicity of bile acids on the electron transport chain of isolated rat liver mitochondria. Hepatology. 1994;19:471–479. doi: 10.1002/hep.1840190228. [DOI] [PubMed] [Google Scholar]
- 11.Shoda J, Kano M, Oda K, Kamiya J, Nimura Y, Suzuki H, Sugiyama Y, Miyazaki H, Todoroki T, Stengelin S, et al. The expression levels of plasma membrane transporters in the cholestatic liver of patients undergoing biliary drainage and their association with the impairment of biliary secretory function. Am J Gastroenterol. 2001;96:3368–3378. doi: 10.1111/j.1572-0241.2001.05339.x. [DOI] [PubMed] [Google Scholar]
- 12.Sokol RJ, Devereaux M, Khandwala RA. Effect of dietary lipid and vitamin E on mitochondrial lipid peroxidation and hepatic injury in the bile duct-ligated rat. J Lipid Res. 1991;32:1349–1357. [PubMed] [Google Scholar]
- 13.Sokol RJ, Devereaux M, Khandwala R, O’Brien K. Evidence for involvement of oxygen free radicals in bile acid toxicity to isolated rat hepatocytes. Hepatology. 1993;17:869–881. [PubMed] [Google Scholar]
- 14.Portincasa P, Moschetta A, Mazzone A, Palasciano G, Svelto M, Calamita G. Water handling and aquaporins in bile formation: recent advances and research trends. J Hepatol. 2003;39:864–874. doi: 10.1016/s0168-8278(03)00294-0. [DOI] [PubMed] [Google Scholar]
- 15.Grattagliano I, Portincasa P, Palmieri VO, Palasciano G. Contribution of canalicular glutathione efflux to bile formation. From cholestasis associated alterations to pharmacological intervention to modify bile flow. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:153–161. doi: 10.2174/1568008054064931. [DOI] [PubMed] [Google Scholar]
- 16.Bilzer M, Lauterburg BH. Effects of hypochlorous acid and chloramines on vascular resistance, cell integrity, and biliary glutathione disulfide in the perfused rat liver: modulation by glutathione. J Hepatol. 1991;13:84–89. doi: 10.1016/0168-8278(91)90868-c. [DOI] [PubMed] [Google Scholar]
- 17.Bouchard G, Tuchweber B, Yousef IM. Bile salt independent flow during bile salt-induced choleresis and cholestasis in the rat: role of biliary thiol secretion. Liver. 2000;20:27–37. doi: 10.1034/j.1600-0676.2000.020001027.x. [DOI] [PubMed] [Google Scholar]
- 18.Huebert RC, Splinter PL, Garcia F, Marinelli RA, LaRusso NF. Expression and localization of aquaporin water channels in rat hepatocytes. Evidence for a role in canalicular bile secretion. J Biol Chem. 2002;277:22710–22717. doi: 10.1074/jbc.M202394200. [DOI] [PubMed] [Google Scholar]
- 19.Calamita G, Mazzone A, Bizzoca A, Cavalier A, Cassano G, Thomas D, Svelto M. Expression and immunolocalization of the aquaporin-8 water channel in rat gastrointestinal tract. Eur J Cell Biol. 2001;80:711–719. doi: 10.1078/0171-9335-00210. [DOI] [PubMed] [Google Scholar]
- 20.Tani T, Koyama Y, Nihei K, Hatakeyama S, Ohshiro K, Yoshida Y, Yaoita E, Sakai Y, Hatakeyama K, Yamamoto T. Immunolocalization of aquaporin-8 in rat digestive organs and testis. Arch Histol Cytol. 2001;64:159–168. doi: 10.1679/aohc.64.159. [DOI] [PubMed] [Google Scholar]
- 21.Calamita G, Ferri D, Gena P, Liquori GE, Cavalier A, Thomas D, Svelto M. The inner mitochondrial membrane has aquaporin-8 water channels and is highly permeable to water. J Biol Chem. 2005;280:17149–17153. doi: 10.1074/jbc.C400595200. [DOI] [PubMed] [Google Scholar]
- 22.Ferri D, Mazzone A, Liquori GE, Cassano G, Svelto M, Calamita G. Ontogeny, distribution, and possible functional implications of an unusual aquaporin, AQP8, in mouse liver. Hepatology. 2003;38:947–957. doi: 10.1053/jhep.2003.50397. [DOI] [PubMed] [Google Scholar]
- 23.Calamita G, Ferri D, Gena P, Carreras FI, Liquori GE, Portincasa P, Marinelli RA, Svelto M. Altered expression and distribution of aquaporin-9 in the liver of rat with obstructive extrahepatic cholestasis. Am J Physiol Gastrointest Liver Physiol. 2008;295:G682–G690. doi: 10.1152/ajpgi.90226.2008. [DOI] [PubMed] [Google Scholar]
- 24.Portincasa P, Calamita G. Water channel proteins in bile formation and flow in health and disease: when immiscible becomes miscible. Mol Aspects Med. 2012;33:651–664. doi: 10.1016/j.mam.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 25.Calamita G, Gena P, Ferri D, Rosito A, Rojek A, Nielsen S, Marinelli RA, Frühbeck G, Svelto M. Biophysical assessment of aquaporin-9 as principal facilitative pathway in mouse liver import of glucogenetic glycerol. Biol Cell. 2012;104:342–351. doi: 10.1111/boc.201100061. [DOI] [PubMed] [Google Scholar]
- 26.Lehmann GL, Larocca MC, Soria LR, Marinelli RA. Aquaporins: their role in cholestatic liver disease. World J Gastroenterol. 2008;14:7059–7067. doi: 10.3748/wjg.14.7059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marinelli RA, Gradilone SA, Carreras FI, Calamita G, Lehmann GL. Liver aquaporins: significance in canalicular and ductal bile formation. Ann Hepatol. 2004;3:130–136. [PubMed] [Google Scholar]
- 28.Lenzen R, Hruby VJ, Tavoloni N. mechanism of glucagon choleresis in guinea pigs. Am J Physiol. 1990;259:G736–G744. doi: 10.1152/ajpgi.1990.259.5.G736. [DOI] [PubMed] [Google Scholar]
- 29.Soria LR, Gradilone SA, Larocca MC, Marinelli RA. Glucagon induces the gene expression of aquaporin-8 but not that of aquaporin-9 water channels in the rat hepatocyte. Am J Physiol Regul Integr Comp Physiol. 2009;296:R1274–R1281. doi: 10.1152/ajpregu.90783.2008. [DOI] [PubMed] [Google Scholar]
- 30.Chojkier M. Regulation of liver-specific gene expression. Prog Liver Dis. 1995;13:37–61. [PubMed] [Google Scholar]
- 31.Muriel P, Suarez OR. Role of lipid peroxidation in biliary obstruction in the rat. J Appl Toxicol. 1994;14:423–426. doi: 10.1002/jat.2550140607. [DOI] [PubMed] [Google Scholar]
- 32.Davies MH, Ngong JM, Pean A, Vickers CR, Waring RH, Elias E. Sulphoxidation and sulphation capacity in patients with primary biliary cirrhosis. J Hepatol. 1995;22:551–560. doi: 10.1016/0168-8278(95)80450-1. [DOI] [PubMed] [Google Scholar]
- 33.Vendemiale G, Grattagliano I, Lupo L, Memeo V, Altomare E. Hepatic oxidative alterations in patients with extra-hepatic cholestasis. Effect of surgical drainage. J Hepatol. 2002;37:601–605. doi: 10.1016/s0168-8278(02)00234-9. [DOI] [PubMed] [Google Scholar]
- 34.Ballatori N, Truong AT. Relation between biliary glutathione excretion and bile acid-independent bile flow. Am J Physiol. 1989;256:G22–G30. doi: 10.1152/ajpgi.1989.256.1.G22. [DOI] [PubMed] [Google Scholar]
- 35.Clottes E, Burchell A. Three thiol groups are important for the activity of the liver microsomal glucose-6-phosphatase system. Unusual behavior of one thiol located in the glucose-6-phosphate translocase. J Biol Chem. 1998;273:19391–19397. doi: 10.1074/jbc.273.31.19391. [DOI] [PubMed] [Google Scholar]
- 36.Morin D, Barthélémy S, Zini R, Labidalle S, Tillement JP. Curcumin induces the mitochondrial permeability transition pore mediated by membrane protein thiol oxidation. FEBS Lett. 2001;495:131–136. doi: 10.1016/s0014-5793(01)02376-6. [DOI] [PubMed] [Google Scholar]
- 37.Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic Biol Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- 38.Harry DS, Owen JS, McIntyre N. Lipids, lipoproteins, and cell membranes. Prog Liver Dis. 1982;7:319–329. [PubMed] [Google Scholar]
- 39.Schachter D. Fluidity and function of hepatocyte plasma membranes. Hepatology. 1984;4:140–151. doi: 10.1002/hep.1840040124. [DOI] [PubMed] [Google Scholar]
- 40.Ozdemirler G, Küçük S, Orhan Y, Aykaç-Toker G, Uysal M. Lipid and protein oxidation in erythrocyte membranes of hypercholesterolemic subjects. Clin Biochem. 2001;34:335–339. doi: 10.1016/s0009-9120(01)00208-9. [DOI] [PubMed] [Google Scholar]
- 41.Owen JS, Bruckdorfer KR, Day RC, McIntyre N. Decreased erythrocyte membrane fluidity and altered lipid composition in human liver disease. J Lipid Res. 1982;23:124–132. [PubMed] [Google Scholar]
- 42.Rafique S, Guardascione M, Osman E, Burroughs AK, Owen JS. Reversal of extrahepatic membrane cholesterol deposition in patients with chronic liver diseases by S-adenosyl-L-methionine. Clin Sci (Lond) 1992;83:353–356. doi: 10.1042/cs0830353. [DOI] [PubMed] [Google Scholar]
- 43.Del Puppo M, Galli Kienle M, Crosignani A, Petroni ML, Amati B, Zuin M, Podda M. Cholesterol metabolism in primary biliary cirrhosis during simvastatin and UDCA administration. J Lipid Res. 2001;42:437–441. [PubMed] [Google Scholar]
- 44.Stramentinoli G, Di Padova C, Gualano M, Rovagnati P, Galli-Kienle M. Ethynylestradiol-induced impairment of bile secretion in the rat: protective effects of S-adenosyl-L-methionine and its implication in estrogen metabolism. Gastroenterology. 1981;80:154–158. [PubMed] [Google Scholar]
- 45.Cynamon HA, Isenberg JN. Erythrocyte lipid alterations in pediatric cholestatic liver disease: relationship to serum bile acids. Hepatogastroenterology. 1986;33:98–100. [PubMed] [Google Scholar]
- 46.Owen JS, Hutton RA, Day RC, Bruckdorfer KR, McIntyre N. Platelet lipid composition and platelet aggregation in human liver disease. J Lipid Res. 1981;22:423–430. [PubMed] [Google Scholar]
- 47.Muriel P, Favari L, Soto C. Erythrocyte alterations correlate with CCl4 and biliary obstruction-induced liver damage in the rat. Life Sci. 1993;52:647–655. doi: 10.1016/0024-3205(93)90456-d. [DOI] [PubMed] [Google Scholar]
- 48.Okano Y, Iida H, Goto M, Sekiya T, Shiojiri H, Hasegawa I, Nozawa Y. Erythrocyte membrane abnormalities in experimental biliary obstruction: comparative studies on erythrocyte membranes from rats with intra- and extrahepatic cholestasis. Jpn J Exp Med. 1980;50:445–451. [PubMed] [Google Scholar]
- 49.Grattagliano I, Giudetti AM, Grattagliano V, Palmieri VO, Gnoni GV, Lapadula G, Palasciano G, Vendemiale G. Structural and oxidative modifications of erythrocyte ghosts in patients with primary biliary cirrhosis: relation with the disease stage and effect of bile acid treatment. Eur J Clin Invest. 2003;33:868–874. doi: 10.1046/j.1365-2362.2003.01238.x. [DOI] [PubMed] [Google Scholar]
- 50.Grattagliano I, Russmann S, Palmieri VO, Jüni P, Bihl F, Portincasa P, Palasciano G, Lauterburg BH. Low membrane protein sulfhydrils but not G6PD deficiency predict ribavirin-induced hemolysis in hepatitis C. Hepatology. 2004;39:1248–1255. doi: 10.1002/hep.20208. [DOI] [PubMed] [Google Scholar]
- 51.Chapman D, Gómez-Fernández JC, Goñi FM. Intrinsic protein--lipid interactions. Physical and biochemical evidence. FEBS Lett. 1979;98:211–223. doi: 10.1016/0014-5793(79)80186-6. [DOI] [PubMed] [Google Scholar]
- 52.Jähnig F. Structural order of lipids and proteins in membranes: evaluation of fluorescence anisotropy data. Proc Natl Acad Sci USA. 1979;76:6361–6365. doi: 10.1073/pnas.76.12.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kakimoto H, Imai Y, Kawata S, Inada M, Ito T, Matsuzawa Y. Altered lipid composition and differential changes in activities of membrane-bound enzymes of erythrocytes in hepatic cirrhosis. Metabolism. 1995;44:825–832. doi: 10.1016/0026-0495(95)90233-3. [DOI] [PubMed] [Google Scholar]
- 54.Taniguchi M, Tanabe F, Ishikawa H, Sakagami T. Experimental biliary obstruction of rat. Initial changes in the structure and lipid content of erythrocytes. Biochim Biophys Acta. 1983;753:22–31. [PubMed] [Google Scholar]
- 55.Ben-Ari Z, Tur-Kaspa R, Schafer Z, Baruch Y, Sulkes J, Atzmon O, Greenberg A, Levi N, Fainaru M. Basal and post-methionine serum homocysteine and lipoprotein abnormalities in patients with chronic liver disease. J Investig Med. 2001;49:325–329. doi: 10.2310/6650.2001.33897. [DOI] [PubMed] [Google Scholar]
- 56.Cabrero C, Duce AM, Ortiz P, Alemany S, Mato JM. Specific loss of the high-molecular-weight form of S-adenosyl-L-methionine synthetase in human liver cirrhosis. Hepatology. 1988;8:1530–1534. doi: 10.1002/hep.1840080610. [DOI] [PubMed] [Google Scholar]
- 57.Duce AM, Ortíz P, Cabrero C, Mato JM. S-adenosyl-L-methionine synthetase and phospholipid methyltransferase are inhibited in human cirrhosis. Hepatology. 1988;8:65–68. doi: 10.1002/hep.1840080113. [DOI] [PubMed] [Google Scholar]
- 58.Portincasa P, Grattagliano I, Testini M, Caruso ML, Wang DQ, Moschetta A, Calamita G, Vacca M, Valentini AM, Renna G, et al. Parallel intestinal and liver injury during early cholestasis in the rat: modulation by bile salts and antioxidants. Free Radic Biol Med. 2007;42:1381–1391. doi: 10.1016/j.freeradbiomed.2007.01.039. [DOI] [PubMed] [Google Scholar]
- 59.Nakamura H, Nakamura K, Yodoi J. Redox regulation of cellular activation. Annu Rev Immunol. 1997;15:351–369. doi: 10.1146/annurev.immunol.15.1.351. [DOI] [PubMed] [Google Scholar]
- 60.Nordberg J, Arnér ES. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med. 2001;31:1287–1312. doi: 10.1016/s0891-5849(01)00724-9. [DOI] [PubMed] [Google Scholar]
- 61.Bindoli A, Rigobello MP. Mitochondrial thioredoxin reductase and thiol status. Methods Enzymol. 2002;347:307–316. doi: 10.1016/s0076-6879(02)47030-9. [DOI] [PubMed] [Google Scholar]
- 62.Rigobello MP, Callegaro MT, Barzon E, Benetti M, Bindoli A. Purification of mitochondrial thioredoxin reductase and its involvement in the redox regulation of membrane permeability. Free Radic Biol Med. 1998;24:370–376. doi: 10.1016/s0891-5849(97)00216-5. [DOI] [PubMed] [Google Scholar]
- 63.Yoshida S, Katoh T, Tetsuka T, Uno K, Matsui N, Okamoto T. Involvement of thioredoxin in rheumatoid arthritis: its costimulatory roles in the TNF-alpha-induced production of IL-6 and IL-8 from cultured synovial fibroblasts. J Immunol. 1999;163:351–358. [PubMed] [Google Scholar]
- 64.Bertini R, Howard OM, Dong HF, Oppenheim JJ, Bizzarri C, Sergi R, Caselli G, Pagliei S, Romines B, Wilshire JA, et al. Thioredoxin, a redox enzyme released in infection and inflammation, is a unique chemoattractant for neutrophils, monocytes, and T cells. J Exp Med. 1999;189:1783–1789. doi: 10.1084/jem.189.11.1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Grattagliano I, Portincasa P, Palmieri VO, Palasciano G. Mutual changes of thioredoxin and nitrosothiols during biliary cirrhosis: results from humans and cholestatic rats. Hepatology. 2007;45:331–339. doi: 10.1002/hep.21519. [DOI] [PubMed] [Google Scholar]
- 66.Kanda T, Foucand L, Nakamura Y, Niot I, Besnard P, Fujita M, Sakai Y, Hatakeyama K, Ono T, Fujii H. Regulation of expression of human intestinal bile acid-binding protein in Caco-2 cells. Biochem J. 1998;330(Pt 1):261–265. doi: 10.1042/bj3300261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sauer P, Stiehl A, Fitscher BA, Riedel HD, Benz C, Klöters-Plachky P, Stengelin S, Stremmel W, Kramer W. Downregulation of ileal bile acid absorption in bile-duct-ligated rats. J Hepatol. 2000;33:2–8. doi: 10.1016/s0168-8278(00)80152-x. [DOI] [PubMed] [Google Scholar]
- 68.Slocum MM, Sittig KM, Specian RD, Deitch EA. Absence of intestinal bile promotes bacterial translocation. Am Surg. 1992;58:305–310. [PubMed] [Google Scholar]
- 69.Dietrich CG, Geier A, Salein N, Lammert F, Roeb E, Oude Elferink RP, Matern S, Gartung C. Consequences of bile duct obstruction on intestinal expression and function of multidrug resistance-associated protein 2. Gastroenterology. 2004;126:1044–1053. doi: 10.1053/j.gastro.2003.12.046. [DOI] [PubMed] [Google Scholar]
- 70.Deitch EA, Bridges WM, Ma JW, Ma L, Berg RD, Specian RD. Obstructed intestine as a reservoir for systemic infection. Am J Surg. 1990;159:394–401. doi: 10.1016/s0002-9610(05)81280-2. [DOI] [PubMed] [Google Scholar]
- 71.Arriero MM, López-Farré A, Fryeiro O, Rodríguez-Feo JA, Velasco S, García-Durán M, Fortes J, De La Pinta JC, Muñoz LE, Celdrán A. Expression of inducible nitric oxide synthase in the liver of bile duct-ligated Wistar rats with modulation by lymphomononuclear cells. Surgery. 2001;129:255–266. doi: 10.1067/msy.2001.110427. [DOI] [PubMed] [Google Scholar]
- 72.McNaughton L, Puttagunta L, Martinez-Cuesta MA, Kneteman N, Mayers I, Moqbel R, Hamid Q, Radomski MW. Distribution of nitric oxide synthase in normal and cirrhotic human liver. Proc Natl Acad Sci USA. 2002;99:17161–17166. doi: 10.1073/pnas.0134112100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hokari A, Zeniya M, Esumi H, Kawabe T, Gershwin ME, Toda G. Detection of serum nitrite and nitrate in primary biliary cirrhosis: possible role of nitric oxide in bile duct injury. J Gastroenterol Hepatol. 2002;17:308–315. doi: 10.1046/j.1440-1746.2002.02689.x. [DOI] [PubMed] [Google Scholar]
- 74.Welsh FK, Ramsden CW, MacLennan K, Sheridan MB, Barclay GR, Guillou PJ, Reynolds JV. Increased intestinal permeability and altered mucosal immunity in cholestatic jaundice. Ann Surg. 1998;227:205–212. doi: 10.1097/00000658-199802000-00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parks RW, Stuart Cameron CH, Gannon CD, Pope C, Diamond T, Rowlands BJ. Changes in gastrointestinal morphology associated with obstructive jaundice. J Pathol. 2000;192:526–532. doi: 10.1002/1096-9896(2000)9999:9999<::AID-PATH787>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 76.Ottesen LH, Harry D, Frost M, Davies S, Khan K, Halliwell B, Moore K. Increased formation of S-nitrothiols and nitrotyrosine in cirrhotic rats during endotoxemia. Free Radic Biol Med. 2001;31:790–798. doi: 10.1016/s0891-5849(01)00647-5. [DOI] [PubMed] [Google Scholar]
- 77.Sanz-Cameno P, Medina J, García-Buey L, García-Sánchez A, Borque MJ, Martín-Vílchez S, Gamallo C, Jones EA, Moreno-Otero R. Enhanced intrahepatic inducible nitric oxide synthase expression and nitrotyrosine accumulation in primary biliary cirrhosis and autoimmune hepatitis. J Hepatol. 2002;37:723–729. doi: 10.1016/s0168-8278(02)00266-0. [DOI] [PubMed] [Google Scholar]
- 78.Wu CT, Eiserich JP, Ansari AA, Coppel RL, Balasubramanian S, Bowlus CL, Gershwin ME, Van De Water J. Myeloperoxidase-positive inflammatory cells participate in bile duct damage in primary biliary cirrhosis through nitric oxide-mediated reactions. Hepatology. 2003;38:1018–1025. doi: 10.1053/jhep.2003.50407. [DOI] [PubMed] [Google Scholar]
- 79.Grattagliano I, Portincasa P, Cocco T, Moschetta A, Di Paola M, Palmieri VO, Palasciano G. Effect of dietary restriction and N-acetylcysteine supplementation on intestinal mucosa and liver mitochondrial redox status and function in aged rats. Exp Gerontol. 2004;39:1323–1332. doi: 10.1016/j.exger.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 80.Arnelle DR, Stamler JS. NO+, NO, and NO- donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Arch Biochem Biophys. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- 81.Stamler JS, Simon DI, Jaraki O, Osborne JA, Francis S, Mullins M, Singel D, Loscalzo J. S-nitrosylation of tissue-type plasminogen activator confers vasodilatory and antiplatelet properties on the enzyme. Proc Natl Acad Sci USA. 1992;89:8087–8091. doi: 10.1073/pnas.89.17.8087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Grattagliano I, Palmieri VO, Portincasa P, Minerva F, Palasciano G. Long-term ursodeoxycholate improves circulating redox changes in primary biliary cirrhotic patients. Clin Biochem. 2011;44:1400–1404. doi: 10.1016/j.clinbiochem.2011.09.008. [DOI] [PubMed] [Google Scholar]
- 83.Albillos A, Rossi I, Cacho G, Martínez MV, Millán I, Abreu L, Barrios C, Escartín P. Enhanced endothelium-dependent vasodilation in patients with cirrhosis. Am J Physiol. 1995;268:G459–G464. doi: 10.1152/ajpgi.1995.268.3.G459. [DOI] [PubMed] [Google Scholar]
- 84.Stumm MM, D’Orazio D, Sumanovski LT, Martin PY, Reichen J, Sieber CC. Endothelial, but not the inducible, nitric oxide synthase is detectable in normal and portal hypertensive rats. Liver. 2002;22:441–450. doi: 10.1034/j.1600-0676.2002.01653.x. [DOI] [PubMed] [Google Scholar]
- 85.Zimmermann H, Kurzen P, Klossner W, Renner EL, Marti U. Decreased constitutive hepatic nitric oxide synthase expression in secondary biliary fibrosis and its changes after Roux-en-Y choledocho-jejunostomy in the rat. J Hepatol. 1996;25:567–573. doi: 10.1016/s0168-8278(96)80218-2. [DOI] [PubMed] [Google Scholar]
- 86.Mathews WR, Kerr SW. Biological activity of S-nitrosothiols: the role of nitric oxide. J Pharmacol Exp Ther. 1993;267:1529–1537. [PubMed] [Google Scholar]
- 87.Zhang Y, Hogg N. S-Nitrosothiols: cellular formation and transport. Free Radic Biol Med. 2005;38:831–838. doi: 10.1016/j.freeradbiomed.2004.12.016. [DOI] [PubMed] [Google Scholar]
- 88.Brookes PS, Levonen AL, Shiva S, Sarti P, Darley-Usmar VM. Mitochondria: regulators of signal transduction by reactive oxygen and nitrogen species. Free Radic Biol Med. 2002;33:755–764. doi: 10.1016/s0891-5849(02)00901-2. [DOI] [PubMed] [Google Scholar]
- 89.Sahaf B, Heydari K, Herzenberg LA, Herzenberg LA. The extracellular microenvironment plays a key role in regulating the redox status of cell surface proteins in HIV-infected subjects. Arch Biochem Biophys. 2005;434:26–32. doi: 10.1016/j.abb.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 90.Nikitovic D, Holmgren A. S-nitrosoglutathione is cleaved by the thioredoxin system with liberation of glutathione and redox regulating nitric oxide. J Biol Chem. 1996;271:19180–19185. doi: 10.1074/jbc.271.32.19180. [DOI] [PubMed] [Google Scholar]
- 91.Stoyanovsky DA, Tyurina YY, Tyurin VA, Anand D, Mandavia DN, Gius D, Ivanova J, Pitt B, Billiar TR, Kagan VE. Thioredoxin and lipoic acid catalyze the denitrosation of low molecular weight and protein S-nitrosothiols. J Am Chem Soc. 2005;127:15815–15823. doi: 10.1021/ja0529135. [DOI] [PubMed] [Google Scholar]
- 92.Sumida Y, Nakashima T, Yoh T, Furutani M, Hirohama A, Kakisaka Y, Nakajima Y, Ishikawa H, Mitsuyoshi H, Okanoue T, et al. Serum thioredoxin levels as a predictor of steatohepatitis in patients with nonalcoholic fatty liver disease. J Hepatol. 2003;38:32–38. doi: 10.1016/s0168-8278(02)00331-8. [DOI] [PubMed] [Google Scholar]
- 93.Okuyama H, Nakamura H, Shimahara Y, Uyama N, Kwon YW, Kawada N, Yamaoka Y, Yodoi J. Overexpression of thioredoxin prevents thioacetamide-induced hepatic fibrosis in mice. J Hepatol. 2005;42:117–123. doi: 10.1016/j.jhep.2004.09.020. [DOI] [PubMed] [Google Scholar]
- 94.Mani AR, Ebrahimkhani MR, Ippolito S, Ollosson R, Moore KP. Metalloprotein-dependent decomposition of S-nitrosothiols: studies on the stabilization and measurement of S-nitrosothiols in tissues. Free Radic Biol Med. 2006;40:1654–1663. [Google Scholar]
- 95.Yagmur E, Trautwein C, Leers MP, Gressner AM, Tacke F. Elevated apoptosis-associated cytokeratin 18 fragments (CK18Asp386) in serum of patients with chronic liver diseases indicate hepatic and biliary inflammation. Clin Biochem. 2007;40:651–655. doi: 10.1016/j.clinbiochem.2006.12.010. [DOI] [PubMed] [Google Scholar]
- 96.Krähenbühl S. Alterations in mitochondrial function and morphology in chronic liver disease: pathogenesis and potential for therapeutic intervention. Pharmacol Ther. 1993;60:1–38. doi: 10.1016/0163-7258(93)90020-e. [DOI] [PubMed] [Google Scholar]
- 97.Krähenbühl S, Krähenbühl-Glauser S, Stucki J, Gehr P, Reichen J. Stereological and functional analysis of liver mitochondria from rats with secondary biliary cirrhosis: impaired mitochondrial metabolism and increased mitochondrial content per hepatocyte. Hepatology. 1992;15:1167–1172. doi: 10.1002/hep.1840150631. [DOI] [PubMed] [Google Scholar]
- 98.Yamauchi H, Koyama K, Otowa T, Ouchi K, Anezaki T, Sato T. Morphometric studies on the rat liver in biliary obstruction. Tohoku J Exp Med. 1976;119:9–25. doi: 10.1620/tjem.119.9. [DOI] [PubMed] [Google Scholar]
- 99.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science. 2012;337:1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gena P, Fanelli E, Brenner C, Svelto M, Calamita G. News and views on mitochondrial water transport. Front Biosci (Landmark Ed) 2009;14:4189–4198. doi: 10.2741/3522. [DOI] [PubMed] [Google Scholar]
- 101.Marchissio MJ, Francés DE, Carnovale CE, Marinelli RA. Mitochondrial aquaporin-8 knockdown in human hepatoma HepG2 cells causes ROS-induced mitochondrial depolarization and loss of viability. Toxicol Appl Pharmacol. 2012;264:246–254. doi: 10.1016/j.taap.2012.08.005. [DOI] [PubMed] [Google Scholar]
- 102.Calamita G, Gena P, Meleleo D, Ferri D, Svelto M. Water permeability of rat liver mitochondria: A biophysical study. Biochim Biophys Acta. 2006;1758:1018–1024. doi: 10.1016/j.bbamem.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 103.Calamita G, Moreno M, Ferri D, Silvestri E, Roberti P, Schiavo L, Gena P, Svelto M, Goglia F. Triiodothyronine modulates the expression of aquaporin-8 in rat liver mitochondria. J Endocrinol. 2007;192:111–120. doi: 10.1677/JOE-06-0058. [DOI] [PubMed] [Google Scholar]
- 104.Soria LR, Marrone J, Calamita G, Marinelli RA. Ammonia detoxification via ureagenesis in rat hepatocytes involves mitochondrial aquaporin-8 channels. Hepatology. 2013;57:2061–2071. doi: 10.1002/hep.26236. [DOI] [PubMed] [Google Scholar]
- 105.Kowaltowski AJ, Castilho RF, Vercesi AE. Mitochondrial permeability transition and oxidative stress. FEBS Lett. 2001;495:12–15. doi: 10.1016/s0014-5793(01)02316-x. [DOI] [PubMed] [Google Scholar]
- 106.Ginn-Pease ME, Whisler RL. Optimal NF kappa B mediated transcriptional responses in Jurkat T cells exposed to oxidative stress are dependent on intracellular glutathione and costimulatory signals. Biochem Biophys Res Commun. 1996;226:695–702. doi: 10.1006/bbrc.1996.1416. [DOI] [PubMed] [Google Scholar]
- 107.Hammond CL, Lee TK, Ballatori N. Novel roles for glutathione in gene expression, cell death, and membrane transport of organic solutes. J Hepatol. 2001;34:946–954. doi: 10.1016/s0168-8278(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 108.Serviddio G, Pereda J, Pallardó FV, Carretero J, Borras C, Cutrin J, Vendemiale G, Poli G, Viña J, Sastre J. Ursodeoxycholic acid protects against secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress. Hepatology. 2004;39:711–720. doi: 10.1002/hep.20101. [DOI] [PubMed] [Google Scholar]
- 109.Turrens JF, Boveris A. Generation of superoxide anion by the NADH dehydrogenase of bovine heart mitochondria. Biochem J. 1980;191:421–427. doi: 10.1042/bj1910421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Mitsuyoshi H, Nakashima T, Sumida Y, Yoh T, Nakajima Y, Ishikawa H, Inaba K, Sakamoto Y, Okanoue T, Kashima K. Ursodeoxycholic acid protects hepatocytes against oxidative injury via induction of antioxidants. Biochem Biophys Res Commun. 1999;263:537–542. doi: 10.1006/bbrc.1999.1403. [DOI] [PubMed] [Google Scholar]
- 111.Wang DQH, Neuschwander-Tetri BA, Portincasa P. The Biliary System. Colloquium Series on Integrated Systems Physiology: From Molecule to Function. San Rafael: Morgan & Claypool Publishers; 2012. pp. 1–148. [Google Scholar]
- 112.Carreras FI, Gradilone SA, Mazzone A, García F, Huang BQ, Ochoa JE, Tietz PS, Larusso NF, Calamita G, Marinelli RA. Rat hepatocyte aquaporin-8 water channels are down-regulated in extrahepatic cholestasis. Hepatology. 2003;37:1026–1033. doi: 10.1053/jhep.2003.50170. [DOI] [PubMed] [Google Scholar]
- 113.Clementi E, Brown GC, Feelisch M, Moncada S. Persistent inhibition of cell respiration by nitric oxide: crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc Natl Acad Sci USA. 1998;95:7631–7636. doi: 10.1073/pnas.95.13.7631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Steffen M, Sarkela TM, Gybina AA, Steele TW, Trasseth NJ, Kuehl D, Giulivi C. Metabolism of S-nitrosoglutathione in intact mitochondria. Biochem J. 2001;356:395–402. doi: 10.1042/0264-6021:3560395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 1997;418:291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 116.Carreras MC, Converso DP, Lorenti AS, Barbich M, Levisman DM, Jaitovich A, Antico Arciuch VG, Galli S, Poderoso JJ. Mitochondrial nitric oxide synthase drives redox signals for proliferation and quiescence in rat liver development. Hepatology. 2004;40:157–166. doi: 10.1002/hep.20255. [DOI] [PubMed] [Google Scholar]
- 117.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C, Bracale R, Valerio A, Francolini M, Moncada S, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 118.Zhou J, Eleni C, Spyrou G, Brüne B. The mitochondrial thioredoxin system regulates nitric oxide-induced HIF-1alpha protein. Free Radic Biol Med. 2008;44:91–98. doi: 10.1016/j.freeradbiomed.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 119.Damdimopoulos AE, Miranda-Vizuete A, Pelto-Huikko M, Gustafsson JA, Spyrou G. Human mitochondrial thioredoxin. Involvement in mitochondrial membrane potential and cell death. J Biol Chem. 2002;277:33249–33257. doi: 10.1074/jbc.M203036200. [DOI] [PubMed] [Google Scholar]
- 120.Lee SR, Kim JR, Kwon KS, Yoon HW, Levine RL, Ginsburg A, Rhee SG. Molecular cloning and characterization of a mitochondrial selenocysteine-containing thioredoxin reductase from rat liver. J Biol Chem. 1999;274:4722–4734. doi: 10.1074/jbc.274.8.4722. [DOI] [PubMed] [Google Scholar]
- 121.Spyrou G, Enmark E, Miranda-Vizuete A, Gustafsson J. Cloning and expression of a novel mammalian thioredoxin. J Biol Chem. 1997;272:2936–2941. doi: 10.1074/jbc.272.5.2936. [DOI] [PubMed] [Google Scholar]
- 122.Lundberg M, Johansson C, Chandra J, Enoksson M, Jacobsson G, Ljung J, Johansson M, Holmgren A. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J Biol Chem. 2001;276:26269–26275. doi: 10.1074/jbc.M011605200. [DOI] [PubMed] [Google Scholar]
- 123.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: implications for diseases in the cardiovascular system. Am J Physiol Heart Circ Physiol. 2007;292:H1227–H1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 124.Berndt C, Lillig CH, Holmgren A. Thioredoxins and glutaredoxins as facilitators of protein folding. Biochim Biophys Acta. 2008;1783:641–650. doi: 10.1016/j.bbamcr.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 125.Holmgren A, Johansson C, Berndt C, Lönn ME, Hudemann C, Lillig CH. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans. 2005;33:1375–1377. doi: 10.1042/BST0331375. [DOI] [PubMed] [Google Scholar]
- 126.Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780:1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- 127.Aon MA, Stanley BA, Sivakumaran V, Kembro JM, O’Rourke B, Paolocci N, Cortassa S. Glutathione/thioredoxin systems modulate mitochondrial H2O2 emission: an experimental-computational study. J Gen Physiol. 2012;139:479–491. doi: 10.1085/jgp.201210772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rhee SG, Chae HZ, Kim K. Peroxiredoxins: a historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic Biol Med. 2005;38:1543–1552. doi: 10.1016/j.freeradbiomed.2005.02.026. [DOI] [PubMed] [Google Scholar]
- 129.Anathy V, Roberson EC, Guala AS, Godburn KE, Budd RC, Janssen-Heininger YM. Redox-based regulation of apoptosis: S-glutathionylation as a regulatory mechanism to control cell death. Antioxid Redox Signal. 2012;16:496–505. doi: 10.1089/ars.2011.4281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Moon KH, Hood BL, Mukhopadhyay P, Rajesh M, Abdelmegeed MA, Kwon YI, Conrads TP, Veenstra TD, Song BJ, Pacher P. Oxidative inactivation of key mitochondrial proteins leads to dysfunction and injury in hepatic ischemia reperfusion. Gastroenterology. 2008;135:1344–1357. doi: 10.1053/j.gastro.2008.06.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Chen T, Pearce LL, Peterson J, Stoyanovsky D, Billiar TR. Glutathione depletion renders rat hepatocytes sensitive to nitric oxide donor-mediated toxicity. Hepatology. 2005;42:598–607. doi: 10.1002/hep.20813. [DOI] [PubMed] [Google Scholar]
- 132.Tu W, Kitade H, Satoi S, Zhang ZT, Kaibori M, Kwon AH, Kamiyama Y, Okumura T. Increased nitric oxide production in hepatocytes is involved in liver dysfunction following obstructive jaundice. J Surg Res. 2002;106:31–36. doi: 10.1006/jsre.2002.6436. [DOI] [PubMed] [Google Scholar]
- 133.Arisawa S, Ishida K, Kameyama N, Ueyama J, Hattori A, Tatsumi Y, Hayashi H, Yano M, Hayashi K, Katano Y, et al. Ursodeoxycholic acid induces glutathione synthesis through activation of PI3K/Akt pathway in HepG2 cells. Biochem Pharmacol. 2009;77:858–866. doi: 10.1016/j.bcp.2008.11.012. [DOI] [PubMed] [Google Scholar]
- 134.Kim YC, Masutani H, Yamaguchi Y, Itoh K, Yamamoto M, Yodoi J. Hemin-induced activation of the thioredoxin gene by Nrf2. A differential regulation of the antioxidant responsive element by a switch of its binding factors. J Biol Chem. 2001;276:18399–18406. doi: 10.1074/jbc.M100103200. [DOI] [PubMed] [Google Scholar]
- 135.Okada K, Shoda J, Taguchi K, Maher JM, Ishizaki K, Inoue Y, Ohtsuki M, Goto N, Takeda K, Utsunomiya H, et al. Ursodeoxycholic acid stimulates Nrf2-mediated hepatocellular transport, detoxification, and antioxidative stress systems in mice. Am J Physiol Gastrointest Liver Physiol. 2008;295:G735–G747. doi: 10.1152/ajpgi.90321.2008. [DOI] [PubMed] [Google Scholar]
- 136.Poupon R. Ursodeoxycholic acid and bile-acid mimetics as therapeutic agents for cholestatic liver diseases: an overview of their mechanisms of action. Clin Res Hepatol Gastroenterol. 2012;36 Suppl 1:S3–12. doi: 10.1016/S2210-7401(12)70015-3. [DOI] [PubMed] [Google Scholar]
- 137.Stanimirov B, Stankov K, Mikov M. Pleiotropic functions of bile acids mediated by the farnesoid X receptor. Acta Gastroenterol Belg. 2012;75:389–398. [PubMed] [Google Scholar]
- 138.Grattagliano I, de Bari O, Bernardo TC, Oliveira PJ, Wang DQ, Portincasa P. Role of mitochondria in nonalcoholic fatty liver disease--from origin to propagation. Clin Biochem. 2012;45:610–618. doi: 10.1016/j.clinbiochem.2012.03.024. [DOI] [PubMed] [Google Scholar]