

Abstract
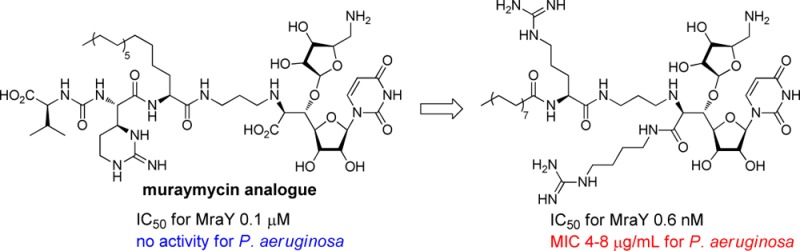
It is urgent to develop novel anti-Pseudomonas agents that should also be active against multidrug resistant P. aeruginosa. Expanding the antibacterial spectrum of muraymycins toward P. aeruginosa was investigated by the systematic structure–activity relationship study. It was revealed that two functional groups, a lipophilic side chain and a guanidino group, at the accessory moiety of muraymycins were important for the anti-Pseudomonas activity, and analogue 29 exhibited antibacterial activity against a range of P. aeruginosa strains with the minimum inhibitory concentration values of 4–8 μg/mL.
Keywords: Antibiotics, drug-resistance, MraY, muraymycin, peptidoglycan, anti-Pseudomonas
Generally, Gram-negative bacterial pathogens are less sensitive to antibacterial drugs in comparison with Gram-positive bacteria because they possess an outer membrane, which acts as a permeability barrier. Among them, Pseudomonas aeruginosa is a common nosocomial pathogen that is intrinsically resistant to a variety of drugs currently used in the clinic, and immuno-compromised patients are at greater risk of infection. The increased rate of acquired multidrug resistance in P. aeruginosa also complicates anti-Pseudomonas chemotherapy. Consequently, it is urgent to develop novel anti-Pseudomonas agents that should also be active against multidrug resistant P. aeruginosa.1−3 In choosing novel antibacterial agents, the target must be essential for their growth, a mechanism of action of the agent should be different from existing drugs. The initial lead compounds should also be amenable to structural changes that allow for optimization of the potency and efficacy.4−8 Muraymycins (MRYs) (Figure 1, 1), isolated from a culture broth of Streptomyces sp.,9,10 are members of a class of naturally occurring 6′-N-alkyl-5′-β-O-aminoribosyl-C-glycyluridine antibiotics.11 The MRYs having a lipophilic side chain have been shown to exhibit excellent antimicrobial activity against Gram-positive bacteria. The MRYs inhibit the formation of lipid II and peptidoglycan and are inhibitors of phospho-MurNAc-pentapeptide transferase (MraY), which is responsible for the formation of lipid I in the peptidoglycan biosynthesis pathway.12−16 The enzymes involved in the biosynthesis of peptidoglycan are essential for bacterial life and represent important targets for antibacterial chemotherapy. Recently, we have investigated the systematic structure–activity relationship (SAR) study of the MRYs.17−19 These studies revealed that the impact of the lipophilic substituent on antibacterial activity was very large, and the lipophilic analogue 2 showed a potent antibacterial activity against a range of Gram-positive bacteria including methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococci (VRE).18 An accessory urea-dipeptide moiety could also be simplified to a large extent, and the truncated analogues 3–10 (Figure 2) retained the antibacterial activity.19 Other biological properties of these analogues were also investigated, and it was found that these analogues are selective inhibitors of MraY and are nontoxic to human liver hepatocellular (HepG2) cells. Consequently, the MraY inhibitors are promising leads for antibacterial agents active against drug-resistant Gram-positive bacterial pathogens. The MraY enzyme is also conserved among Gram-negative bacteria; however, the natural MRYs are not effective to these Gram-negative bacteria, presumably as a consequence of outer membrane impermeability. Here we describe our efforts to expand antibacterial spectrum of the MRYs toward P. aeruginosa.
Figure 1.
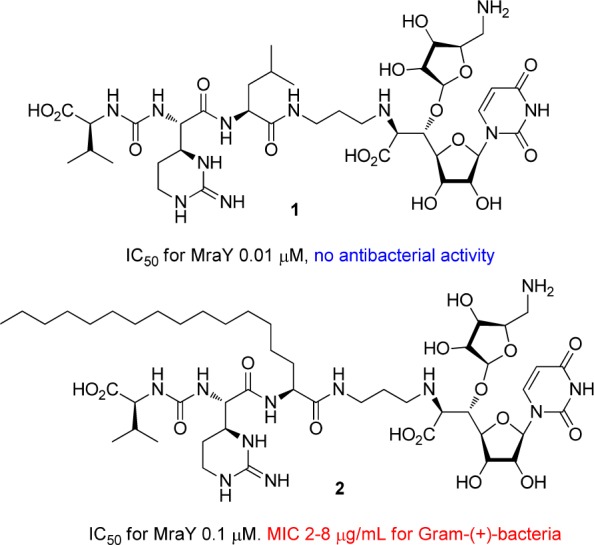
Structures and biological activity of muraymycin D2 and its lipophilic analogue.
Figure 2.
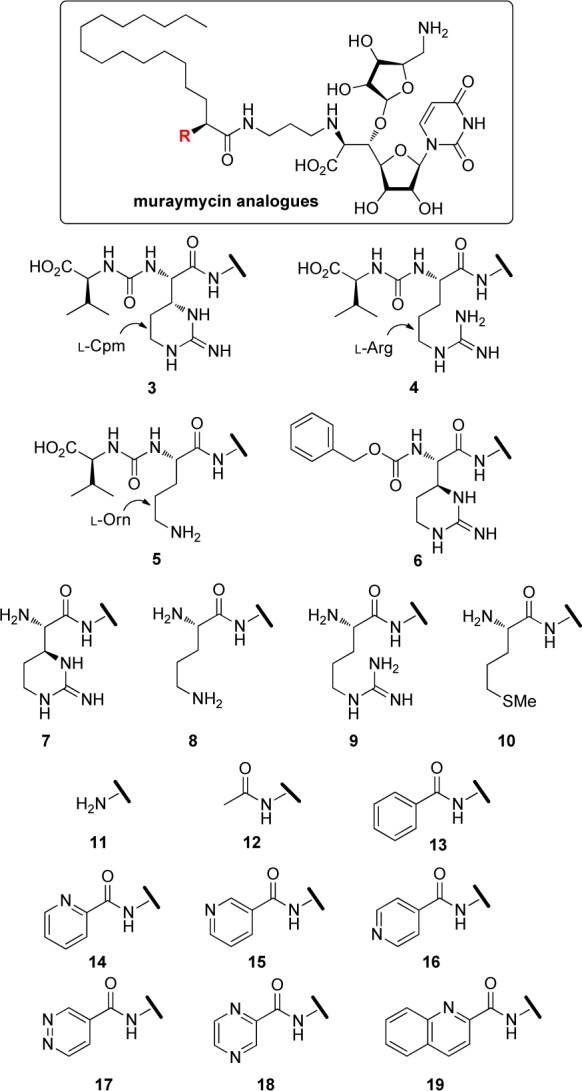
Structures of muraymycin analogues.
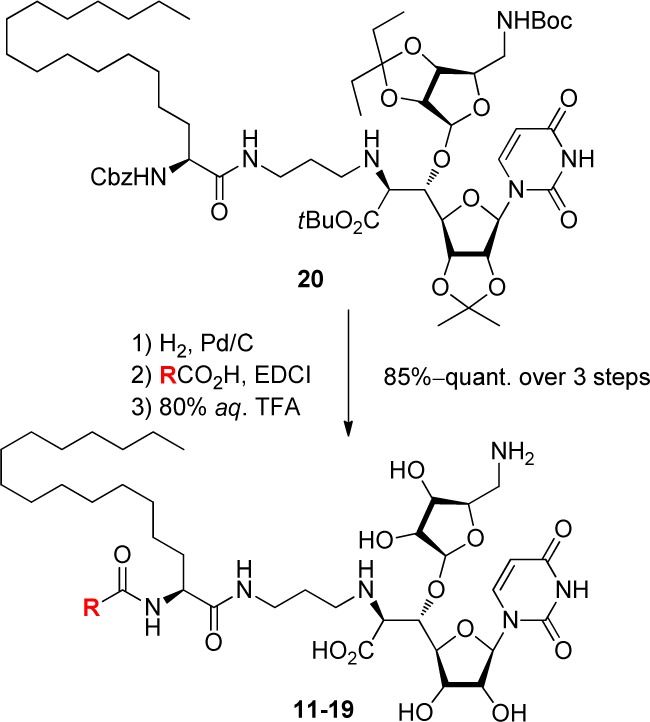
It is expected that chemical modification of our MRY analogues 2–10 modulates outer membrane permeability of P. aeruginosa and makes it possible to expand antibacterial activity toward this pathogen. However, antibacterial evaluation of 2–10 for P. aeruginosa has not been investigated yet. We initiated this program by revisiting anti-Pseudomonas activity of 2–10. In addition, simple acyl derivatives 11–19 were also planned to evaluate (Figure 2). As shown in Scheme 1, these analogues were easily obtained by the Cbz deprotection of 6′-N-aminopropyl-5′-β-O-aminoribosyl-C-glycyluridine 20,15 acylation of the liberated amine, and global deprotection by aqueous TFA over 3 steps.
Scheme 1. Synthesis of Acyl Analogues 11–19.
First, the inhibitory activity of the analogues on the purified MraY enzyme (Staphylococcus aureus) was examined by fluorescence-based MraY assay20,21 using UDP-MurNAc-dansylpentapeptide, where the formation of dansylated lipid I was monitored by fluorescence enhancement (excitation at 355 nm and emission at 535 nm), and the results are summarized in Table 1. As a result, most of the analogues exhibited strong inhibitory activity with a nanomolar range of the IC50s. Anti-Pseudomonas activity of 1–19 was next evaluated against a range of P. aeruginosa strains.22 As expected, MRY D2 (1) showed a very weak activity, and its lipophilic analogue 2 did not show any activity. Analogues 7–12 exhibited a moderate activity with the MIC values of 8–64 μg/mL. Analogue 9, which possesses the guanidino and the amino functional groups, was the most active among those tested (MIC values of 8–32 μg/mL). It is a great contrast that the analogues 3 and 4 also have guanidine groups but show no anti-Pseudomonas activity. Since these analogues have the terminal carboxylic acid, it would partially neutralize the positive charge of the guanidino group. Newly synthesized analogues 13–19, which have an aromatic ring at the accessory moiety, were inactive to a range of P. aeruginosa strains. As a result, analogues possessing net positive charges at the accessory moiety tend to have the expanded antibacterial activity toward P. aeruginosa although there are some exceptions.
Table 1. Screening of Muraymycin Analogues 1–19 as Anti-Pseudomonas Agents.
| MIC (μg/mL)a |
||||||
|---|---|---|---|---|---|---|
| compound | MraY IC50 (nM) | P. aeruginosa PAO1 | P. aeruginosa YY165 (ΔmexB) | P. aeruginosa ATCC 25619 | P. aeruginosa SR 27156 | P. aeruginosa ATCC 27853 |
| 1 | 2.8 | >64 | 64 | 64 | 32 | >64 |
| 2 | 1.1 | >64 | >64 | >64 | >64 | >64 |
| 3 | 0.8 | >64 | >64 | >64 | >64 | >64 |
| 4 | 0.7 | >64 | >64 | >64 | >64 | >64 |
| 5 | 0.8 | >64 | >64 | >64 | >64 | >64 |
| 6 | 4.2 | >64 | >64 | >64 | >64 | >64 |
| 7 | 2.4 | >64 | 32 | 64 | 64 | >64 |
| 8 | 3.8 | 64 | 32 | 32 | 32 | 64 |
| 9 | 2.2 | 16 | 8 | 8 | 16 | 32 |
| 10 | 8.5 | >64 | 16 | 16 | 64 | 64 |
| 11 | 2.6 | >64 | 32 | 32 | >64 | >64 |
| 12 | 2.7 | 64 | 32 | 32 | 64 | >64 |
| 13 | 29.6 | >64 | >64 | >64 | >64 | >64 |
| 14 | 6.4 | >64 | >64 | 64 | >64 | >64 |
| 15 | 9.6 | >64 | >64 | 64 | >64 | >64 |
| 16 | 8.1 | >64 | >64 | 64 | >64 | >64 |
| 17 | 13.5 | >64 | 64 | 64 | >64 | >64 |
| 18 | 33.1 | >64 | >64 | 64 | >64 | >64 |
| 19 | 105 | >64 | >64 | >64 | >64 | >64 |
| doripenem | n.d. | 0.5 | 0.25 | 0.063 | 0.031 | 0.5 |
MICs were determined by a microdilution broth method as recommended by the CLSI with cation-adjusted Mueller–Hinton broth (CA-MHB). Serial 2-fold dilutions of each compound were made in appropriate broth, and the plates were inoculated with 5 × 104 CFU of each strain in a volume of 0.1 mL. Plates were incubated at 35 °C for 20 h, and then MICs were scored.
A revisit of the antibacterial evaluation of muraymycin analogues gave us a direction for a further molecular design for expanding the antibacterial spectrum to P. aeruginosa. Thus, necessity of a lipophilic side chain and a guanidino group at the accessory moiety was implied to be important for the anti-Pseudomonas activity from the SAR in Table 1. Therefore, we planned to diversify the relative positions of these two functional groups to the core aminoribosyluridine moiety in order to optimize the orientation of the three moieties as shown in Figure 3. For example, the lipophilic side chain and the guanidino group were linked in a linear manner to give analogue 21. Interconversion of the lipophilic side chain and the guanidino group gave N-acyl-l-arginine analogues 23–26. Analogues 3–19 contains l-pentadecylglycin residue, the preparation of which required a number of steps. The molecular design of 23–26 enables us to use a commercially available l-Arg derivative and simple fatty acids. Analogue 27 is a branched version of 26, where the guanidino group located in the accessory moiety is moved to the carboxylic acid at the 6′-position of the uridine core structure. An interconversion of the branching functional groups of 27 gave 28. Analogue 29 is a hybrid-type of 26–28. Preparation of 21–26 is summarized in Scheme 2. The key intermediate 30(13−15) for the synthesis of muraymycin D2 and its derivatives was used as a starting material. The trichloroethoxycarbonyl (Troc) group was removed by Zn in MeOH, and the liberated amine was acylated with the corresponding carboxylic acids A–F (see Supporting Information) by EDCI and HOBt in CH2Cl2. Finally, the global deprotection by aqueous TFA successfully gave 21–26, respectively. Scheme 3 illustrates the preparation of analogues 27–29. Another key intermediate carboxylic acid 31(23,24) was coupled with Boc-protected 4-aminobutyl guanidine G (see Supporting Information) by EDCI and HOBt to give 32 in 89% yield. After the Cbz protecting group was removed by hydrogenolysis catalyzed by Pd/C, reductive alkylation of the resulting amine with TrocHNCH2CH2CHO afforded 34 in 89% yield. Removal of the Troc group (Zn and MeOH), acylation with palmitic acid (EDCI and HOBt), and deprotection (80% aqueous TFA) provided 27 in 50% yield over three steps. In a manner similar to the synthesis of 27, the analogue 28 and the hybrid-type analogue 29 were prepared.
Figure 3.
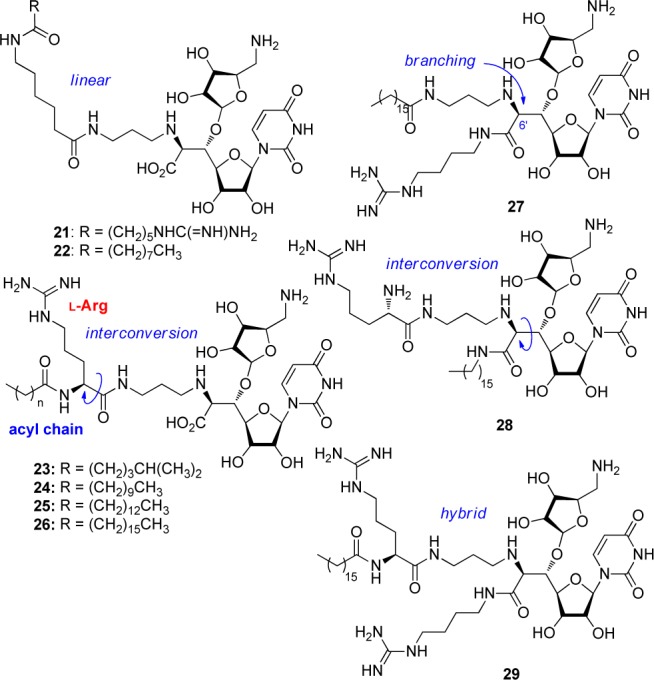
Molecular design of analogues 21–29.
Scheme 2. Synthesis of Analogues 21–26.

Scheme 3. Synthesis of Branched Analogues 27–29.

The MraY inhibitory activity of 21–29 was evaluated (Table 2). The linear analogues 21 and 22 reduced the inhibitory activity. As for interconverted analogues 23–26, the length of the lipophilic side chain is important for the MraY inhibitory activity. Thus, the analogue 23 with the short side chain largely reduced the activity with the IC50 value of 225 nM. Increasing the length of the side chain resulted in an increase of the inhibitory activity, and the length and the activity were well-correlated. Analogue 26 with C16 side chain exhibited the IC50 value of 1.6 nM. Analogues 27 and 29 exhibited strong MraY inhibitory activity (IC50 = 0.14 nM for 27 and 0.60 nM for 29, respectively). The anti-Pseudomonas activity of this series of the compounds was then evaluated (Table 2). The analogues 21 and 22 did not show any activity. The analogues 23 and 24, which have the shorter lipophilic side chain, exhibited no antibacterial activity, whereas 25 and 26 with the longer side chain showed moderate activity. As was indicated by the first screening shown in Table 1, two functional groups, a lipophilic side chain and a guanidino group, at the accessory moiety were important for the anti-Pseudomonas activity. The antibacterial activity was also well-correlated to the length of the lipophilic side chain. The branching analogues 27 and 28 have a similar anti-Pseudomonas activity to 26. Finally, the hybrid-type analogue 29 was the strongest activity among those tested in this study. It is worthy to mention that P. aeruginosa YY165 is slightly more sensitive to these analogues than other strains tested in this study. P. aeruginosa YY165 is a deletion mutant of MexB, which is an inner membrane transporter component of the MexAB-OprM multidrug efflux system that confers multidrug resistance. The sensitivity of analogues to P. aeruginosa YY165 indicates that they could be a substrate of the MexAB-OprM multidrug efflux system and excluded outside the cell. The analogues 27 and 29 are almost equipotent in terms of MraY inhibitory and anti-Pseudomonas activities. Selective toxicity is a key concern in the chemotherapy for infectious diseases. Since the chemical structure of 27 and 29 contains the long lipophilic side chain and positively charged guanidine functionality, it is suspected that the analogues 27 and 29 act as detergents, which are sometimes cytotoxic. Accordingly, their cytotoxic activity against human hepatocellular liver carcinoma (HepG2) cells was then evaluated. Analogue 27 showed cytotoxicity with an IC50 value of 4.5 μg/mL. Under these conditions, 29 exhibited less cytotoxicity (IC50 34 μg/mL). With the observed higher therapeutic index than 27, analogue 29 could be a lead for further development as anti-Pseudomonas agent although much improvement to reduce the toxicity is necessary. Finally, the metabolic stability of 26, 27, and 29 was briefly evaluated by treatment of these analogues with human or rat liver microsome at 37 °C for 30 min and LC–MS/MS analysis of remaining analogues (Table 3). Around 90% of each analogue was unaffected by human liver microsomes, and these analogues were revealed to be metabolically stable especially to human liver microsomes.
Table 2. MraY Inhibitory and Anti-Pseudomonas Activity of Analogues 21–29.
| MIC (μg/mL)a |
||||||
|---|---|---|---|---|---|---|
| compound | MraY IC50 (nM) | P. aeruginosa PAOl | P. aeruginosa YY165 (ΔmexB) | P. aeruginosa ATCC 25619 | P. aeruginosa SR 27156 | P. aeruginosa ATCC 27853 |
| 21 | 1491 | >64 | >64 | >64 | >64 | >64 |
| 22 | 97.3 | >64 | >64 | >64 | >64 | >64 |
| 23 | 225 | >64 | >64 | >64 | >64 | >64 |
| 24 | 9.8 | >64 | >64 | 32 | >64 | >64 |
| 25 | 2.1 | 32 | 16 | 16 | 16 | 32 |
| 26 | 1.6 | 16 | 8 | 16 | 16 | 32 |
| 27 | 0.14 | 16 | 8 | 8 | 8 | 16 |
| 28 | 12.2 | 32 | 16 | 16 | 16 | 16 |
| 29 | 0.60 | 4 | 4 | 8 | 8 | 8 |
| doripenem | n.d. | 0.5 | 0.25 | 0.063 | 0.031 | 0.5 |
MICs were determined by a microdilution broth method as recommended by the CLSI with cation-adjusted Mueller–Hinton broth (CA-MHB). Serial 2-fold dilutions of each compound were made in appropriate broth, and the plates were inoculated with 5 × 104 CFU of each strain in a volume of 0.1 mL. Plates were incubated at 35 °C for 20 h and then MICs were scored.
Table 3. Metabolic Stability of Muraymycin Analogues 26, 27, and 29a.
| 26 | 27 | 29 | |
|---|---|---|---|
| human | 89.5%b | 90.0% | 86.9% |
| rat | 81.1% | 80.7% | 69.8% |
Rat and human liver microsomes were incubated at 37 °C in Tris-HCl buffer (pH 7.4) containing 50 mM Tris-HCl, 150 mM KCl, 10 mM MgCl2, 1 mM β-NADPH, and 2 μM compounds. The protein concentration was 0.5 mg/mL, and the final volume was 0.2 mL. Incubations were terminated by the addition of 2-fold volume of ice-cold acetonitrile/methanol 1:1, v/v) after 0 and 30 min of incubation. Samples were centrifuged at 3000 rpm for 10 min prior to LC–MS/MS analysis of the supernatants. Experiments were conducted in duplicate.
Percentage of the remaining material after treatment with liver microsomes.
In conclusion, antibacterial spectrum of the MRYs toward P. aeruginosa was investigated by the systematic SAR study of the MRYs. It was revealed that two functional groups, a lipophilic side chain and a guanidino group, at the accessory moiety of MRYs were important for the anti-Pseudomonas activity. The knowledge obtained from our SAR study of the MRYs would provide future direction toward the expansion of the antibacterial spectrum of the MRYs to find more potent and selective agents active against P. aeruginosa.
Acknowledgments
We thank Ms. S. Oka and Ms. A. Tokumitsu (Center for Instrumental Analysis, Hokkaido University) for measurement of the mass spectra. We are also thankful for Ms. M. Okane and Mr. K. Uotani (Shionogi Techno Advance Research Co., Ltd.) for MIC measurement and Mr. K. Sekiguchi (Shionogi Co., Ltd.) for evaluating metabolic stability.
Glossary
ABBREVIATIONS
- Cbz
benzyloxycarbonyl
- EDCI
1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride
- HOBt
1-hydroxy-1H-benzotriazol
- HepG2
human hepatocellular liver carcinoma
- IC50
50% inhibitory concentration
- MIC
minimum inhibitory concentration
- MRY
muraymycin
- MraY
phospho-MurNAc-pentapeptide transferase
- MRSA
methicillin-resistant Staphylococcus aureus
- P. aeruginosa
Pseudomonas aeruginosa
- SAR
structure–activity relationship
- TFA
trifluoroacetic acid
- Troc
trichloroethoxycarbonyl
- C55–P
undecaprenyl–phosphate
- VRE
vancomycin-resistant Enterococci
Supporting Information Available
Experimental procedures for the synthesis and characterization of synthesized compounds, fluorescence-based MraY assay, antibacterial activity evaluation, and metabolic stability testing. This material is available free of charge via the Internet at http://pubs.acs.org.
This research was supported by JSPS Grant-in-Aid for Challenging Exploratory Research (Grant Number 22659020 to S.I.) and Scientific Research (B) (Grant Number 25293026 to S.I.).
The authors declare no competing financial interest.
Supplementary Material
References
- Tam V. H.; Chang K. T.; Abdelraouf K.; Brioso C. G.; Ameka M.; McCaskey L. A.; Weston J. S.; Caeiro J. P.; Garey K. W. Prevalence, resistance mechanisms, and susceptibility of multidrug-resistant bloodstream isolates of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2010, 54, 1160–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page M. G.; Heim J. Prospects for the next anti-Pseudomonas drug. Curr. Opin. Pharmacol. 2009, 9, 558–565. [DOI] [PubMed] [Google Scholar]
- Scheld M.; Spellberg B.; Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48, 1–12. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holms D. J.; Pompliano D. L. Drugs for bad bugs: confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 2007, 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Talbot G. H.; Bradley J.; Edwards J. E. Jr.; Gilbert D.; Scheld M.; Bartlett J. G. Bad bugs need drugs: an update on the development pipeline from the antimicrobial availability task force of the Infectious Diseases Society of America. Clin. Infect. Dis. 2006, 42, 657–668. [DOI] [PubMed] [Google Scholar]
- Overbye K. M.; Barrett J. F. Antibiotics: where did we go wrong?. Drug Discovery Today 2005, 10, 45–52. [DOI] [PubMed] [Google Scholar]
- Monagham R. L.; Barrett J. F. Antibacterial drug discovery-then, now and the genomics future. Biochem. Pharmacol. 2006, 71, 901–909. [DOI] [PubMed] [Google Scholar]
- Walsh C. Where will new antibiotics come from?. Nat. Rev. Microbiol. 2003, 1, 65–70. [DOI] [PubMed] [Google Scholar]
- McDonald L. A.; Barbieri L. R.; Carter G. T.; Lenoy E.; Lotvin J.; Petersen P. J.; Siegel M. M.; Singh G.; Williamson R. T. Structures of the muraymycins, novel peptidoglycan biosynthesis inhibitors. J. Am. Chem. Soc. 2002, 124, 10260–10261. [DOI] [PubMed] [Google Scholar]
- Carter G. T.; Lotvin J. A.; McDonald L. A.. WO 2002085310 A2.
- Winn M.; Goss R. J. M.; Kimura K.; Bugg T. D. H. Antimicrobial nucleoside antibiotics targeting cell wall assembly: Recent advances in structure–function studies and nucleoside biosynthesis. Nat. Prod. Rep. 2010, 27, 279–304. [DOI] [PubMed] [Google Scholar]
- Bugg T. D. H.; Lloyd A. J.; Roper D. I. Phospho-MurNAc-pentapeptide translocase (MraY) as a target for antibacterial agents and antibacterial proteins. Infect. Dis. Drug Targets 2006, 6, 85–106. [DOI] [PubMed] [Google Scholar]
- Kimura K.; Bugg T. D. H. Recent advances in antimicrobial nucleoside antibiotics targeting cell wall biosynthesis. Nat. Prod. Rep. 2003, 20, 252–273. [DOI] [PubMed] [Google Scholar]
- Bouhss A.; Mengin-Lecreulx D.; Le Beller D.; Van Heijenoort J. Topological analysis of the MraY protein catalyzing the first membrane step of peptidoglycan synthesis. Mol. Microbiol. 1999, 34, 576–585. [DOI] [PubMed] [Google Scholar]
- Bouhss A.; Trunkfield A. E.; Bugg T. D.; Mengin-Lecreulx D. The biosynthesis of peptidoglycan lipid-linked intermediates. FEMS Microbiol. Rev. 2008, 32, 208–33. [DOI] [PubMed] [Google Scholar]
- Al-Dabbagh B.; Henry X.; El Ghachi M.; Auger G.; Blanot D.; Parquet C.; Mengin-Lecreulx D.; Bouhss A. Active site mapping of MraY, a member of the polyprenyl-phosphate N-acetylhexosamine 1-phosphate transferase superfamily, catalyzing the first membrane step of peptidoglycan biosynthesis. Biochemistry 2008, 47, 8919–8928. [DOI] [PubMed] [Google Scholar]
- Tanino T.; Ichikawa S.; Shiro M.; Matsuda A. Total synthesis of (−)-muraymycin D2 and its epimer. J. Org. Chem. 2010, 75, 1366–1377. [DOI] [PubMed] [Google Scholar]
- Tanino T.; Ichikawa S.; Al-Dabbagh B.; Bouhss A.; Matsuda A. Synthesis and biological evaluation of muraymycin analogues as potential anti-drug-resistant bacterial. ACS Med. Chem. Lett. 2010, 1, 258–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanino T.; Al-Dabbagh B.; Mengin-Lecreulx D.; Bouhss A.; Oyama H.; Ichikawa S.; Matsuda A. Mechanistic analysis of muraymycin analogues: a guide to the design of MraY inhibitors. J. Med. Chem. 2011, 54, 8421–8439. [DOI] [PubMed] [Google Scholar]
- Bouhss A.; Crouvoisier M.; Blanot D.; Mengin-Lecreulx D. Purification and characterization of the bacterial MraY translocase catalyzing the first membrane step of peptidoglycan biosynthesis. J. Biol. Chem. 2004, 279, 29974. [DOI] [PubMed] [Google Scholar]
- Stachyra T.; Dini C.; Ferrari P.; Bouhss A.; van Heijenoort J.; Mengin-Lecreulx D.; Blanot D.; Biton J.; Le Beller D. Fluorescence detection-based functional assay for high-throughput screening for MraY. Antimicrob. Agents Chemother. 2004, 48, 897–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maki H.; Miura K.; Yamano Y.; Katanosin B. and plusbacin A3, inhibitors of peptidoglycan synthesis in methicillin-resistant Staphylococcus aureus. Antimicrob. Agents Chemother. 2001, 45, 1823–1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano S.; Ichikawa S.; Matsuda A. Total synthesis of caprazol, a core structure of the caprazamycin antituberculosis antibiotics. Angew. Chem., Int. Ed. 2005, 44, 1854–1856. [DOI] [PubMed] [Google Scholar]
- Hirano S.; Ichikawa S.; Matsuda A. Development of a highly β-selective ribosylation reaction without using neighboring group participation: total synthesis of (+)-caprazol, a core structure of caprazamycins. J. Org. Chem. 2007, 72, 9936–9946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.