

Abstract

A novel thiazolopyrimidinone series of PI3K-beta selective inhibitors has been identified. This chemotype has provided an excellent tool compound, 18, that showed potent growth inhibition in the PTEN-deficient breast cancer cell line MDA-MB-468 under anchorage-independent conditions, and it also demonstrated pharmacodynamic effects and efficacy in a PTEN-deficient prostate cancer PC-3 xenograft mouse model.
Keywords: PI3K-beta inhibitor, PTEN-deficient, phosphatidylinositol 3-kinase, homology model, structure–activity relationship
Since 2006, numerous small molecule phosphatidylinositol 3-kinase (PI3K) inhibitors have entered a wide range of clinical trials, primarily as targeted anticancer agents.1,2 These molecules are either class I pan-PI3K inhibitors (with or without mTOR activity) or PI3K-alpha selective or PI3K-delta selective inhibitors. Despite many years of effort, it remains unclear what PI3K target selectivity profile is required of an inhibitor to provide a safe and effective agent for a specific patient population. Recent preclinical studies have shown that, in a PTEN-loss context, tissue-specific deletion of the PI3K-beta isoform in the prostate specifically reduces PI3K signaling and blocks the formation of aggressive prostate tumors.3 Driven by these intriguing data, a couple of academic groups have published their efforts on identifying novel beta-isoform selective small molecule tools or using an existing tool molecule to understand the biology and the pathway in depth.4 In previous communications,5 we have reported on the discovery of two novel series of PI3K-beta inhibitors, imidazo[1,2-a]-pyrimidin-5(1H)-ones and 1,2,4-triazolo[1,5-a]pyrimidin-7(3H)-ones, which are exemplified by compounds 1 and 2 (Figure 1). Although these are excellent tool molecules for understanding PI3K-beta biology at a cellular level, they are limited from in vivo target validation due to the poor rodent pharmacokinetic profile.6
Figure 1.

Thiazolopyrimidinone, a novel series of PI3K-beta inhibitors by design.
The modeling and SAR results from our two previous series have suggested that a potent and selective PI3K-beta inhibitor could be designed from a bicyclic core structure bearing substituents designed to make three key binding interactions: (a) a carbonyl group to interact with the back-pocket Tyr-839; (b) a morpholine to act as a hinge binder; and (c) a lipophilic group that can induce a selectivity-pocket formed by Met-779 and Trp-787 (Figure 2A). We soon discovered that thiazolopyrimidinones with a substituted benzyl group at the N1-position met the above requirements (Figure 1).
Figure 2.
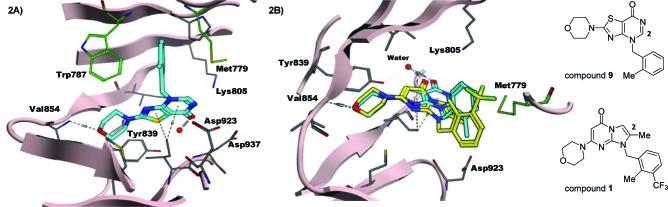
(A) Compound 9 in the PI3K-beta homology model; (B) overlay of compounds 9 and 1.
Compound 9 was docked in our PI3K-beta homology model based on public and in house unpublished structural information.7 Figure 2A depicts the preferred pose of compound 9 in the binding site as viewed from below the plane of the thiazolopyrimidinone core. The morpholine-oxygen could accept an H-bond from the hinge Val-854 while the carbonyl group of the thiazolopyrimidinone core could interact with the back-pocket Tyr-839, via a network of H-bonds involving a bridge water molecule. Furthermore, the N1-o-toluene group of compound 9 may induce a conformational switch in the P-loop at the top of the ATP binding site, creating a lipophilic pocket lined by Met-779 and Trp-787, which we believe to be responsible for beta isoform selectivity.
In addition, the 2-position of the thiazolopyrimidinone is in the proximity of Met-779, Lys-805, Asp-923, and Asp-937, which are reasonably flexible residues that could provide other lipophilic or H-bond interactions. We predicted that a variety of substituents at the 2-position could not only be tolerated but also enhance potency. This predicted improvement had been witnessed in the imidazopyrimidinone series.8
Figure 2B depicts an overlay of docked poses of compounds 9 and 1 in the homology model of PI3K beta, as viewed from above the plane of the thiazolopyrimidinone core. The interactions between the compounds and the binding site are the same as described above; however, for visual clarity, parts of the P-loop including the residue Trp-787 are not shown in Figure 2B. From the overlay, it appears that the benzene ring of compound 9 occupies the space where the 2-methyl and 3-CF3 groups on the benzyl tail of compound 1 are. The ortho-methyl group of compound 9 points toward the benzene ring of compound 1, and a small lipophilic group adjacent to the methyl group might be able to increase potency. This overlay indicates that even though the two ortho-methyl groups in the two molecules align in different directions, we could still predict that, in the thiazolopyrimidinone series, 2,3-disubstituted benzyl groups should provide potent and selective PI3K-beta inhibitors.
The synthesis of thiazolopyrimidinones is exemplified by the preparation of compounds 16, 18, and 20 (Scheme 1). Methyl 4-amino-1,3-thiazole-5-carboxylate, prepared as described in the literature,9 was heated in formamide at 150 °C overnight to provide [1,3]thiazolo[4,5-d]pyrimidin-7(4H)-one (3).10 Deprotonation of 3 with 2 equiv of lithium bis(trimethylsilyl)amide (LiHMDS) in anhydrous tetrahydrofuran (THF) at 0 °C, followed by addition of 1-(bromomethyl)-2-methyl-3-(trifluoromethyl)benzene, afforded the desired product 16 as the major regioisomer, which was separable from the minor regioisomer either by flash column chromatography on silica gel or by reversed phase HPLC.11
Scheme 1. Synthesis of Compounds 16, 18, and 20.

Reagents and conditions: (a) HCONH2, 150 °C, 80%; (b) LiHMDS (2 equiv), 1-(bromomethyl)-2-methyl-3-(trifluoromethyl)benzene, THF, 58%; (c) NaOH, MeOH, 75 °C; (d) propionyl chloride, then NaOH, 30%; (e) AcCl, DMAP, Py, 80 °C, 82%; (f) K2CO3, 1-(bromomethyl)-2-methyl-3-(trifluoromethyl)benzene, DMF, 90 °C, 81%; (g) K2CO3, sodium perborate hydrate, MeOH/THF/H2O 1:1:1, 55 °C, 32–50%.
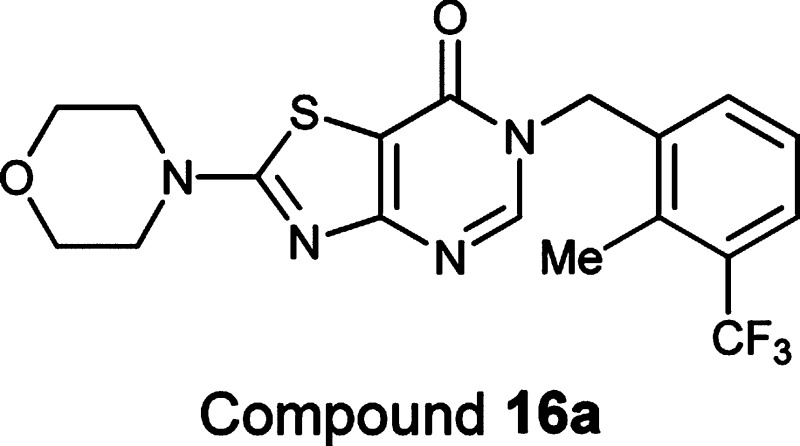
Since the above-mentioned alkylation is hindered by any substitution at the 2-position, two methods that are complementary in many cases have been developed to introduce a variety of functional groups (R2 in Figure 1). For example, compound 20 was prepared via method A. Hydrolysis of compound 16 in aqueous NaOH with gentle heat (65–75 °C) afforded amino-amido thiazole 4, which was treated with propionyl chloride followed by ring closure under basic conditions to provide compound 20. Compound 18 was first prepared in a similar alkylation procedure to that of 16 but in low yield.12 It was later prepared in large scale using method B.13 4-Amino-2-(4-morpholinyl)-1,3-thiazole-5-carbonitrile (5), prepared following the literature procedures,14 was mixed with acetyl chloride and DMAP in pyridine at 80 °C overnight to afford compound 6. Alkylation of compound 6 with 1-(bromomethyl)-2-methyl-3-(trifluoromethyl)benzene in the presence of K2CO3 in DMF at 90 °C gave compound 7. Subsequent cyclization of 7 under mild oxidative conditions with sodium perborate provided compound 18 in good yield.
To determine if the SAR of this series was consistent with the modeling results, and tracked with the previously reported imidazopyrimidinone and triazolopyrimidinone series, we initially explored substitutions at the benzylic group at the N1-position while maintaining the critical morpholine-hinge interaction. Indeed, preference for substitution at the 2- or 3-position was similar to that of imidazo- and triazolopyrimidinones. Compared to unsubstituted benzyl analogue 8, 2-Me and 3-Cl analogues (9 vs 8 and 14 vs 8) increased potency by 8- and 4-fold, respectively. In agreement with our prediction based on the overlay of compounds 9 and 1 in a PI3K-beta homology model, 2,3-disubstituted analogues such as 16 and 17 were single-digit nM PI3K-beta inhibitors. In addition, the thiophene analogue was about 5-fold less potent than the benzene analogue with a similar substitution pattern (13 vs 12), while both alpha-Me benzyl analogue 10 and phenethyl analogue 11 were slightly less potent than 8.
Replacement of the benzene ring with a polar heteroaryl group such as 2-pyridine or 3-pyridine was not tolerated; neither was the polar substitution on the benzene ring, such as CN, CO2H, NO2, etc. (structures or data not shown).
This series has also demonstrated good to excellent isoform selectivity versus the alpha and gamma enzymes, that is especially profound in more potent analogues such as compounds 16 and 17, although selectivity versus the delta enzyme was a more modest 10- to 20-fold in most cases (Table 1).
Table 1. SAR of R1 and Biochemical Activity in PI3K Isoforms15,16.
| PI3K enzyme IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| no. | R1 | R1′ | α | β | δ | γ |
| 8 | Ph | H | 7.9 | 0.32 | 2.0 | 6.3 |
| 9 | (2-Me)Ph | H | 2.5 | 0.025 | 0.25 | 2.5 |
| 10 | Ph | Me | >4.0 | 0.40 | 0.50 | 5.0 |
| 11 | Bn | H | >2.0 | 0.79 | 1.0 | 3.2 |
| 12 | (2-Cl)Ph | H | 2.5 | 0.16 | >0.79 | 7.9 |
| 13 | 2-Cl-3-thieno | H | 3.2 | 0.79 | 2.5 | 4.0 |
| 14 | (3-Cl)Ph | H | >3.2 | 0.079 | 0.32 | >0.79 |
| 15 | (2,3-diCl)Ph | H | 1.0 | 0.010 | 0.079 | 1.3 |
| 16 | (2-Me,3-CF3)Ph | H | 4.0 | 0.003 | 0.063 | 1.6 |
| 17 | (2-Me,3-Cl)Ph | H | 2.5 | 0.003 | 0.040 | 2.5 |
These SAR results support our hypothesis of how a PI3K-beta selective inhibitor might interact with the enzyme in a PI3K-beta homology model, in which the substituted benzene group could induce a lipophilic pocket formed by Met-779 and Trp-787.
Compounds with substitution at the 2-position of the thiazolopyrimidinone ring were prepared, since the model suggested available space and possible H-bond interactions with flexible residues such as Met-779, Lys-805, Asp-923, and Asp-937 in this region. The robustness of the synthesis of this chemical series has allowed us to explore a variety of functional groups at the 2-position to explore SAR with great detail and in depth comprehension.
As demonstrated in Table 2, various substituents on the pyrimidinone ring, in combination with our best R1 groups, provided compounds with improved enzymatic potency, without erosion of selectivity versus the other PI3K isoforms. For example, the addition of a methyl group at the 2-position of compounds 16 (PI3K-beta IC50 of 0.003 μM) and 17 (PI3K-beta IC50 of 0.003 μM) led to extremely potent compounds 18 and 19 (PI3K-beta IC50 values of 0.0006 and 0.0005 μM, respectively). While small alkyl groups increased potency (20 and 21 vs 16) and Bn was tolerated (23), a bulky group such as Ph reduced potency by 16-fold (22 vs 16). Polar functional groups were well-tolerated, and some of them increased potency significantly. For example, 2-CH2NH2, 2-OH, and 2-SH analogues were as potent (25 and 27) as or slightly less potent (29) than 16, while 2-OMe, 2-SMe, and 2-NH2 analogues (28, 30, and 32) were 10-fold more potent than 16. Compound 31 was found to be the most potent inhibitor in this series.17
Table 2. SAR of R2 and Biochemical Activity in PI3K Isoforms18.
| PI3K enzyme IC50 (μM) |
||||||
|---|---|---|---|---|---|---|
| no. | R1 | R2 | α | β | δ | γ |
| 16 | 2-Me,3-CF3 | H | 4.0 | 0.003 | 0.063 | 1.6 |
| 17 | 2-Me,3-Cl | H | 2.5 | 0.003 | 0.040 | 2.5 |
| 18 | 2-Me,3-CF3 | Me | 2.5 | 0.0006 | 0.020 | 0.79 |
| 19 | 2-Me,3-Cl | Me | 2.0 | 0.0005 | 0.010 | 1.0 |
| 20 | 2-Me,3-CF3 | Et | >4.0 | 0.0005 | 0.013 | 1.3 |
| 21 | 2-Me,3-CF3 | c-Pr | 1.3 | 0.0005 | 0.016 | 1.0 |
| 22 | 2-Me,3-CF3 | Ph | >3.2 | 0.050 | 0.50 | 4.0 |
| 23 | 2-Me,3-CF3 | Bn | 2.5 | 0.003 | 0.063 | 2.0 |
| 24 | 2-Me,3-CF3 | CH2OH | 0.63 | 0.0002 | 0.003 | 0.25 |
| 25 | 2-Me,3-CF3 | CH2NH2 | >2.5 | 0.003 | 0.16 | >7.9 |
| 26 | 2-Me,3-Cl | CH2OH | 0.50 | 0.0001 | 0.002 | 0.25 |
| 27 | 2-Me,3-Cl | OH | 50 | 0.008 | 0.50 | >6.3 |
| 28 | 2-Me,3-Cl | OMe | 1.6 | 0.0003 | 0.010 | 0.40 |
| 29 | 2-Me,3-Cl | SH | 2.0 | 0.005 | 0.050 | >3.2 |
| 30 | 2-Me,3-Cl | SMe | 2.0 | 0.0003 | 0.010 | 2.5 |
| 31 | 2-Me,3-Cl | SCH2-CO2H | 0.40 | 0.00005 | 0.002 | 0.16 |
| 32 | 2-Me,3-Cl | NH2 | 0.25 | 0.0002 | 0.003 | 0.50 |
A select set of potent inhibitors was tested in the PTEN-deficient MDA-MB-468 breast cell line to assess the inhibition of downstream phosphorylation of AKT and antiproliferative activity under anchorage-independent conditions.19 As expected, they were very potent in both assays. In Table 3, Compound 26 was a particularly potent inhibitor with EC50 value of 6 nM and gIC50 value of 8 nM in pAKT and growth inhibition assays, respectively.
Table 3. Cellular Activitya.

| PI3Kβ IC50 (μM) | MDA-MB-468 (μM) |
||||
|---|---|---|---|---|---|
| no. | R1 | R2 | pAKT EC50 | proliferation gIC50 | |
| 18 | 2-Me,3-CF3 | Me | 0.0006 | 0.024 | 0.103 |
| 19 | 2-Me,3-Cl | Me | 0.0005 | 0.019 | 0.032 |
| 20 | 2-Me,3-CF3 | Et | 0.0005 | 0.016 | 0.023 |
| 21 | 2-Me,3-CF3 | c-Pr | 0.0005 | 0.009 | 0.043 |
| 26 | 2-Me,3-Cl | CH2OH | 0.0001 | 0.006 | 0.008 |
| 28 | 2-Me,3-Cl | OMe | 0.0003 | 0.006 | 0.021 |
| 32 | 2-Me,3-Cl | NH2 | 0.0002 | 0.003 | 0.026 |
IC50 values given are means of at least two experiments
The thiazolopyrimidinone series, represented by compound 18, is selective over the other PI3K isoforms in class I lipid kinases, and also some class II, III, and IV lipid kinases available in house, including C2B, VSP34, FRAP1 (mTOR), and DNA-PK, as shown in Figure 3. Furthermore, compound 18 was tested against more than 30 protein kinases for dose response curves and was found to be inactive (IC50 > 10 μM). At 10 μM concentration, compound 18 showed <30% inhibition against a panel of 290 kinases screened.20 The clean selectivity profile of compound 18 provided us with a high level of confidence that any biological effects observed with this tool compound would be directly associated with PI3K-beta inhibition.
Figure 3.
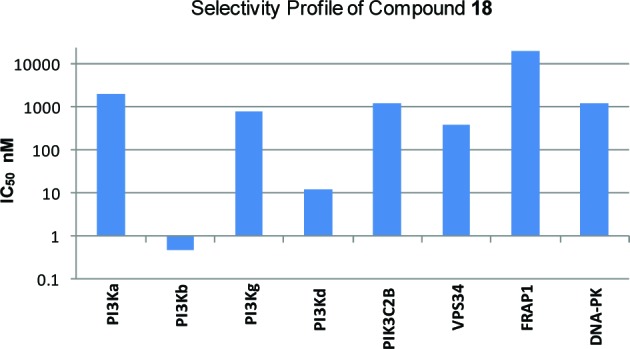
Selectivity profile of compound 18 vs other lipid kinases.
Mouse pharmacokinetic (PK) studies (All animal studies were conducted after review by the GSK Institutional Animal Care and Use Committee and in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals.) were carried out with three potent inhibitors (see Table 4),21 and they showed low to moderate clearance.22 To enable in vivo biological evaluations, all three compounds were subjected to high dose (100 mg/kg) oral suspension mouse PK studies. We observed good oral exposure of all three, measured by DNAUC. Considering overall profile including potency, selectivity, oral exposure in mice, and the ease of synthesis, we selected compound 18 for in vivo pharmacodynamic and efficacy studies.
Table 4. Mouse PKa–c Profile of Compounds 18, 21, and 26.
| no. | T1/2 | Vdss | Cl | PO DNAUC | F%d |
|---|---|---|---|---|---|
| 18 | 1.20 | 2.80 | 29.6 | 278 | 49 |
| 21 | 0.75 | 2.60 | 48.6 | 192 | 54 |
| 26 | 1.90 | 3.35 | 29.4 | 264 | 21 |
The data are the average of two animals (male CD mice).
Units: T1/2, h; Vdss, L/kg; Cl, mL/(min kg); DNAUC, ng/(h mL mg kg).
iv: studies were carried out at 0.3 mg/kg dose.
Based on noncrossover studies.
Compound 18 was evaluated in a PTEN-deficient PC-3 prostate carcinoma xenograft mouse model.23 As shown in Figure 4, compound 18 demonstrated a time dependent inhibition of pAKT with similar levels of pAKT inhibition observed from 1 to 6 h (53% to 64% with 100 mg/kg and 63% to 72% with 300 mg/kg). By 8 h, the levels of inhibition were reduced at both the 100 and 300 mg/kg doses (46% and 48%, respectively) with further reduction by 10 h (33% and 22%, respectively) and a return to baseline by 24 h. The changes in pAKT levels were consistent with the drug concentrations in both blood and tumor samples with similar compound levels from 1 to 6 h, with a decrease at 8 h followed by a further decrease at 10 and 24 h. Higher levels (1.2- to 2-fold) of compound were observed at the 300 mg/kg dose compared to the 100 mg/kg dose, resulting in overall slightly greater levels of pAKT inhibition with the 300 mg/kg dose compared to the 100 mg/kg dose. The blood concentrations of compound 18 at both doses at the 10 h time point exceeded EC50 in the pAKT assay and gIC50 in the growth inhibition assay.
Figure 4.
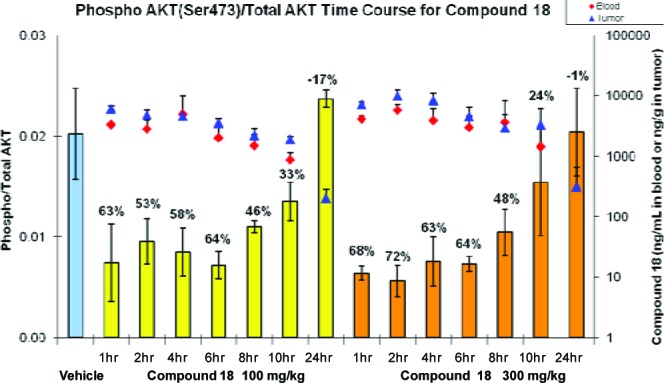
Pharmacodynamic effect of compound 18 in the PC-3 xenograft mouse model. Numbers above the bars indicate the percent inhibition of phosphor AKT/total AKT relative to vehicle.
To further establish the antitumor activity in an in vivo setting, compound 18 was administered once-a-day orally for 21 consecutive days in the same xenograft model (Figure 5). At both 100 and 300 mg/kg doses, complete tumor growth inhibition relative to vehicle treated mice was observed with no effect on body weight. The pharmacodynamic and efficacy data from the PC-3 xenograft model (Figures 4 and 5) indicate that approximately 63% to 24% inhibition of pAKTser473 from 1 to 10 h, respectively, results in tumor growth inhibition. These data do not, however, rule out the possibility that other unknown activities of PI3K beta may be contributing to effects on tumor growth.
Figure 5.
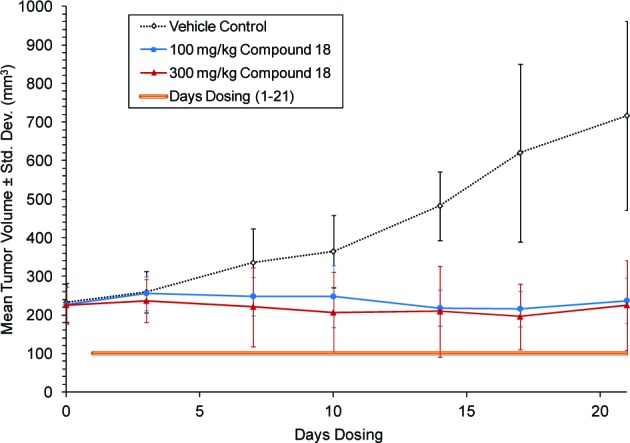
Effect of compound 18 on tumor growth in the PC-3 xenograft mouse model.
In summary, we have designed a novel series of thiazolopyrimidinone PI3K-beta inhibitors based on our previous findings of how to best optimize potency and selectivity. Many compounds demonstrated subnanomolar potency against the beta enzyme, and several compounds have achieved nanomolar potency in cell-based assays (21, 26, 28, 31). After further profiling, compound 18 emerged as an excellent tool molecule for in vivo studies, as it is potent and selective and displays good oral exposure in mouse. To the best of our knowledge, these studies constitute the first time that an in vivo pharmacodynamic effect and efficacy have been demonstrated with an orally bioavailable small molecule PI3K-beta selective inhibitor in a PTEN-deficient xenograft model, a significant milestone toward validating PI3K-beta as a potential target for treatment of proliferative disorders.
Acknowledgments
The authors thank Dr. Minghui Wang for his help with structural confirmation with NMR techniques throughout the program.
Glossary
Abbreviations
- PI3K
phosphoinositide 3-kinase
- PTEN
phosphotase and tensin homologue
- DNAUC
dose-normalized area under the curve
Supporting Information Available
Biological assays, biological data, and experimental procedures. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- According to Thomson Reuters Pharma, there are 22 small molecule PI3K inhibitors in clinical trials.
- Astra Zeneca was developing AZD-6482 as an intravenous platelet aggregation inhibitor for the potential treatment of thrombosis. A phase I trial was completed in May 2008; however, by July 2010, the program had been discontinued. The compound is described in WO2009093972.
- a Wee S.; Wiederschain D.; Maira S.-M.; Loo A.; Miller C.; DeBeaumont R.; Stegmeier F.; Yao Y. M.; Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc. Natl. Acad. Sci. U.S.A. 2008, 105, 13057–13062. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Jia S.; Liu Z.; Zhang S.; Liu P.; Zhang L.; Lee S. H.; Zhang J.; Signoretti S.; Loda M.; Roberts T. M.; Zhao J. J. Essential roles of PI(3)K-p110β in cell growth, metabolism and tumorigenesis. Nature 2008, 454, 776–779. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Lee S. H.; Poulogiannis G.; Pyne S.; Jia S.; Zou L.; Signoretti S.; Loda M.; Cantley L. C.; Roberts T. M. A constitutively activated form of the p110β isoform of PI3-kinase induces prostatic intraepithelial neoplasia in mice. Proc. Natl. Acad. Sci. U.S.A. 2010, 107, 11002–11007. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Hill K. M.; Kalifa S.; Das J. R.; Bhatti T.; Gay M.; Williams D.; Taliferro-Smith L.; De Marzo A. M. The role of PI3-kinase p110β in AKT signally, cell survival, and proliferation in human prostate cancer cells. The Prostate 2010, 70, 755–764. [DOI] [PubMed] [Google Scholar]
- a Kim J.; Hong Se.; Hong Su. Discovery of new aminopyrimidine-based phosphoinositide 3-kinase beta (PI3Kβ) inhibitors with selectivity over PI3Kα. Biol. Med. Chem. Lett. 2011, 21, 6977–6981. [DOI] [PubMed] [Google Scholar]; b After this paper was submitted, an article from Dana-Farber Cancer Institute and Harvard Medical School was published:Ni J.; Liu Q.; Xie S.; Carlson C.; Von T.; Vogel K.; Riddle S.; Benes C.; Eck M.; Roberts T.; Gray N.; Zhao J. Functional Characterization of an Isoform-Selective Inhibitor of PI3K-p110β as a Potential Anticancer Agent. Cancer Discovery 2012, 2, 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lin H.; Erhard K.; Hardwicke M. A.; Luengo J. I.; Mack J. F.; McSurdy-Freed J.; Plant R.; Raha K.; Rominger C. M.; Sanchez R.; Schaber M. D.; Schulz M.; Spengler M. D.; Tedesco R.; Xie R.; Zeng J. J.; Rivero R. A. Synthesis and structure-activity relationships of imidazo[1,2-a]pyrimidin-5(1H)-ones as a novel series of beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 2262230–2234and references therein. [DOI] [PubMed] [Google Scholar]; b Sanchez R.; Erhard K.; Hardwicke M. A.; Lin H.; McSurdy-Freed J.; Plant R.; Kaushik R.; Rominger C.; Schaber M.; Spengler M.; Tedesco R.; Rivero R. Synthesis and structure activity relationships of 1,2,4-triazolo[1,5-a]pyrimidin-7(3H)-ones as novel series of potent beta isoform selective phosphatidylinositol 3-kinase inhibitors. Bioorg. Med. Chem. Lett. 2012, 2293198–3202. [DOI] [PubMed] [Google Scholar]
- Clearance of the evaluated analogues exceeded the hepatic blood flow of mouse or rat.
- Knight Z. A.; Gonzalez B.; Feldman M. E.; Zunder E. R.; Goldenberg D. D.; William O.; Loewith R.; Stokoe D.; Balla A.; Toth B.; Balla T.; Weiss W. A.; Williams R. L.; Shokat K. M. A pharmacological map of the PI3-K family defines a role for p110α in insulin signaling. Cell 2006, 125, 733–747and references therein. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The methyl group at the 2-position greatly improved the potency of both compounds 1 and 2 compared to their unmethylated counterparts. See ref 54.
- Reid W.; Kuhnt D. Thiazolsynthesen mit N-Cyanimidsäureestern oder 3-Cyanisoharnstoffen und Thioglycolsäurederivaten. Liebigs Ann. Chem. 1986, 4, 780–784. [Google Scholar]
- Depending on the scale, the reaction can last for 1–3 days, or it can be carried out under microwave irradiation at 200 °C for 30 min to 1 h.
- Some analogues were prepared with an additional
1 equiv of MgiPrCl in this step to improve the ratio of
the desired regioisomer. The following is the minor regioisomer, whose
carbonyl group has a distinctive IR absorption at 1630 cm–1, while the carbonyl group of the major isomer has IR absorption
at 1610 cm–1.
- Compound 18 was first synthesized by regioselective alkylation of 5-methyl-2-(4-morpholinyl)[1,3]thiazolo[4,5-d]pyrimidin-7(4H)-one, which was prepared by the condensation of 4-amino-2-(4-morpholinyl)-1,3-thiazole-5-carboxamide with acetic anhydride in acetic acid. See experimental details in this patent: Lin H.; Luengo J. I.; Ralph R. A.; Schulz M. J.; Xie R.; Zeng J. PCT Int. Appl. (2010), WO2010135504 A1 20101125.
- Compound 18 prepared from the alkylation method was identical to that prepared from method B illustrated in Scheme 1. This result supports the structural assignment of compound 16 and all others prepared from the alkylation method.
- Thomae D.; Perspicace E.; Xu Z.; Henryon D.; Schneider S.; Hesse S.; Kirsh G.; Seck P. One-pot synthesis of new 2,4,5-trisubstituted 1,3-thiazoles and 1,3-selenazoles. Tetrahedron 2009, 65, 2982–2988. [Google Scholar]
- Inhibition of PI3K-isoforms was measured using a continuous read time-resolved fluorescence resonance energy transfer displacement assay:Gray A.; Olsson H.; Batty I. H.; Priganica L.; Downes C. P. Nonradioactive methods for the assay of phosphoinositide 3-kinase and phosphoinositide phosphatase and selective detection of signaling lipids in cell and tissue extracts. Anal. Biochem. 2003, 313, 234–245. [DOI] [PubMed] [Google Scholar]
- Compounds were routinely tested only once in the PI3K bundle; replicate analyses were done only by special requests, as the potency of the pharmacological standards (which were run on every plate) was consistently reproducible.
- We hypothesized that the extreme potency of compound 31 might come from the interaction of its extended acid group with catalytic Lys805, which appears to have reasonable flexibility.
- The synthesis of compounds 25 and 27–32 can be found in this patent: Lin H.; Luengo J. I.; Ralph R. A.; Schulz M. J.; Xie R.; Zeng J. PCT Int. Appl. (2010), WO2010135504 A1 20101125.
- See notes 19 and 21 in ref 54.
- See single shot results of compound 18 from Reaction Biology Corporation (RBC) in the Supporting Information.
- Xiang H.; McSurdy-Freed J.; Moorthy G. S.; Hugger E.; Bambal R.; Han C.; Ferrer S.; Gargallo D.; Davis C. B. Preclinical drug metabolism and pharmacokinetic evaluation of GW844520, a novel anti-malarial mitochondrial electron transport inhibitor. J. Pharm. Sci. 2006, 95, 2657–72. [DOI] [PubMed] [Google Scholar]
- Both compounds 18 and 26 were subjected to dog and monkey PK studies and showed high clearance in these two species. The clearance values of all three compounds in Table 4 exceeded the rate of rat hepatic flow as well.
- Female nude mice bearing PC-3 prostate carcinoma xenografts were administered compound 18 at 100 or 300 mg/kg and euthanized using carbon dioxide at the indicated time points. Vehicle-treated mice were euthanized at the 2 h time point. Blood was collected for compound concentration determination. The tumor was excised; half was snap frozen in liquid nitrogen for subsequent compound concentration determination, and the other half was immediately processed by Medicon in 1 mL of Meso-Scale Discovery (MSD) lysis buffer with protease inhibitors (Roche complete protease cocktail, catalog # 04 693 116 001) and phosphatase inhibitors (Sigma, catalog # P2850 and P-5726). After centrifugation, the supernatant was serially diluted and the ratio of phosphoAKT (Ser473) to total AKT was measured using MSD Multi-Spot assay plates (whole cell lysate kit: Phospho(ser473), Total AKT Assay, catalog # K15100D-3).

Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.