


Abstract
DNA double-strand breaks (DSBs) can cause chromosomal rearrangements and extensive loss of heterozygosity (LOH), hallmarks of cancer cells. Yet, how such events are normally suppressed is unclear. Here we identify roles for the DNA damage checkpoint pathway in facilitating homologous recombination (HR) repair and suppressing extensive LOH and chromosomal rearrangements in response to a DSB. Accordingly, deletion of Rad3ATR, Rad26ATRIP, Crb253BP1 or Cdc25 overexpression leads to reduced HR and increased break-induced chromosome loss and rearrangements. We find the DNA damage checkpoint pathway facilitates HR, in part, by promoting break-induced Cdt2-dependent nucleotide synthesis. We also identify additional roles for Rad17, the 9-1-1 complex and Chk1 activation in facilitating break-induced extensive resection and chromosome loss, thereby suppressing extensive LOH. Loss of Rad17 or the 9-1-1 complex results in a striking increase in break-induced isochromosome formation and very low levels of chromosome loss, suggesting the 9-1-1 complex acts as a nuclease processivity factor to facilitate extensive resection. Further, our data suggest redundant roles for Rad3ATR and Exo1 in facilitating extensive resection. We propose that the DNA damage checkpoint pathway coordinates resection and nucleotide synthesis, thereby promoting efficient HR repair and genome stability.
INTRODUCTION
DNA double-strand breaks (DSBs) are potentially lethal lesions, which can arise from exposure to DNA damaging agents or through endogenous metabolic errors. DSBs are normally efficiently repaired by the non-homologous end-joining (NHEJ) or homologous recombination (HR) repair pathways. However, incorrectly repaired DSBs can give rise to a wide range of chromosomal rearrangements, which can lead to oncogene activation or tumor suppressor loss through loss of heterozygosity (LOH) (reviewed in (1)).
The DNA damage checkpoint pathway plays a key role in maintaining genome stability in response to DNA damage. While originally identified as an intracellular signal transduction pathway that detects DNA lesions and blocks cell cycle progression until DNA repair is completed (2), the DNA damage checkpoint pathway is now understood to promote genome stability through a wide range of processes including transcriptional regulation of repair genes (3); regulation of nucleotide synthesis (4); interaction with, and post-translational modification of DNA repair proteins (5); relocalization of repair proteins (6) and regulation of the formation of DNA repair centres (7) (reviewed in (8)).
Central to the DNA damage checkpoint are the phosphatidylinositol 3′ kinase-like kinases, ataxia telangiectasia mutated (ATM) in humans (Hs) (Tel1 in Schizosaccharomyces pombe (Sp) and Saccharomyces cerevisiae (Sc)) and ataxia telangiectasia and Rad3-related ATRHs (Rad3Sp/Mec1Sc), which localize to DNA damage and play important roles as the initial sensor kinases (9). During checkpoint activation, the checkpoint loading complex, a modified form of the replication factor C (RFC) heteropentamer in which the Rfc1 subunit is replaced by a checkpoint-specific subunit Rad17Sp/Hs (Rad24Sc), recognizes single-stranded DNA (ssDNA)/double-stranded DNA junctions generated at damage sites (10,11). This recruits the 9-1-1 checkpoint sliding clamp complex, a heterotrimer composed of Rad9Sp/Hs, Hus1Sp/Hs, Rad1Sp/Hs (Ddc1Sc, Mec3Sc and Rad17Sc), which structurally resembles proliferating cellnuclear antigen (PCNA), the processivity factor for DNA replication (12–14). Both the Rad17 checkpoint loading complex and the 9-1-1 complex are required for activation of the ATRHs/Rad3Sp/Mec1Sc checkpoint kinase. The ATRHs/Rad3Sp/Mec1Sc kinase is recruited through its interaction between ATRIPHs/Rad26Sp/Ddc2Sc and replication protein A (RPA) (15), and colocalization of Ddc1Sc with Mec1Sc is necessary and sufficient for checkpoint activation (16).
The checkpoint signal is transduced through recruitment and activation of the effector kinases, Chk1Sp/Hs/Sc and Chk2Hs/Cds1Sp/Rad53Sc. This is achieved through ATRHs/Rad3Sp/Mec1Sc-dependent phosphorylation of the checkpoint clamp Rad9Sp/Hs/Ddc1Sc and recruitment of TopBP1Hs/Rad4/Cut5Sp/Dpb11Sc (17,18). Effector kinase activity is regulated by mediator proteins. In fission yeast, activation of Chk1 in response to DNA damage is mediated by Crb253BP1, while activation of Cds1Chk2 kinase in response to replication stress is mediated by Mrc1 (19–21).
The cell cycle is subsequently targeted by the checkpoint effector kinases. In fission yeast, Cdc25 is phosphorylated by Chk1 or Cds1Chk2 in response to DNA damage or replication stress, respectively (22,23). This results in Cdc25 nuclear export through the binding of Rad24, a 14-3-3 protein, thus preventing activation of nuclear Cdc2CDK1 kinase, thereby resulting in G2 arrest (24,25). Accordingly, checkpoint inactivation can be achieved through overexpression of Cdc25 (26).
In agreement with a central role for the DNA damage checkpoint in maintaining genome stability, its disruption has been shown to result in elevated levels of spontaneous and break-induced chromosomal rearrangements in both yeast and humans (27–32). Further, DNA damage checkpoint genes have been shown to function as tumor suppressors, in accordance with their role in maintaining genome stability (33). Despite a reasonable understanding of DNA damage checkpoint signalling, less is known about how this pathway coordinates repair in response to DNA damage.
In this study, we have examined the roles of the DNA integrity checkpoint genes in facilitating DSB repair and genome stability in fission yeast. We show that loss of the DNA damage checkpoint can lead to strikingly increased levels of break-induced chromosomal rearrangements and extensive LOH. Our findings identify distinct roles for DNA damage checkpoint genes in promoting efficient HR and genome stability in response to a DSB through both facilitating nucleotide synthesis and extensive resection.
MATERIALS AND METHODS
Yeast strains, media and genetic methods
All S. pombe strains were cultured, manipulated and stored as previously described (34). All strain genotypes are listed in Supplementary Table S1. The construction of Ch16-RMGAH is as described in (35).
Serial dilution assays
Log phase cultures of OD 0.2 (595 nm) of the strains indicated were spotted onto Ye5S plates with the indicated concentrations of bleocin. Plates were incubated at 32°C for two days before analysis.
Site-specific DSB assay
The DSB assay was performed as described previously (34). The percentage of colonies undergoing NHEJ/SCC (arg+ G418R/HygR ade+ his+), gene conversion (GC) (arg+ G418S/HygS ade+ his+), Ch16 loss (arg− G418S/HygS ade− his−) or LOH (arg+ G418S/HygS ade− his−; HygR ade− G418S his− for Ch16-YAMGH) were calculated. To determine the levels of break-induced GC, Ch16 loss and LOH, background events at 48h-T in a blank vector assay were subtracted from break-induced events at 48h-T in cells transformed with pREP81X-HO. Each experiment was performed three times using three independently derived strains for all mutants tested. More than 1000 colonies were scored for each time point. Southern blots were performed as previously described (34). It has been previously estimated that every cell will have incurred at least one HO endonuclease-induced DSB during this assay (36).
Rapidly inducible DSB resection and SSA repair assay
Rapid HO induction using the urg promoter together with analysis of DSB resection and single-strand annealing (SSA) repair was performed as previously described (37,38).
Pulsed field gel electrophoresis
Pulsed field gel electrophoresis (PFGE) analysis was performed as described previously (39).
Comparative genome hybridization
Comparative genome hybridization (CGH) analysis was performed as previously described (35).
RESULTS
Rad3ATR is a suppressor of break-induced LOH
To identify suppressors of break-induced LOH, a colony-sectoring screen was performed following ethyl methanesulfonate (EMS) mutagenesis of a strain carrying a modified non-essential minichromosome (Ch16-RMGAH). Ch16-RMGAH encodes an arg3 marker on the left arm of the minichromosome, and a MATa target site, together with an adjacent kanMX6 gene encoding G418 resistance, an ade6-M216, allele which complements the ade6-M210 allele present on the homologous chromosome ChrIII, and a his3 marker on the right arm (Figure 1A). These cells are heterozygous for these markers. Following HO endonuclease-induced cleavage at the MATa site, extensive break-induced LOH resulting from loss of the distal chromosome arm would be expected to result in arg+ G418S ade− his− cells, which can be detected when occurring at increased levels as pink sectored colonies when grown on arg- plates in the presence of low levels of adenine (35) (Supplementary Figure S1). Following mutagenesis of the strain carrying Ch16-RMGAH, mutants loh1-loh7 exhibited elevated levels of break-induced sectoring and were isolated from the screen. The mutants loh2-1, loh3-1 and loh4-1 corresponded to mutations in rad57+, rad52+ and rad51+, respectively, as previously described (35); our unpublished results.
Figure 1.
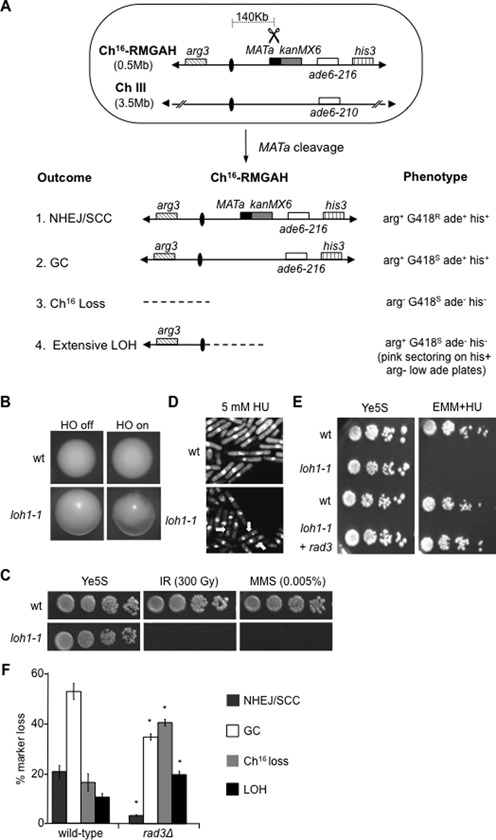
Rad3ATR suppresses break-induced extensive LOH. (A) Schematic of the minichromosome Ch16-RMGAH. The relative positions of the arg3 marker (diagonal stripes), centromeres (ovals), the MATa site (black), the kanMX6 resistance marker (gray), the complementary ade6 heteroalleles (ade6-M216 and ade6-M210; white) and the his3 marker (vertical stripes) on Ch16-RMGAH and ChIII are shown as described previously (35). The sizes of the ChIII and Ch16 are shown. In Ch16-RMYAH, kanMX6 is replaced by hph. Derepression of pREP81X-HO (not shown) generates a DSB at the MATa target site (scissors). Possible outcomes resulting from DSB induction, together with schematics of the minichromosome, and expected phenotypes are shown. (B) Colony sectoring of wild-type or loh1–1 arg+ G418S ade− his− colonies grown on Edinburgh minimal medium (EMM) plus uracil, histidine and low adenine (5 mg/l) without arginine (arg- plates) thus facilitating detection of extensive LOH (LOH) in the presence (HO off) or absence (HO on) of thiamine. (C) Ten-fold serial dilutions of wild-type (WT) Ch16-RMGAH (TH2130) or loh1–1 (TH4089) strains on Ye5S plates, Ye5S plates exposed to 300 Gy IR, or 0.005% MMS as indicated. (D) 4',6-diamidino-2-phenylindole (DAPI) stained wild-type Ch16-RMGAH (TH2130) or loh1–1 (TH4089) strains either untreated or following exposure to 5 mM HU for 6 h. ‘Cut’ phenotypes indicated (yellow arrows). (E) Serial dilutions of wild-type Ch16-RMGAH (TH2130), loh1–1 (TH4089) with pREP41X empty vector or pREP41X-rad3 (TH4093) on Ye5S and 10 mM HU EMM plates without thiamine, to derepress pREP41X expression. (F) Percentage DSB-induced marker loss of Ch16-RMGAH in wild-type (TH2130) and rad3Δ (TH2941) backgrounds. The levels of NHEJ/sister chromatid conversion (SCC), GC, Ch16 loss and extensive LOH are shown. Data are the mean of three experiments and standard errors of the mean are indicated. The asterisk (*) represents significant difference compared to wild-type.
Here we investigated the mutant loh1-1 and found it exhibited increased break-induced sectoring (Figure 1B), and acute sensitivity to ionizing radiation (IR), and methyl methanesulfonate (MMS) (Figure 1C). Further analysis indicated loh1-1 exhibited a ‘cut’ (cells untimely torn) phenotype in the presence of hydroxyurea (HU), which depletes nucleotide pools and disrupts DNA replication (Figure 1D). A ‘cut’ phenotype can arise from a DNA integrity checkpoint defect in which instead of arresting mitosis prior to the completion of DNA replication, unreplicated DNA is divided into two daughter cells (26). These findings strongly suggested loh1-1 encoded a mutation in a checkpoint gene. Accordingly, a cross between rad3Δ and loh1-1 was unable to generate progeny with wild-type sensitivity to DNA damaging agents, and the HU sensitivity of loh1-1 could be rescued by expression of a plasmid encoding rad3 (Figure 1E). Sequence analysis confirmed loh1-1 encoded a W1700X mutation in the rad3+ gene, in which a stop codon was introduced. This mutation lies in the FRAP-ATM-TRRAP (FAT) domain, a kinase domain that is conserved through the phosphatidylinositol 3-kinase-related kinase family (40). Similar findings were obtained for loh5-1 and loh7-1, which were found to encode W1701X and W253X mutations in the rad3+ gene (our unpublished results).
To further assess the role of Rad3ATR in suppressing break-induced LOH, a DSB assay was performed to quantitate levels of marker loss in a rad3Δ background compared to wild-type following break induction in a non-essential minichromosome. Following HO endonuclease-induced cleavage at the MATa site in a wild-type strain carrying Ch16-RMGAH, 20.5% of cells were repaired by NHEJ or sister chromatid conversion (SCC) and maintained all the minichromosome markers (arg+ G418R ade+ his+); 52.7% of cells were repaired by interchromosomal GC leading to loss of the G418R cassette adjacent to the break site on the minichromosome (arg+ G418S ade+ his+); 16.3% of colonies failed to repair the break and lost the non-essential minichromosome (arg− G418S ade− his−) and 10.3% underwent break-induced extensive LOH resulting in loss of the distal minichromosome arm (arg+ G418S ade− his−) (Figure 1A and F).
DSB induction in a rad3Δ background confirmed a role for Rad3ATR in both promoting efficient HR repair and suppressing Ch16 loss and break-induced LOH, as previously described (44). The rad3Δ strain exhibited significantly reduced NHEJ/SCC (3.3% P = 0.01) and GC (34.7% P = 0.02) compared to wild-type. This was accompanied by a significant increase in both Ch16 loss (40.5% P < 0.01) and break-induced extensive LOH (19.6% P < 0.01) (Figure 1F). No significant loss of viability was observed following DSB induction in this non-essential minichromosome in a rad3Δ background (our unpublished results).
We identified isochromosome formation as the predominant mechanism of break-induced extensive LOH in arg+ G418S ade− his− colonies associated with failed HR repair, resulting in a chromosomal element of 388 kb (35). Analysis of 18 arg+ G418S ade− his− colonies from a rad3Δ background indicated that the majority (78%) were of an identical size to that of a previously characterized isochromosome (388 kb; Figure 2A, left panel, compare lanes 2–4). The remaining four rad3Δ arg+ G418S ade− his− colonies displayed a truncated minichromosome of a smaller size to those corresponding to isochromosomes (Figure 2A, left panel, lane 5). Southern blot analysis, using a probe derived from Spcc4b3.18, which anneals directly distal to the centromere on the right arm of Ch16-RMGAH and ChIII (Figure 2A, right panel), showed annealing to the parental minichromosome, but failed to anneal to the chromosomal elements associated with extensive LOH, indicating that these smaller chromosomal elements had lost the entire broken chromosome arm (Figure 2A, right panel).
Figure 2.
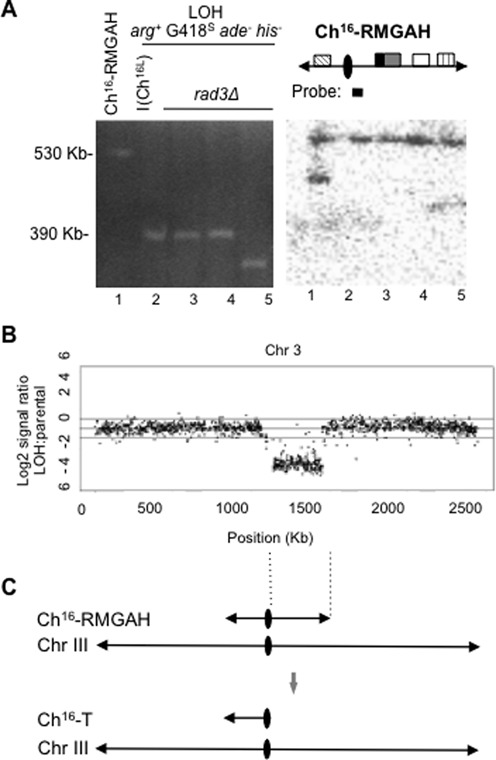
Break-induced extensive LOH in rad3Δ results from extensive resection, and predominantly isochromosome formation (A). Left panel: PFGE analysis from rad3Δ Ch16-RMGAH parental strain (TH2941; lane 1), individual arg+ G418S ade− his− (LOH) colonies from wild-type (a CGH confirmed isochromosome I(Ch16L); lane 2) and rad3Δ (lanes 3–5) backgrounds following DSB induction are shown. Right panel: Southern blot analysis of the PFGE, probed with Spcc4b3.18, which anneals directly distal the centromere on Ch16-RMGAH and ChIII (as indicated) (B). CGH of wild-type Ch16-RMGAH (TH2125) and an arg+ G418S ade− his− (LOH) strain (TH8399) carrying a truncated minichromosome that is shorter than the known isochromosome (TH4313) (Figure 2A, lane 1) previously characterized by CGH (35). The Log2 of the LOH:parental signal ratio across the and chromosome III (from which the minichromosome is derived) is shown. (C) A schematic of the structure of the smaller chromosomal element arising following DSB induction in a rad3Δ background as related to the CGH data. CGH analysis of an isochromosome with a duplicated left arm is presented in Supplementary Figure S2 for comparison.
CGH analysis of an arg+ G418S ade− his− strain carrying a smaller non-isochromosomal element and a parental strain carrying Ch16-RMGAH showed reduced Log2 hybridization ratios across the right arm of the minichromosome, thus confirming the absence of the right arm of the minichromosome in these LOH colonies (Figure 2B). CGH analysis also failed to show increased ratios across the intact left arm of the minichromosome, indicating that in contrast to the previously characterized isochromosomes, this region had not been duplicated in these less frequent and shorter chromosomal elements and were therefore not isochromosomes (Figure 2B and C; (35)). These findings support a model in which failed HR repair results in extensive end processing leading to Ch16 loss or extensive LOH through the formation of isochromosomes or smaller chromosomal elements in a rad3Δ background. These less frequently occurring shorter chromosomal elements are likely to have arisen from de novo telomere addition at or near the centromere of the minichromosome.
Using a wild-type strain carrying Ch16-MGH, which in contrast to Ch16-RMYAH contains an ade6-M216 heteroallele, ∼30 kb centromere-proximal to the break site, we have previously identified LOH events resulting in retention of the ade6-M216 heteroallele, while losing a G418R marker adjacent to the break site and a his3 gene ∼30 kb distal to the break site (Supplementary Figure S3A) (39). These LOH events were associated with DSB repair by HR, and included break-induced replication (BIR) and allelic cross-overs (39). However, isochromosome formation (in which the entire broken arm is lost) cannot be detected in this assay. Using this Ch16-MGH based assay, no increase in LOH events associated with DSB repair (and retention of the ade6-M216 heteroallele) was observed in a rad3Δ background (Supplementary Figure S3B and C). This contrasts with a role for Rad3ATR in suppressing break-induced LOH leading to isochromosome formation, and further supports a role for Rad3ATR in suppressing extensive LOH associated with failed HR repair.
The DNA damage checkpoint pathway promotes HR and suppresses break-induced LOH and minichromosome loss
To test a general role of the DNA damage checkpoint pathway in suppressing break-induced LOH, levels of marker loss were additionally examined in other checkpoint-deficient strains. Like loss of Rad3ATR, loss of the checkpoint sensor Rad26ATRIP, the checkpoint adaptor Crb253BP1 or overexpression of Cdc25 (OPcdc25) led to reduced HR repair, and increased levels of Ch16 loss and LOH. In a rad26Δ background, GC was significantly reduced (32.7% P = 0.01), while levels of Ch16 loss (35.6% P = 0.01) and break-induced LOH (15.8% P = 0.05) were significantly increased, compared to wild-type (Figure 3A). Similarly, in a crb2Δ background break-induced NHEJ/SCC (3.6% P < 0.01) and GC (25.6% P < 0.01) were significantly reduced while Ch16 loss (49.8% P < 0.01) and LOH (20.5% P < 0.01) were significantly increased compared to wild-type (Figure 3A). OPcdc25 encodes cdc25 under the control of the strong constitutive adh promoter, leading to its over-production and subsequently to checkpoint loss (26). DSB induction in an OPcdc25 background resulted in significantly reduced NHEJ/SCC (12.4% P = 0.03), significantly reduced GC (36.8% P = 0.03), and significantly increased Ch16 loss (30.4% P = 0.02) and break-induced LOH (18.9%; P < 0.01) compared to wild-type (Figure 3A). Further analysis of at least 16 of the arg+ G418S ade− his− colonies from the rad26Δ, crb2Δ or OPcdc25 backgrounds indicated that they carried a truncated minichromosome of an identical size to that of a known isochromosome (388 kb) (our unpublished results). These findings support a general role for the DNA damage checkpoint pathway in facilitating efficient HR repair and suppressing break-induced chromosomal rearrangements and LOH.
Figure 3.
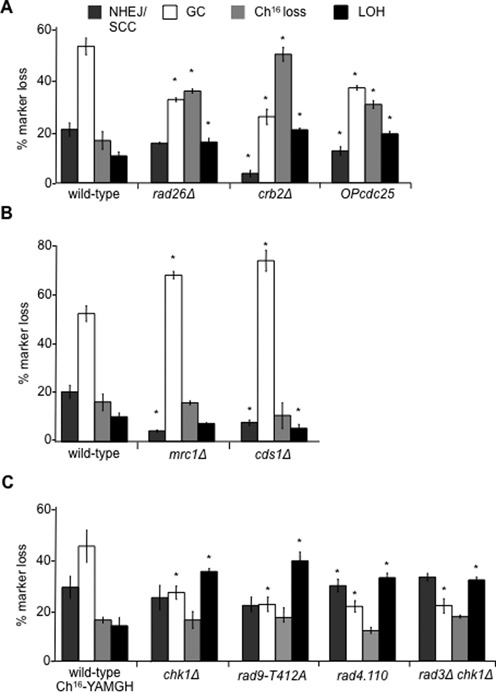
The DNA damage checkpoint promotes HR and suppresses break-induced LOH. (A) Percentage DSB-induced marker loss of Ch16-RMGAH in wild-type (TH2130), rad26Δ (TH3410), crb2Δ (TH3383) and OPcdc25 (TH3395) backgrounds. (B) The DNA replication checkpoint does not suppress break-induced LOH. Percentage DSB-induced marker loss of Ch16-RMGAH in wild-type (TH2130), mrc1Δ (TH3253) and cds1Δ (TH3256) backgrounds. (C) An additional role for Chk1 activation in promoting HR and suppressing break-induced LOH. Percentage DSB-induced marker loss of Ch16-YAMGH in wild-type (TH3317), chk1Δ (TH3153), rad9-T412A (TH5381), rad4.110 (TH4481) and rad3Δchk1Δ (TH3623) backgrounds. For (A), (B) and (C) the levels of NHEJ/SCC, GC, Ch16 loss and extensive LOH are shown. Data are the mean of three experiments and standard errors of the mean are indicated. The asterisk (*) represents P < 0.05 compared to wild-type.
The DNA replication checkpoint does not suppress break-induced LOH
A possible role for the DNA replication checkpoint in DSB repair was also analysed in mrc1Δ or cds1Δ backgrounds. In contrast to the DNA damage checkpoint mutants, levels of GC were significantly increased in mrc1Δ (69.3%; P < 0.01), while levels of NHEJ/SCC (4.4%; P = 0.01) were significantly reduced compared to wild-type (Figure 3B). Similarly, levels of GC were significantly increased in cds1Δ (75.3%; P < 0.01), while levels of NHEJ/SCC (7.9%; P = 0.01) and LOH (5.4%; P < 0.01) were reduced compared to wild-type (Figure 3B). Thus, in contrast to the DNA damage checkpoint pathway, disrupting the DNA replication checkpoint resulted in a hyper-recombinant phenotype.
Chk1+ activation is required to suppress break-induced LOH
To test the role of the DNA damage checkpoint effector kinase Chk1 in suppressing break-induced LOH, the chk1::ura4 mutant background was established using Ch16-YAMGH in which the chk1+ gene present on the minichromosome was deleted with a hygromycin resistance marker. While NHEJ/SCC levels in chk1Δ (24.1%) were similar to wild-type Ch16-YAMGH (27.8%), levels of GC were significantly reduced in a chk1Δ background (26.0% P < 0.01), compared to wild-type Ch16-YAMGH (43.3%). However, levels of break-induced LOH (33.9%) were significantly increased in chk1Δ compared to wild-type Ch16-YAMGH (13.3% P < 0.01) and rad3Δ (19.6% P < 0.01) backgrounds, thus suggesting an additional role for Chk1 in suppressing break-induced LOH, to that of Rad3ATR. The further increase in levels of break-induced LOH in the chk1Δ background was associated with reduced levels of Ch16 loss (15.7%), but this was not significantly different to wild-type Ch16-YAMGH (16.3% P = 0.9) (Figure 3C). Further PFGE analysis of the chk1Δ HygR ade− G418S his− colonies indicated that LOH had resulted from isochromosome formation (our unpublished results).
Chk1 activation requires Rad9 phosphorylation on T412/S423 to promote association with Rad4TOPBP1 (17). Therefore, we tested levels of break-induced LOH in rad9-T412A and rad4-110 mutant backgrounds in which Chk1 activation is abrogated. Both resembled the DSB profile of chk1Δ with increased break-induced LOH. DSB induction in a rad9-T412A background resulted in significantly reduced GC (21.5% P = 0.01) and significantly increased break-induced LOH (39.8% P = 0.02) compared to wild-type (Figure 3C). Similarly, DSB induction in a rad4-110 temperature-sensitive background at the semi-permissive temperature of 30°C resulted in significantly elevated levels of NHEJ/SCC (34.5% P = 0.03), significantly reduced GC (20.8% GC P < 0.01) and significantly increased LOH (32.8% P < 0.01) compared to wild-type (Figure 3C).
These results support a role for Chk1 activation in suppressing break-induced LOH, which is functionally distinct from Rad3ATR. DSB repair in a rad3Δchk1Δ double mutant exhibited a similar DSB repair profile to the chk1Δ single mutant (Figure 3C). These findings indicate Rad3ATR and Chk1 function in the same pathway to suppress break-induced LOH and to facilitate efficient Ch16 loss. However, Chk1 performs an additional Rad3ATR -independent role in suppressing break-induced LOH.
A distinct role for Rad17 and the 9-1-1 complex in suppressing break-induced LOH
Another component of the DNA damage checkpoint is Rad17 that functions as part of the RFC-checkpoint loading complex to load the 9-1-1 complex onto sites of damaged DNA (13,14). Mutant loh6-1, isolated from the screen, was found to encode a nonsense (W72X) mutation in the rad17+ gene (Supplementary Figure S4; our unpublished results). DSB induction in a rad17Δ background resulted in a striking DSB repair profile, which suggested a distinct role for Rad17 in facilitating extensive resection leading to Ch16 loss and suppressing break-induced LOH compared to Rad3ATR. rad17Δ had significantly reduced levels of GC (34.4% P = 0.03) and Ch16 loss (0.8%, P < 0.01) and significantly increased levels of break-induced LOH (59.1% P = 0.03) compared to wild-type (Figure 4A).
Figure 4.
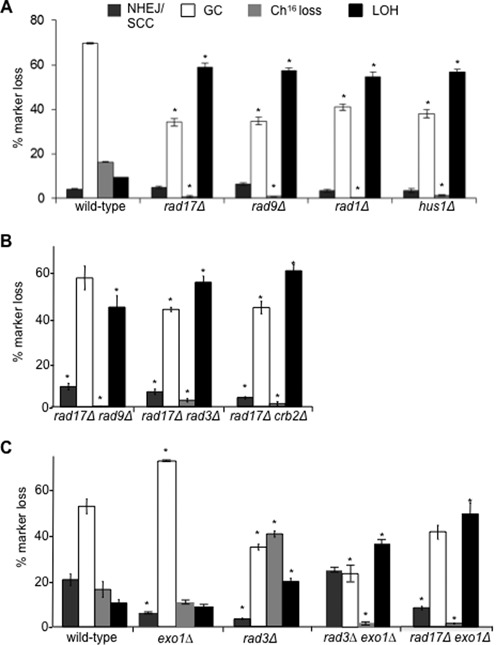
An additional role for Rad17 and the 9-1-1 complex in promoting HR and suppressing break-induced LOH. (A) Percentage DSB-induced marker loss of Ch16-RMYAH in wild-type (TH4104, TH4121, TH4122, TH4125), rad17Δ (TH7427-TH7430), rad9Δ (TH7588-TH7591), rad1Δ (TH7493, TH7494, TH7495) and hus1Δ (TH7431-TH7434) backgrounds. (B) Percentage DSB-induced marker loss of Ch16-RMGAH in rad17Δ rad9Δ (TH3454), rad17Δ rad3Δ (TH3455) and rad17Δ crb2Δ (TH3529) backgrounds. (C) Rad3ATR and Exo1 function redundantly to suppress break-induced LOH. Percentage DSB-induced marker loss of Ch16-RMGAH in wild-type (TH2130), exo1Δ (TH3378), rad3Δ (TH2941), rad3Δexo1Δ (TH3382) and rad17Δ exo1Δ (TH3701) backgrounds. For (A), (B) and (C) the levels of NHEJ/SCC, GC, Ch16 loss and extensive LOH are shown. Data are the mean of three experiments and standard errors of the mean are indicated. The asterisk (*) represents significant difference compared to wild-type.
The DSB repair profiles of rad9Δ, rad1Δ and hus1Δ mutants were also examined, and were found to be very similar to those observed for rad17Δ: GC was significantly reduced in rad9Δ (34.7% P < 0.01), rad1Δ (41.1% P < 0.01) and hus1Δ (38.1% P = 0.01) backgrounds compared to wild-type (69.6%). Ch16 loss was also significantly reduced in rad9Δ (1.0% P < 0.01), rad1Δ (0.19% P < 0.01) and hus1Δ (1.4% P < 0.01) backgrounds compared to wild-type (16.5%). In contrast, break-induced LOH was dramatically and significantly increased in rad9Δ (57.4% P < 0.01), rad1Δ (54.8% P = 0.01) and hus1Δ (56.8% P < 0.01) backgrounds compared to wild-type (9.6%) (Figure 4A).
PFGE analysis of individual LOH colonies (arg+ G418S/HygS ade− his−) derived from each of the rad17Δ, rad9Δ, rad1Δ or hus1Δ mutant backgrounds confirmed the majority to have a chromosomal element of equivalent size to a known isochromosome. Additionally, shorter chromosomal elements as described earlier were observed in 5 out of 19 (26%) rad17Δ LOH colonies, 4 out of 20 (20%) of rad9Δ LOH colonies, 4 out of 19 (21%) of rad1Δ LOH colonies and 4 out of 17 (24%) of hus1Δ LOH colonies (our unpublished results). CGH analysis of one rad17Δ arg+ G418S ade− his− colony indicated the shorter chromosomal element resulted from loss of the broken minichromosome arm while the intact arm was not duplicated (our unpublished results), thus resembling the shorter truncated chromosomal elements observed in the rad3Δ LOH colonies (Figure 2B and C).
Given the strikingly similar DSB-induced marker loss profiles observed in rad17Δ, rad9Δ, rad1Δ and hus1Δ, we tested the possibility that they functioned in the same pathway by epistasis analysis. Break-induced marker loss in a rad17Δrad9Δ double mutant was very similar to the single mutants, rad17Δ and rad9Δ (Figure 4B), supporting roles for the Rad17 loading clamp and 9-1-1 complex acting in the same pathway to suppress break-induced LOH. A double mutant rad17Δrad3Δ was also constructed. This strain exhibited similar levels of break-induced LOH (50.2%) and levels of Ch16 loss (2.7%) to a rad17Δ mutant (49.3% and 1.4%, respectively, Figure 4B). Thus Rad17 performs an additional role in extensive resection and suppression of break-induced LOH to that of Rad3ATR. As increased levels of break-induced extensive LOH correlated with reduced levels of Ch16 loss these findings support a role for Rad17 and the 9-1-1 complex in facilitating extensive end-processing required for Ch16 loss. The S. cerevisiae crb2+ homologue, RAD9, has been shown to limit the amount of ssDNA produced at uncapped telomeres (41). We found rad17Δcrb2Δ double mutants exhibited similar levels of SCC (3.5%), GC (39.9%), Ch16 loss (1.2%) and break-induced LOH (50.2%) as those observed in a rad17Δ single mutant (Figure 4B) . These results suggested that Crb253BP1 was not limiting extensive resection in a rad17Δ mutant background in S. pombe.
Analysis of spontaneous isochromosome formation within the Ch16 minichromosome indicates that they contain the breakpoint in centromere repeats, showing that isochromosomes are produced by centromere rearrangements. Rad3ATR has been previously shown to suppress spontaneous isochromosome formation associated with increased centromeric recombination (42). We therefore tested whether deletion of rad9+ exhibited increased spontaneous centromeric recombination. However, deletion of rad9+ did not significantly increase the rate of spontaneous recombination between the ade6B and ade6X heteroalleles integrated into the centromere (Supplementary Figure S5A and B), consistent with the idea that Rad9Sp affects resection of DSB-induced ends, generated outside the centromere.
Rad3ATR and Exo1 act redundantly to suppress break-induced LOH
In S. cerevisiae, Mec1 is required for the inhibitory phosphorylation of Exo1 (43). We therefore tested the relationship between Rad3ATR and Exo1. DSB induction in an exo1Δ background resulted in significantly reduced levels of NHEJ/SCC (7.6% P = 0.01), and significantly elevated levels of GC (69.8% P = 0.02) when compared to wild-type (Figure 4C). DSB induction in a rad3Δ exo1Δ background resulted in significantly reduced GC in a rad3Δ exo1Δ background (28.3% P < 0.01) compared to wild-type (52.7%). A dramatic decrease in Ch16 loss was observed in the rad3Δ exo1Δ double mutant (1.24% P = 0.02) in comparison to wild-type (16.3%) exo1Δ (11.4% P < 0.01) and rad3Δ (40.5% P < 0.01) single mutants. Importantly, in the rad3Δexo1Δ double mutant the level of break-induced LOH was strikingly increased (41.6% P < 0.01) compared to wild-type (10.3%; Figure 4C). These data are consistent with the idea that Rad3 phosphorylates Exo1, thus inactivating it. PFGE analysis of the arg+ G418S ade− his− colonies derived from the rad3Δ exo1Δ mutant background confirmed that LOH had arisen predominantly through isochromosome formation, with smaller chromosomal elements observed in 3/17 (18%) of the individually isolated LOH colonies examined (our unpublished results). This contrasts with the finding that the rad17Δexo1Δ double mutant did not affect the levels of break-induced LOH compared to rad17Δ (Figure 4C).
Deleting spd1+ suppresses HR defects of rad3Δ,rad26Δ but not 9-1-1 mutants
We previously identified a role for Rad3ATR in facilitating efficient HR repair by inducing nucleotide synthesis in response to DSBs. This allows efficient DNA synthesis during HR, preventing LOH. Rad3ATR induces Ddb1-Cul4Cdt2 ubiquitin ligase dependent degradation of the ribonucleotide reductase (RNR) inhibitor Spd1 to increase nucleotide pools (44). Transactivation of Cdt2 is required for the recruitment of Spd1 to the Ddb1-Cul4Cdt2 complex and requires Rad3ATR and Chk1 (45).
Given the contrasting repair profiles of rad3Δ and rad9Δ, rad1Δ or hus1Δ deletion strains, we investigated the role of the 9-1-1 complex in Cdt2 accumulation and thus dNTP synthesis. Deletion of rad3+, rad26+, rad17+, rad9+, rad1+ and hus1+ each abolished nuclear accumulation of Cdt2 in response to DNA damage (Supplementary Figure S6A and B). These findings are consistent with a common role for the DNA damage checkpoint pathway in facilitating dNTP synthesis through Cdt2 transactivation.
To further test the role of DNA damage checkpoint genes in dNTP synthesis, we tested whether deleting spd1+, an inhibitor of ribonucleotide reductase (46), might suppress the DNA damage sensitivity of other checkpoint mutants by increasing cellular nucleotide pools. We found that deletion of spd1+ could partially suppress the bleocin sensitivity of rad3Δ and rad26Δ (Figure 5A). In contrast, deletion of spd1+ was unable to suppress the bleocin sensitivity of rad17Δ, rad9Δ, rad1Δ or hus1Δ (Figure 5A).
Figure 5.
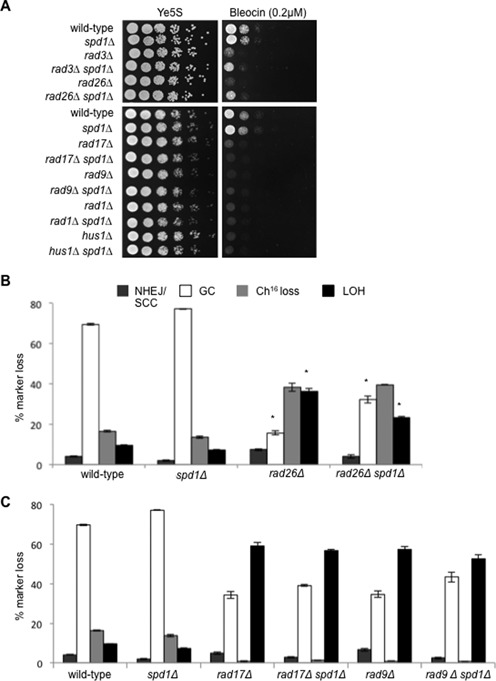
spd1Δ suppresses the repair defect of rad3Δ and rad26Δ. (A) Five-fold serial dilutions of wild-type (TH2094), spd1Δ (TH4355), rad3Δ (TH7329), rad3Δspd1Δ (TH8295), rad26Δ (TH7330) and rad26Δspd1Δ (TH8194) strains (top panel) and wild-type (TH2094), spd1Δ (TH4355), rad17Δ (TH7331), rad17Δspd1Δ (TH7794), rad9Δ (TH7414), rad9Δspd1Δ (TH7146), rad1Δ (TH7333), rad1Δspd1Δ (TH8249), hus1Δ (TH8296) and hus1Δspd1Δ (TH8195) strains (bottom panel) grown on Ye5S (untreated) and Ye5S + 0.2 μg/ml bleocin. (B) Percentage DSB-induced marker loss in wild-type (TH4121, TH4122, TH4104), spd1Δ (TH4077-TH4079) rad26Δ (TH7424-TH7426) and rad26Δspd1Δ (TH7585-TH7587) backgrounds. Means ± standard errors of three experiments are shown. Asterisk (*) represents significant difference compared to rad26Δ and rad26Δspd1Δ mutants. (C) Percentage DSB-induced marker loss in wild-type (TH4121, TH4122, TH4104), spd1Δ (TH4077-TH4079), rad17Δ (TH7429-TH7430), rad17Δspd1Δ (TH7566-TH7568), rad9Δ (TH7589-TH7591) and rad9Δspd1Δ (TH7464-TH7466) backgrounds. Means ± standard errors of three experiments are shown.
To confirm that suppression of bleocin sensitivity by spd1Δ correlated with increased HR, DSB assays were performed on these strains. Consistent with this, DSB induction in a rad26Δ spd1Δ background resulted in significantly increased levels of GC (32.4%, P = 0.02) and significantly reduced levels of LOH (23.4%, P = 0.02), compared to rad26Δ (GC 15.6%; LOH 36.3%, respectively) (Figure 5B), as was previously observed for rad3Δ spd1Δ (44). These findings are consistent with roles for both Rad3ATR and Rad26ATRIP in facilitating efficient HR by promoting nucleotide synthesis. In contrast, deletion of spd1+ in rad17Δ, rad9Δ, rad1Δ or hus1Δ backgrounds did not result in suppression of HR or a reduction in LOH compared to the parental strains following DSB induction (Figure 5C and our unpublished results). Together these results indicate a role for Rad3ATR Rad26ATRIP, Rad17 and the 9-1-1 complex in DNA damage induced dNTP synthesis, while Rad17 and the 9-1-1 complex also perform an additional function from that of Rad3ATR Rad26ATRIP that cannot be suppressed by spd1+ deletion.
Role for Rad17 and the 9-1-1 complex in facilitating DSB end resection and SSA
To further test a role for the 9-1-1 complex in DSB resection, we utilized a strain in which DSB-induced extensive resection facilitates SSA of two overlapping regions of the LEU2 gene containing sequence homology, placed either side of a break site (Figure 6A). The HO endonuclease was placed under the control of the endogenous urg promoter, which is rapidly inducible with uracil, generating a unique DSB at the HO cut site (HO-cs) (37,38). DSB induction in wild-type rad3Δ, rad17Δ and rad9Δ backgrounds was observed genetically by loss of histidine auxotrophy and found to be comparable between the mutants (Figure 6B). The repair kinetics was next determined by Southern blot analysis of the levels of loss of a 6.2 kb band and the appearance of a shorter 3.1 kb band containing the reformed LEU2 gene resulting from SSA (Figure 6A). In a wild-type or rad3Δ background DSB induction resulted in almost complete loss of the upper 6.2 kb band, and generation of a much stronger 3.1 kb band after 360 min, consistent with efficient extensive resection and SSA repair (Figure 6C and D). In contrast, DSB induction in a rad17Δ or rad9Δ background resulted in formation of a weaker 3.1 kb band consistent with reduced extensive resection and SSA repair in these backgrounds (Figure 6C and D). These findings support roles for Rad17 and the 9-1-1 complex in extensive resection and SSA repair.
Figure 6.
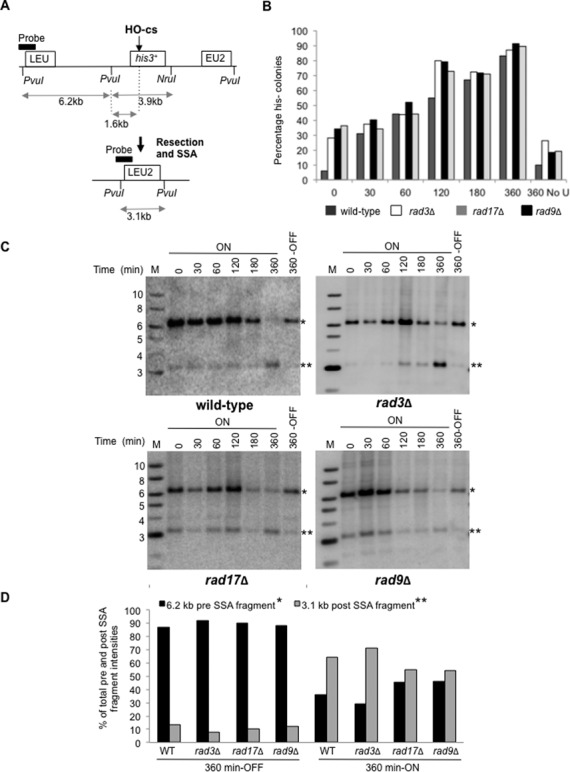
A role for Rad17 and the 9-1-1 complex in SSA repair. (A) A schematic of a resection and SSA assay as previously described (37). (B) Graph of HOcs-HIS SSA genetic colony assay showing loss of his3+ marker following induction of Purg1lox-HO-endonuclease in wild-type (TH7184), rad3Δ (TH8091) rad17Δ (TH8040) and rad9Δ (TH8050) backgrounds. The genetic assay was repeated independently at least three times. Error bars are ± standard deviation of the mean. (C) Physical analysis of HO-endonuclease cutting and repair by Southern hybridization in wild-type (TH7184), rad3Δ (TH8091) rad17Δ (TH8040) and rad9Δ(TH8050) cells. Genomic DNA extracted after Purg1lox induction at intervals shown, digested with PvuI and NruI, blotted and hybridized to probe as indicated in (A). Marker lane (M) and band sizes (kb) are indicated. The 6.2 kb pre-SSA fragment (*) and 3.1 kb post-SSA fragment (**) are indicated. (D) Graph of band intensities at 360 min without HO induction (OFF) or with HO induction (ON) for blots shown in (C). Blots were scanned using a personal molecular imagerTM (PMITM) and Quantity One Software (Bio-rad). Relative intensities of 6.2 kb pre-SSA fragment and 3.1 kb post-SSA fragments are shown, and were normalized by calculating the intensities of pre- and post-SSA bands as a percentage of the total intensities for these bands for each time point. M indicates DNA size marker and kb sizes of marker bands shown. 360 OFF refers to cells grown in EMM+L+H.
DISCUSSION
Here we establish roles for the DNA damage checkpoint pathway in facilitating efficient HR, and suppressing break-induced chromosomal rearrangements associated with failed HR repair. We define distinct yet overlapping functions for the DNA damage checkpoint genes in facilitating both extensive resection and nucleotide synthesis thereby promoting HR repair. These findings suggest that the DNA damage checkpoint pathway plays an important role in coordinating these processes in addition to promoting cell cycle arrest in response to DSBs.
A common role for the DNA damage checkpoint pathway was identified in facilitating nucleotide synthesis in response to DNA damage. Consistent with this, we found rad3+, rad26+, rad17+, rad9+, rad1+ and hus1+ genes to be required for transactivating Cdt2 expression in response to DNA damage. Checkpoint activation has previously been shown to lead to Cdt2 transactivation, which in turn activates the Ddb1-Cul4Cdt2 ubiquitin ligase complex leading to degradation of Spd1, an RNR inhibitor in fission yeast (45). The resulting increase in nucleotide synthesis following RNR activation has been shown to promote HR repair by facilitating gap filling of resected ssDNA ends (44). Accordingly, we found increased nucleotide synthesis resulting from spd1+ deletion could partially suppress the DNA damage sensitivity and HR deficiency of rad26Δ, as well as that of rad3Δ, as previously described (44). However, spd1+ deletion was unable to suppress the DNA damage sensitivity and HR deficiency of rad17Δ rad9Δ, rad1Δ or hus1Δ, consistent with an additional role for Rad17 and the 9-1-1 complex in the DNA damage response.
An additional role for Rad17 and the 9-1-1 complex in extensive resection was identified. Deletion of rad17+ rad9+, rad1+ and hus1+ genes resulted in a remarkable reduction in break-induced Ch16 loss and a concomitant increase in chromosomal rearrangements, predominantly through isochromosome formation. Given that Ch16 loss was previously shown to arise from extensive resection from the break site (35), these findings suggest roles for the Rad17 and the 9-1-1 complex in facilitating efficient resection through centromeric DNA (Figure 7A). Further, using a physical assay, we confirmed a role for Rad17 and the 9-1-1 complex in resection and SSA repair, strongly supporting the genetic data for the 9-1-1 complex in facilitating extensive resection. Moreover, rad17Δ functioned epistatically with rad9Δ, consistent with a role for Rad17 in loading the 9-1-1 complex (18). As no increase in spontaneous centromere recombination was observed in a rad9Δ background compared to wild-type, these findings further support a role for Rad17 and the 9-1-1 complex in DSB metabolism. Consistent with these findings, roles for homologues of Rad17 and the 9-1-1 complex in DSB resection have been reported previously (41,47–49).
Figure 7.
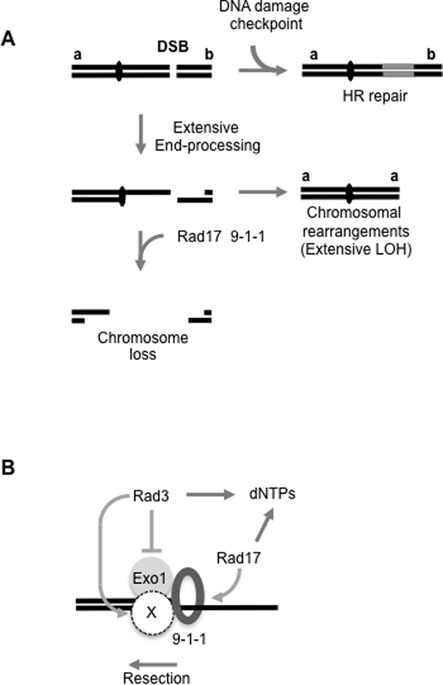
(A) Model for roles for the DNA damage checkpoint pathway in suppressing extensive LOH and chromosomal rearrangements associated with failed DSB repair. The DNA damage checkpoint pathway promotes efficient HR repair. Failed HR leads to extensive end processing and to chromosome loss or rearrangements. Rad17 and the 9-1-1 complex further suppress break-induced LOH by promoting extensive end processing through the centromere, resulting in loss of the broken chromosome. This is supported by the findings that Rad17 and the 9-1-1 complex are required for extensive resection, removal of the unrepaired broken minichromosome and suppression of extensive LOH. (B) Model for the roles of the DNA damage checkpoint proteins and Exo1 in facilitating extensive resection in S. pombe. Following DSB induction, the 9-1-1 complex (ring) is loaded by Rad17. The 9-1-1 complex facilitates processivity of Exo1 and nuclease X. Rad3ATR, together with other checkpoint proteins (not shown), promotes dNTP synthesis, promotes nuclease X and additionally inhibits Exo1. This model is supported by the findings that the rad3Δ exo1Δ double mutant phenocopies the DSB repair profile of rad17Δ, leading to high levels of extensive LOH and low levels of minichromosome loss, while rad3Δ or exo1Δ do not; as exo1Δ was not equivalent to rad17Δ or loss of the 9-1-1 complex, this suggests that the 9-1-1 complex additionally provides processivity to another nuclease (X), which requires Rad3 for activity. All checkpoint genes tested are required for transactivating Cdt2 expression, an initial step in damage-induced dNTP synthesis. See the text for details.
Isochromosomes were previously determined to have arisen from extensive resection resulting from failed HR leading to BIR within the centromere, and to duplication of the intact minichromosome arm (35). We speculate that the striking increase in break-induced isochromosomes and reduced chromosome loss observed in the absence of Rad17 or the 9-1-1 complex may reflect the increased stability of DSB repair intermediates arising through reduced resection efficiency, thereby facilitating BIR. Together these findings underline the importance of efficient DSB resection in maintaining genome stability.
We further identified deletions of rad3+ or exo1+ to be epistatic with deletion of rad17+ suggesting that Rad3, Exo1, Rad17 and the 9-1-1 complex function in the same pathway to facilitate extensive resection and Ch16 loss. In contrast to the single mutants, simultaneous deletion of rad3+ and exo1+ was found to be functionally equivalent to deletion of rad17+, resulting in very high levels of break-induced LOH and low levels of Ch16 loss. These findings suggest a role for Rad3ATR in inhibiting Exo1 activity, consistent with findings in S. cerevisiae (43). Thus in the absence of Rad3, reduced GC leads to increased levels of Exo1-dependent resection resulting in increased levels of Ch16 loss and LOH. However, in the absence of both Rad3 and Exo1, extensive resection becomes inefficient, resulting in reduced Ch16 loss and very high levels of LOH. As the repair profile of the rad3Δ exo1Δ double mutant is similar to those observed in rad17Δ, rad9Δ, rad1Δ or hus1Δ backgrounds, these findings suggest the 9-1-1 complex functions to promote efficient resection through supporting Exo1 activity. In this respect, the 9-1-1 complex may function analogously to structurally related PCNA to provide processivity to Exo1. That the phenotype associated with loss of Exo1 was not equivalent to the loss of Rad17 or the 9-1-1 complex strongly suggests that the 9-1-1 complex additionally provides processivity to another nuclease (X) that acts redundantly with Exo1 to promote extensive resection (Figure 7B). As rad3Δ exo1Δ exhibits a phenotype equivalent to rad17Δ while exo1Δ does not suggest that Rad3ATR may additionally promote nuclease X activity, which is also facilitated by the 9-1-1 complex. A likely candidate for nuclease X is Dna2, which is required for extensive resection, functions in a parallel pathway to Exo1 (50,51), and can be targeted by Rad3ATR, albeit through Cds1Chk2 (52).
Our data further identified a distinct role for Chk1 activation in facilitating HR and suppressing break-induced chromosomal rearrangements. As Chk1 activation requires Rad3ATR-dependent phosphorylation, and Rad3ATR activation requires the Rad17 and the 9-1-1 complex (reviewed in (53)), these data suggest that Rad17-dependent loading of the 9-1-1 complex may facilitate Rad3ATR activation and thus Chk1 activation. Yet, we previously found that in contrast to rad3Δ the DNA damage sensitivity of chk1Δ could not be suppressed by spd1Δ (44). Chk1 may therefore function like the 9-1-1 complex to support both Rad3ATR- and Exo1-dependent extensive resection. However, rad17Δ and chk1Δ backgrounds exhibit distinct DSB repair profiles suggesting that the relationship between these checkpoint proteins is more complex.
In contrast to the DNA damage checkpoint genes, deletion of the replication checkpoint genes mrc1+ and cds1+ resulted in a hyper-recombinant phenotype, exhibiting significantly elevated levels of break-induced GC compared to wild-type. These findings indicate a clear demarcation of the DNA damage and replication checkpoint functions, with the former facilitating efficient DSB repair by HR. One possible explanation for this ‘hyper-rec’ phenotype associated with the replication checkpoint mutants is a role for Mrc1 in promoting sister chromatid cohesion in S. cerevisiae (54). As sister chromatid cohesion limits recombination between homologous chromosomes (55), disrupting sister chromatid cohesion through such mutations could facilitate increased levels of interchromosomal GC.
We have identified roles for the DNA damage checkpoint pathway, including homologues of the haploinsufficient tumor suppressors, Rad3ATR, Crb253BP1 and Chk1 in suppressing break-induced LOH (56–58). Our data suggest that these homologues may function to suppress tumorigenesis through promoting efficient HR thereby suppressing extensive resection, chromosomal rearrangements and extensive LOH. In addition, we found that overexpression of Cdc25, which abrogates the DNA damage checkpoint, resulted in inefficient HR repair, increased levels of break-induced chromosome loss and LOH. Reduced HR efficiency following Cdc25 overexpression may have arisen from inappropriate cyclin-dependent kinase (CDK) dependent activation of CtIP and thus extensive resection, as suggested from studies in S. cerevisiae (59), or alternatively through a reduced G2-phase and accelerated entry into mitosis through increased CDK activity. In humans, CDC25 orthologues can function as oncogenes and are frequently over expressed in high-grade tumours with poor prognosis (reviewed in (60)). Our findings suggest a mechanistic explanation for these observations.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENT
We thank the laboratory of Antony Carr for strains and reagents.
FUNDING
Medical Research Council [R06538 to H.T.P., E.B., T.K., L.H., S.H., R.D., C.W., C.P., T.H.]; Cancer Research UK [C9546/A6517 to S.M., J.B.]; A*STAR, Singapore (to B.W.); Grant-in-Aid for Scientific Research from the Japan Society for the Promotion of Science (to T.N.). Source of open access funding: MRC (T.H.).
Conflict of interest. None declared.
REFERENCES
- 1.Kasparek T.R., Humphrey T.C. DNA double-strand break repair pathways, chromosomal rearrangements and cancer. Semin. Cell Dev. Biol. 2011;22:886–897. doi: 10.1016/j.semcdb.2011.10.007. [DOI] [PubMed] [Google Scholar]
- 2.Weinert T.A., Hartwell L.H. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science. 1988;241:317–322. doi: 10.1126/science.3291120. [DOI] [PubMed] [Google Scholar]
- 3.Bashkirov V.I., Bashkirova E.V., Haghnazari E., Heyer W.D. Direct kinase-to-kinase signaling mediated by the FHA phosphoprotein recognition domain of the Dun1 DNA damage checkpoint kinase. Mol. Cell. Biol. 2003;23:1441–1452. doi: 10.1128/MCB.23.4.1441-1452.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao X., Muller E.G., Rothstein R. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell. 1998;2:329–340. doi: 10.1016/s1097-2765(00)80277-4. [DOI] [PubMed] [Google Scholar]
- 5.Bashkirov V.I., King J.S., Bashkirova E.V., Schmuckli-Maurer J., Heyer W.D. DNA repair protein Rad55 is a terminal substrate of the DNA damage checkpoints. Mol. Cell. Biol. 2000;20:4393–4404. doi: 10.1128/mcb.20.12.4393-4404.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Martin S.G., Laroche T., Suka N., Grunstein M., Gasser S.M. Relocalization of telomeric Ku and SIR proteins in response to DNA strand breaks in yeast. Cell. 1999;97:621–633. doi: 10.1016/s0092-8674(00)80773-4. [DOI] [PubMed] [Google Scholar]
- 7.Lisby M., Mortensen U.H., Rothstein R. Colocalization of multiple DNA double-strand breaks at a single Rad52 repair centre. Nat. Cell Biol. 2003;5:572–577. doi: 10.1038/ncb997. [DOI] [PubMed] [Google Scholar]
- 8.Harper J.W., Elledge S.J. The DNA damage response: ten years after. Mol. Cell. 2007;28:739–745. doi: 10.1016/j.molcel.2007.11.015. [DOI] [PubMed] [Google Scholar]
- 9.Abraham R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. doi: 10.1101/gad.914401. [DOI] [PubMed] [Google Scholar]
- 10.Ellison V., Stillman B. Biochemical characterization of DNA damage checkpoint complexes: clamp loader and clamp complexes with specificity for 5’ recessed DNA. PLoS Biol. 2003;1:E33. doi: 10.1371/journal.pbio.0000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Green C.M., Erdjument-Bromage H., Tempst P., Lowndes N.F. A novel Rad24 checkpoint protein complex closely related to replication factor C. Curr. Biol. 2000;10:39–42. doi: 10.1016/s0960-9822(99)00263-8. [DOI] [PubMed] [Google Scholar]
- 12.Thelen M.P., Venclovas C., Fidelis K. A sliding clamp model for the Rad1 family of cell cycle checkpoint proteins. Cell. 1999;96:769–770. doi: 10.1016/s0092-8674(00)80587-5. [DOI] [PubMed] [Google Scholar]
- 13.Majka J., Burgers P.M. Yeast Rad17/Mec3/Ddc1: a sliding clamp for the DNA damage checkpoint. Proc. Natl. Acad. Sci. U.S.A. 2003;100:2249–2254. doi: 10.1073/pnas.0437148100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bermudez V.P., Lindsey-Boltz L.A., Cesare A.J., Maniwa Y., Griffith J.D., Hurwitz J., Sancar A. Loading of the human 9-1-1 checkpoint complex onto DNA by the checkpoint clamp loader hRad17-replication factor C complex in vitro. Proc. Natl. Acad. Sci. U.S.A. 2003;100:1633–1638. doi: 10.1073/pnas.0437927100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zou L., Elledge S.J. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]
- 16.Bonilla C.Y., Melo J.A., Toczyski D.P. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell. 2008;30:267–276. doi: 10.1016/j.molcel.2008.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Furuya K., Poitelea M., Guo L., Caspari T., Carr A.M. Chk1 activation requires Rad9 S/TQ-site phosphorylation to promote association with C-terminal BRCT domains of Rad4TOPBP1. Genes Dev. 2004;18:1154–1164. doi: 10.1101/gad.291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Navadgi-Patil V.M., Burgers P.M. The unstructured C-terminal tail of the 9-1-1 clamp subunit Ddc1 activates Mec1/ATR via two distinct mechanisms. Mol. Cell. 2009;36:743–753. doi: 10.1016/j.molcel.2009.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saka Y., Esashi F., Matsusaka T., Mochida S., Yanagida M. Damage and replication checkpoint control in fission yeast is ensured by interactions of Crb2, a protein with BRCT motif, with Cut5 and Chk1. Genes Dev. 1997;11:3387–3400. doi: 10.1101/gad.11.24.3387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alcasabas A.A., Osborn A.J., Bachant J., Hu F., Werler P.J., Bousset K., Furuya K., Diffley J.F., Carr A.M., Elledge S.J. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 2001;3:958–965. doi: 10.1038/ncb1101-958. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka K., Russell P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat. Cell Biol. 2001;3:966–972. doi: 10.1038/ncb1101-966. [DOI] [PubMed] [Google Scholar]
- 22.Furnari B., Rhind N., Russell P. Cdc25 mitotic inducer targeted by chk1 DNA damage checkpoint kinase. Science. 1997;277:1495–1497. doi: 10.1126/science.277.5331.1495. [DOI] [PubMed] [Google Scholar]
- 23.Boddy M.N., Furnari B., Mondesert O., Russell P. Replication checkpoint enforced by kinases Cds1 and Chk1. Science. 1998;280:909–912. doi: 10.1126/science.280.5365.909. [DOI] [PubMed] [Google Scholar]
- 24.Zeng Y., Forbes K.C., Wu Z., Moreno S., Piwnica-Worms H., Enoch T. Replication checkpoint requires phosphorylation of the phosphatase Cdc25 by Cds1 or Chk1. Nature. 1998;395:507–510. doi: 10.1038/26766. [DOI] [PubMed] [Google Scholar]
- 25.Lopez-Girona A., Furnari B., Mondesert O., Russell P. Nuclear localization of Cdc25 is regulated by DNA damage and a 14–3–3 protein. Nature. 1999;397:172–175. doi: 10.1038/16488. [DOI] [PubMed] [Google Scholar]
- 26.Enoch T., Nurse P. Mutation of fission yeast cell cycle control genes abolishes dependence of mitosis on DNA replication. Cell. 1990;60:665–673. doi: 10.1016/0092-8674(90)90669-6. [DOI] [PubMed] [Google Scholar]
- 27.Myung K., Datta A., Kolodner R.D. Suppression of spontaneous chromosomal rearrangements by S phase checkpoint functions in Saccharomyces cerevisiae. Cell. 2001;104:397–408. doi: 10.1016/s0092-8674(01)00227-6. [DOI] [PubMed] [Google Scholar]
- 28.Myung K., Chen C., Kolodner R.D. Multiple pathways cooperate in the suppression of genome instability in Saccharomyces cerevisiae. Nature. 2001;411:1073–1076. doi: 10.1038/35082608. [DOI] [PubMed] [Google Scholar]
- 29.Myung K., Kolodner R.D. Induction of genome instability by DNA damage in Saccharomyces cerevisiae. DNA Repair (Amst) 2003;2:243–258. doi: 10.1016/s1568-7864(02)00216-1. [DOI] [PubMed] [Google Scholar]
- 30.Myung K., Kolodner R.D. Suppression of genome instability by redundant S-phase checkpoint pathways in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4500–4507. doi: 10.1073/pnas.062702199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Admire A., Shanks L., Danzl N., Wang M., Weier U., Stevens W., Hunt E., Weinert T. Cycles of chromosome instability are associated with a fragile site and are increased by defects in DNA replication and checkpoint controls in yeast. Genes Dev. 2006;20:159–173. doi: 10.1101/gad.1392506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Callen E., Jankovic M., Difilippantonio S., Daniel J.A., Chen H.T., Celeste A., Pellegrini M., McBride K., Wangsa D., Bredemeyer A.L., et al. ATM prevents the persistence and propagation of chromosome breaks in lymphocytes. Cell. 2007;130:63–75. doi: 10.1016/j.cell.2007.06.016. [DOI] [PubMed] [Google Scholar]
- 33.Khanna K.K., Lavin M.F., Jackson S.P., Mulhern T.D. ATM, a central controller of cellular responses to DNA damage. Cell Death Differ. 2001;8:1052–1065. doi: 10.1038/sj.cdd.4400874. [DOI] [PubMed] [Google Scholar]
- 34.Prudden J., Evans J.S., Hussey S.P., Deans B., O’Neill P., Thacker J., Humphrey T. Pathway utilization in response to a site-specific DNA double-strand break in fission yeast. EMBO J. 2003;22:1419–1430. doi: 10.1093/emboj/cdg119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tinline-Purvis H., Savory A.P., Cullen J.K., Dave A., Moss J., Bridge W.L., Marguerat S., Bahler J., Ragoussis J., Mott R., et al. Failed gene conversion leads to extensive end processing and chromosomal rearrangements in fission yeast. EMBO J. 2009;28:3400–3412. doi: 10.1038/emboj.2009.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hope J.C., Mense S.M., Jalakas M., Mitsumoto J., Freyer G.A. Rqh1 blocks recombination between sister chromatids during double strand break repair, independent of its helicase activity. Proc. Natl. Acad. Sci. U.S.A. 2006;103:5875–5880. doi: 10.1073/pnas.0601571103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Watt S., Mata J., Lopez-Maury L., Marguerat S., Burns G., Bahler J. urg1: a uracil-regulatable promoter system for fission yeast with short induction and repression times. PLoS One. 2008;3:e1428. doi: 10.1371/journal.pone.0001428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Watson A.T., Werler P., Carr A.M. Regulation of gene expression at the fission yeast Schizosaccharomyces pombe urg1 locus. Gene. 2011;484:75–85. doi: 10.1016/j.gene.2011.05.028. [DOI] [PubMed] [Google Scholar]
- 39.Cullen J.K., Hussey S.P., Walker C., Prudden J., Wee B.Y., Dave A., Findlay J.S., Savory A.P., Humphrey T.C. Break-induced loss of heterozygosity in fission yeast: dual roles for homologous recombination in promoting translocations and preventing de novo telomere addition. Mol. Cell. Biol. 2007;27:7745–7757. doi: 10.1128/MCB.00462-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang X., Sun Y., Chen S., Roy K., Price B.D. The FATC domains of PIKK proteins are functionally equivalent and participate in the Tip60-dependent activation of DNA-PKcs and ATM. J. Biol. Chem. 2006;281:15741–15746. doi: 10.1074/jbc.M513172200. [DOI] [PubMed] [Google Scholar]
- 41.Lydall D., Weinert T. Yeast checkpoint genes in DNA damage processing: implications for repair and arrest. Science. 1995;270:1488–1491. doi: 10.1126/science.270.5241.1488. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura K., Okamoto A., Katou Y., Yadani C., Shitanda T., Kaweeteerawat C., Takahashi T.S., Itoh T., Shirahige K., Masukata H., et al. Rad51 suppresses gross chromosomal rearrangement at centromere in Schizosaccharomyces pombe. EMBO J. 2008;27:3036–3046. doi: 10.1038/emboj.2008.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morin I., Ngo H.P., Greenall A., Zubko M.K., Morrice N., Lydall D. Checkpoint-dependent phosphorylation of Exo1 modulates the DNA damage response. EMBO J. 2008;27:2400–2410. doi: 10.1038/emboj.2008.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Moss J., Tinline-Purvis H., Walker C.A., Folkes L.K., Stratford M.R., Hayles J., Hoe K.L., Kim D.U., Park H.O., Kearsey S.E., et al. Break-induced ATR and Ddb1-Cul4(Cdt)(2) ubiquitin ligase-dependent nucleotide synthesis promotes homologous recombination repair in fission yeast. Genes Dev. 2010;24:2705–2716. doi: 10.1101/gad.1970810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu C., Poitelea M., Watson A., Yoshida S.H., Shimoda C., Holmberg C., Nielsen O., Carr A.M. Transactivation of Schizosaccharomyces pombe cdt2+ stimulates a Pcu4-Ddb1-CSN ubiquitin ligase. EMBO J. 2005;24:3940–3951. doi: 10.1038/sj.emboj.7600854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Liu C., Powell K.A., Mundt K., Wu L., Carr A.M., Caspari T. Cop9/signalosome subunits and Pcu4 regulate ribonucleotide reductase by both checkpoint-dependent and -independent mechanisms. Genes Dev. 2003;17:1130–1140. doi: 10.1101/gad.1090803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aylon Y., Kupiec M. The checkpoint protein Rad24 of Saccharomyces cerevisiae is involved in processing double-strand break ends and in recombination partner choice. Mol. Cell. Biol. 2003;23:6585–6596. doi: 10.1128/MCB.23.18.6585-6596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dubrana K., van Attikum H., Hediger F., Gasser S.M. The processing of double-strand breaks and binding of single-strand-binding proteins RPA and Rad51 modulate the formation of ATR-kinase foci in yeast. J. Cell Sci. 2007;120:4209–4220. doi: 10.1242/jcs.018366. [DOI] [PubMed] [Google Scholar]
- 49.Zubko M.K., Guillard S., Lydall D. Exo1 and Rad24 differentially regulate generation of ssDNA at telomeres of Saccharomyces cerevisiae cdc13–1 mutants. Genetics. 2004;168:103–115. doi: 10.1534/genetics.104.027904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhu Z., Chung W.H., Shim E.Y., Lee S.E., Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mimitou E.P., Symington L.S. DNA end resection: Many nucleases make light work. DNA Repair (Amst) 2009;8:983–995. doi: 10.1016/j.dnarep.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu J., Sun L., Shen F., Chen Y., Hua Y., Liu Y., Zhang M., Hu Y., Wang Q., Xu W., et al. The intra-S phase checkpoint targets Dna2 to prevent stalled replication forks from reversing. Cell. 2012;149:1221–1232. doi: 10.1016/j.cell.2012.04.030. [DOI] [PubMed] [Google Scholar]
- 53.Navadgi-Patil V.M., Burgers P.M. A tale of two tails: activation of DNA damage checkpoint kinase Mec1/ATR by the 9-1-1 clamp and by Dpb11/TopBP1. DNA Repair (Amst) 2009;8:996–1003. doi: 10.1016/j.dnarep.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu H., Boone C., Brown G.W. Genetic dissection of parallel sister-chromatid cohesion pathways. Genetics. 2007;176:1417–1429. doi: 10.1534/genetics.107.072876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Covo S., Westmoreland J.W., Gordenin D.A., Resnick M.A. Cohesin Is limiting for the suppression of DNA damage-induced recombination between homologous chromosomes. PLoS Genet. 2010;6:e1001006. doi: 10.1371/journal.pgen.1001006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fang Y., Tsao C.C., Goodman B.K., Furumai R., Tirado C.A., Abraham R.T., Wang X.F. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO J. 2004;23:3164–3174. doi: 10.1038/sj.emboj.7600315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ward I.M., Difilippantonio S., Minn K., Mueller M.D., Molina J.R., Yu X., Frisk C.S., Ried T., Nussenzweig A., Chen J. 53BP1 cooperates with p53 and functions as a haploinsufficient tumor suppressor in mice. Mol. Cell. Biol. 2005;25:10079–10086. doi: 10.1128/MCB.25.22.10079-10086.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lam M.H., Liu Q., Elledge S.J., Rosen J.M. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell. 2004;6:45–59. doi: 10.1016/j.ccr.2004.06.015. [DOI] [PubMed] [Google Scholar]
- 59.Ira G., Pellicioli A., Balijja A., Wang X., Fiorani S., Carotenuto W., Liberi G., Bressan D., Wan L., Hollingsworth N.M., et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature. 2004;431:1011–1017. doi: 10.1038/nature02964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Boutros R., Lobjois V., Ducommun B. CDC25 phosphatases in cancer cells: key players? Good targets. Nat. Rev. Cancer. 2007;7:495–507. doi: 10.1038/nrc2169. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.