

Abstract
Autotaxin is an extracellular phospholipase D that catalyzes the hydrolysis of lysophosphatidyl choline (LPC) to bioactive lipid lysophosphatidic acid (LPA). LPA has been implicated in many pathological processes relevant to cancer, including cell migration and invasion, proliferation, and survival. The most potent autotaxin inhibitor described to date is the LPA analogue S32826 (IC50 5.6 nM). S32826 and many other autotaxin inhibitors are notably lipophilic, creating a need to improve their physical properties. Polymers are becoming an increasingly useful tool in the delivery of drugs and have the potential to improve the properties of small molecules. Herein we report the synthesis of a S32826 dendrimer conjugate and its biological evaluation. The conjugate was found to inhibit autotaxin activity using two different substrates and to decrease the migration of an ovarian cancer cell line modified to overexpress autotaxin. Furthermore, the conjugate potentiated activation of caspase 3/7 induced by carboplatin.
Keywords: Autotaxin, dendrimer, S32826, ovarian cancer
Autotaxin (ATX) is an extracellular enzyme which is a member of the ecto-nucleotide pyrophosphatase/phosphodiesterase (ENPP) family and is also known as ENPP-2.1,2 Autotaxin is a 125 kDa glycoprotein, originally identified as an autocrine motility factor released by human melanoma cells.1 It was later found to have phospholipase D activity and to play a major role in the production of lysophosphatidic acid (LPA) from both normal human cells and cancer cells3 by catalyzing the hydrolysis of lysophosphatidyl choline (LPC).3,4 LPA acts via G protein coupled receptors on the cell surface to activate a variety of signaling pathways.5,6 These LPA-activated signaling pathways are implicated in a number of biological processes, such as cell migration, proliferation, and survival, that are associated with the pathogenesis of many cancers, including ovarian cancer.7 Additionally, LPA-activated signaling pathways have been implicated in other human diseases, including chronic inflammation,8 fibrotic diseases,9,10 and thrombosis.11 Patients with ovarian cancer often present with an accumulation of ascites fluid in the intraperitoneal cavity which contains LPA at concentrations up to 80 μM.12 Autotaxin is also found in the ascites fluid of patients with ovarian cancer.13 Autotaxin is overexpressed in ovarian cancers that are resistant to chemotherapy,14 and it has also been shown to delay apoptosis induced by carboplatin in ovarian cancer cells.15 Together with the well-established role of LPA in ovarian cancer cell migration and invasion,16 this suggests that inhibition of autotaxin has therapeutic potential in the treatment of ovarian cancer.
The observation of product inhibition of autotaxin by LPA has led to the discovery of a large number of LPA analogues which inhibit autotaxin.17 However, compounds based on the structure of LPA are often lipophilic and may have poor bioavailability, have poor solubility, or be degraded by endogenous hydrolytic pathways. Furthermore, LPA analogues may act downstream of autotaxin on LPA receptors due to their structural similarity with LPA. One of the most potent LPA analogues described to date is S32826 (Figure 1).18 This compound exhibited nanomolar inhibition of autotaxin in vitro and reduced the amount of LPA present in plasma and ascites ex vivo. However, when tested in vivo, no inhibition of LPA production in plasma was observed a few minutes after administration.18 Nonetheless, S32826 offers the opportunity to develop autotaxin inhibitors lacking affinity for LPA receptors.18 More recently there have been a number of nonlipid autotaxin inhibitors identified, of which two, HA150 and PF8380, have been shown to lower LPA levels in vivo.(19,20) However, the effect of these compounds on LPA receptors has not yet been fully investigated. Thus, there remains a need to identify specific autotaxin inhibitors with appropriate druglike properties.
Figure 1.

Structure of S32826.
Dendrimers have rapidly emerged as promising drug delivery systems due to their unique structural features. They are highly branched macromolecules of low polydispersity with a multifunctional central core molecule.21,22 The multiple surface groups of dendrimers offer the opportunity to covalently attach multiple drug molecules per dendrimer. The larger hydrodynamic volume of polymers has the potential to increase the plasma half-life of a drug molecule, increasing the probability of tumor accumulation.23,24 The conjugation of drugs to dendrimers can also increase their metabolic stability,21,25 and improve their pharmacokinetic properties. For example, a PAMAM-cisplatin conjugate shows improved solubility and reduced toxicity when compared to unconjugated cisplatin.26
These observations, suggesting that dendrimers have the ability to improve the half-life and metabolic stability of drug molecules, prompted us to evaluate the feasibility of conjugating autotaxin inhibitors to dendrimers. In particular, we wished to confirm that the conjugation of an autotaxin inhibitor to a dendrimer would not impair its ability to antagonize autotaxin. This is particularly significant because many autotaxin inhibitors display undesirable physical and pharmaceutical properties that might be ameliorated by conjugation to a dendrimer. We chose to investigate S32826 because, unlike many other autotaxin inhibitors, this compound has relatively low potency for LPA receptors. This facilitates the evaluation of the relative importance of these drug targets and avoids potentially unwanted affects mediated by antagonizing LPA1.27 S32826 was attached to a third generation (G3) PAMAM dendrimer in order to achieve a high hydrodynamic volume while not encountering the issues of higher toxicity associated with higher generations of dendrimers.22,28
Herein we report the synthesis of a novel drug conjugate of the potent autotaxin inhibitor, S32826 with a third generation PAMAM dendrimer. The conjugate inhibits autotaxin in vitro, suppresses autotaxin-induced cell migration, and potentiates apoptosis induced by carboplatin.
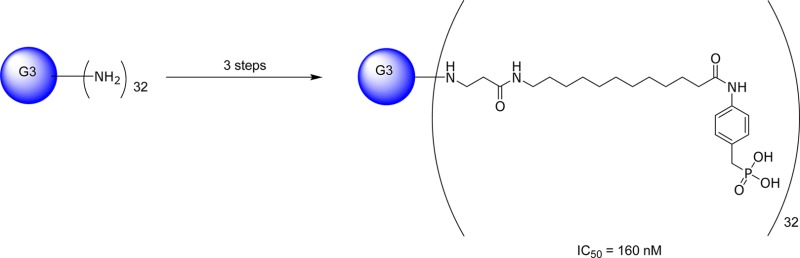
The S32826-dendrimer conjugate was synthesized via an effective three step process (Scheme 1). The potent autotaxin inhibitor S32826 was covalently conjugated to the terminal amino groups of G3 PAMAM dendrimer via an amide bond. In brief, the commercially available [4-(13-aminotridecanoylamino)benzyl]phosphonic acid diethyl ester 1 underwent amide formation with acryloyl chloride to generate the intermediate acrylamide phosphonate 2, which could then be conjugated to the dendrimer. Reaction conditions were chosen to maximize the stoichiometry of coupling, anticipating that this would yield a more potent and uniform product. The aqueous solubility of compound 4 was >90 μM. We also synthesized a derivative imitating the linkage to the dendrimer by changing the alkyl to an acetamide group to investigate the effect of changing the functional at the terminal end of S32826.
Scheme 1. Synthetic Route for Preparation of Compound 4.

Reagents and conditions: (i) acryloyl chloride, Et3N, CH2Cl2, 0 °C, 4 h; (ii) G3 PAMAM dendrimer, EtOH, 100 °C, 48 h; (iii) Me3SiI, MeOH, rt, 8 h.
The structure of the conjugate was supported by 1H NMR, IR spectroscopy, and elemental analysis. The 1H NMR spectrum showed conjugation to be complete, and residual unconjugated compound 1 was undetectable (absence of the acrylamide peaks at 6.08, 6.02, and 5.53 ppm). IR analysis of the final compound 4 shows the presence of a peak at 2860 cm–1 which was not present in compound 3, this peak is most likely to correspond to the P–OH exposed after phosphate ester hydrolysis, further supporting the structure of the final compound.
Elemental analysis was performed on compound 4 to determine the amount of S32826 coupled to the dendrimer. The mass fraction of phosphorus (present in S32826 and not the dendrimer) in the conjugate was compared to that which would be obtained with the fully substituted PAMAM dendrimer in which all 32 terminal amines was coupled to a S32826 molecule.
To establish the stoichiometry of coupling of the drug to the G3 PAMAM dendrimer, we measured the mass fraction of the phosphorus (only present in the drug molecule, not the dendrimer) compared to carbon, hydrogen, and nitrogen in the final compound 4. These data suggested essentially complete coupling of the 32 terminal amines in the dendrimer, with a ratio of phosphorus to nitrogen which corresponds closely to the theoretical ratio for maximum conjugation. The ratio of phosphorus to carbon and hydrogen also gave similar ratios to that expected of maximum conjugation (Table 1). Although almost complete conjugation was achieved, steric considerations suggest it is unlikely that all of the pharmacophores can bind to autotaxin molecules simultaneously. Our primary objective was to determine if conjugating autotaxin to a dendrimer would impair its activity. Several lines of evidence show it retained activity.
Table 1. Estimation of Stoichiometry of Coupling of S32826 to PAMAM Dendrimer.
| phosphorus
mass fraction compared to C, H, or N |
||
|---|---|---|
| element | theoretical | found |
| C | 0.075 | 0.0740 |
| H | 0.500 | 0.550 |
| N | 0.430 | 0.400 |
First, the ability of the conjugate to inhibit autotaxin catalytic activity was evaluated using autotaxin obtained from two separate sources, and the hydrolysis of two different substrates, FS329 or bis-pNPP, was measured.17 Compound 4 showed a moderate decrease in apparent potency after coupling to the dendrimer (IC50 = 160 nM), compared to the free compound S32826 (IC50 = 9 nM).30 Comparable potencies were measured using bis-pNPP as a substrate (S32826, IC50 = 30 nM; compound 4, IC50 = 1.7 μM). The unconjugated dendrimer had no measurable effect on autotaxin activity in either assay. The conjugate showed an approximately 10–60-fold reduction in potency compared to the free drug. This suggests that conjugation to the dendrimer has moderately impaired the affinity of individual pharmacophores, but nevertheless the conjugate has retained notable pharmacological activity. Acylation of the terminal end of S32826 to mimic the linkage led to a compound moderate decrease in potency in the bis-pNPP assay (IC50 = 50 nM), suggesting the decrease in potency is due to conjugation to the dendrimer rather than introduction of an amide.
Second, autotaxin is known to catalyze the production of LPA, which in turn is known to regulate cell migration in many cancer cell types including ovarian cancer.31−33 Autotaxin inhibitors have been shown to inhibit migration. To validate the ability of compound 4 to inhibit autotaxin in a biological assay, the effect of compound 4 on wound healing was evaluated using 3E3 ovarian cancer cells. These cells were derived from Ovcar-3 cells and engineered to overexpress autotaxin.15 3E3 cells were grown to confluence, the monolayer was wounded, and cell migration was measured in serum free media, supplemented with LPC (0.5 μM), containing the autotaxin inhibitors S32826 or compound 4. Both autotaxin inhibitors reduced wound closure almost 2-fold compared to the wound closure measured with cells exposed to vehicle alone (Figure 2).
Figure 2.

Effect of autotaxin inhibition on wound closure. (A) Effect of inhibition of autotaxin with either vehicle (0.5% DMSO), S32826 or compound 4 (0.65 μM or 1.3 μM), on wound closure. The wound in a 3E3 cell monolayer was viewed by phase contrast microscopy immediately after wounding or after 16 h. (B) Wound closure was measured and expressed as a percentage of complete wound closure (mean ± S.D., n = 3–5, DMSO).* P < 0.05, ** P < 0.01, *** P < 0.001 denote results that differ significantly from vehicle alone (paired t test).
Third, in contrast to the plethora of data showing that inhibition of autotaxin inhibits cell migration, there is a relative paucity of data showing that inhibition of autotaxin promotes cell death. LPA has previously been shown to inhibit cell death induced by cisplatin,34 and autotaxin inhibits apoptosis induced by paclitaxel.35 We have previously shown in cells engineered to overexpress autotaxin, that inhibition of autotaxin with the inhibitor ccPA (16:1) potentiates cell death induced by carboplatin. Previous work has shown that expression of autotaxin delays apoptosis induced by carboplatin, while apoptosis was accelerated after inhibition of autotaxin either by siRNA or with a small molecule inhibitor.15 To investigate the ability of compound 4 to potentiate apoptosis induced by carboplatin, caspase 3/7 activity was measured in 3E3 and 3V5 cells. The cells were treated with carboplatin, in the presence or absence of LPC, and either vehicle, S32826 or compound 4. Exposure to carboplatin for 18 h increased caspase 3/7 activity in 3E3 cells, and this could be substantially repressed by the addition of the substrate of autotaxin, LPC. However, the inclusion of either the free drug, S32826, or compound 4 prevented the suppression of caspase activity induced by LPC (Figure 3A). The autotaxin inhibitors had no measurable effect on caspase 3/7 activity on their own (Figure 3A). In contrast, in 3V5 cells, (which had been transfected with the empty vector) addition of LPC was unable to suppress the increase in caspase activity induced by carboplatin (Figure 3B) and addition of S32826 or compound 4 with carboplatin did not significantly alter the caspase 3/7 activity from that measured in the presence of carboplatin alone (Figure 3B). This confirms that the conjugate retains its activity when conjugated to dendrimer. Autotaxin is overexpressed in tumors that are relatively resistant to chemotherapy,14 and we found that autotaxin can contribute to resistance to carboplatin.15 Thus, autotaxin inhibitors may be useful in the treatment of ovarian cancer as agents to overcome resistance to chemotherapy, which may in part be mediated by autotaxin. This is particularly significant because the emergence of drug resistant is the key cause of treatment failure in patients with ovarian cancer and the resulting poor 5-year survival rate of approximately 40%.We conclude that, in principle, autotaxin inhibitors may be conjugated to dendrimers and retain the pharmacologic activity. The compound we have described may be useful to evaluate the therapeutic value of specifically inhibiting autotaxin. It will be useful to evaluate this approach using the other autotaxin inhibitors and determine their utility in the treatment of ovarian cancer.
Figure 3.

Caspase 3/7 activity was measured in 3E3 cells (A, ectopically expressing autotaxin) or 3V5 cells (B, transfected with vector) following incubation with 10 μM compound 4, 300 nM S32826, with or without 300 μM carboplatin for 18 h. Caspase 3/7 activity was normalized for cell number measured by staining with SRB. The results (mean ± S.D., n = 3–5) differed significantly from cells treated with LPC and carboplatin where shown: * P < 0.05, ** P < 0.01, *** P < 0.001 vs (one-way anova). RLU, relative luminescence units.
Glossary
Abbreviations
- LPA
lysophosphatidic acid
- LPC
lysophosphatidyl choline
- ATX
autotaxin
- ENPP
ectonucleotide pyrophosphatase/phosphodiesterase
- PAMAM
polyamidoamine
- Bis-pNPP
bis-p-nitrophenyl phosphate
Supporting Information Available
Supporting Information includes experimental procedures for chemical synthesis, biochemical and cellular assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
Experimental work was performed by N.F. and T.H.-B. The manuscript was written through contributions of all authors, and all approved the final version of the manuscript.
Funding was provided by the Medical Research Council, U.K.
The authors declare no competing financial interest.
Supplementary Material
References
- Stracke M. L.; Krutzsch H. C.; Unsworth E. J.; Arestad A.; Cioce V.; Schiffmann E.; Liotta L. A. Identification, purification, and partial sequence-analysis of autotaxin, a novel motility-stimulating protien. J. Biol. Chem. 1992, 26742524–2529. [PubMed] [Google Scholar]
- Clair T.; Lee H. Y.; Liotta L. A.; Stracke M. L. Autotaxin is an exoenzyme possessing 5′-nucleotide phosphodiesterase/ATP pyrophosphatase and ATPase activities. J. Biol. Chem. 1997, 2722996–1001. [DOI] [PubMed] [Google Scholar]
- Umezu-Goto M.; Kishi Y.; Taira A.; Hama K.; Dohmae N.; Takio K.; Yamori T.; Mills G. B.; Inoue K.; Aoki J.; Arai H. Autotaxin has lysophospholipase D activity leading to tumor cell growth and motility by lysophosphatidic acid production. J. Cell Biol. 2002, 1582227–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokumura A.; Majima E.; Kariya Y.; Tominaga K.; Kogure K.; Yasuda K.; Fukuzawa K. Identification of human plasma lysophospholipase D, a lysophosphatidic acid-producing enzyme, as autotaxin, a multifunctional phosphodiesterase. J. Biol. Chem. 2002, 2774239436–39442. [DOI] [PubMed] [Google Scholar]
- Anliker B.; Chun J. Lysophospholipid G protein-coupled receptors. J. Biol. Chem. 2004, 2792020555–20558. [DOI] [PubMed] [Google Scholar]
- Hurst J. H.; Mendpara N.; Hooks S. B. Regulator of G-protein signalling expression and function in ovarian cancer cell lines. Cell. Mol. Biol. Lett. 2009, 141153–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang X. J.; Schummer M.; Mao M. L.; Yu S. X.; Tabassam F. H.; Swaby R.; Hasegawa Y.; Tanyi J. L.; LaPushin R.; Eder A.; Jaffe R.; Erickson J.; Mills G. B. Lysophosphatidic acid is a bioactive mediator in ovarian cancer. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2002, 15821–3257–264. [DOI] [PubMed] [Google Scholar]
- Kanda H.; Newton R.; Klein R.; Morita Y.; Gunn M. D.; Rosen S. D. Autotaxin, an ectoenzyme that produces lysophosphatidic acid, promotes the entry of lymphocytes into secondary lymphoid organs. Nature Immunol. 2008, 94415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradère J.-P.; Klein J.; Grès S.; Guigné C.; Neau E.; Valet P.; Calise D.; Chun J.; Bascands J.-L.; Saulnier-Blache J.-S.; Schanstra J. P. LPA1 Receptor Activation Promotes Renal Interstitial Fibrosis. J. Am. Soc. Nephrol. 2007, 18123110–3118. [DOI] [PubMed] [Google Scholar]
- Tager A. M.; LaCamera P.; Shea B. S.; Campanella G. S.; Selman M.; Zhenwen Z.; Polosukhin V.; Wain J.; Karimi-Shah B. A.; Kim N. D.; Hart W. K.; Pardo A.; Blackwell T. S.; Yan X.; Chun J.; Luster A. D. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat. Med. 2008, 14145–54. [DOI] [PubMed] [Google Scholar]
- Pamuklar Z.; Federico L.; Liu S.; Umezu-Goto M.; Dong A.; Panchatcharam M.; Fulerson Z.; Berdyshev E.; Natarajan V.; Fang X.; van Meeteren L. A.; Moolenaar W. H.; Mills G. B.; Morris A. J.; Smyth S. S. Autotaxin/Lysopholipase D and Lysophosphatidic Acid Regulate Murine Hemostasis and Thrombosis. J. Biol. Chem. 2009, 284117385–7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Gaudette D. C.; Boynton J. D.; Frankel A.; Fang X. J.; Sharma A.; Hurteau J.; Casey G.; Goodbody A.; Mellors A.; Holub B. J.; Mills G. B. Characterization of an ovarian cancer activating factor in ascites from ovarian cancer patients. Clin. Cancer Res.: Off. J. Am. Assoc. Cancer Res. 1995, 1101223–1232. [PubMed] [Google Scholar]
- Tokumura A.; Kume T.; Fukuzawa K.; Tahara M.; Tasaka K.; Aoki J.; Arai H.; Yasuda K.; Kanzaki H. Peritoneal fluids from patients with certain gynecologic tumor contain elevated levels of bioactive lysophospholipase D activity. Life Sci. 2007, 80181641–1649. [DOI] [PubMed] [Google Scholar]
- Jazaeri A. A.; Avvtrey C. S.; Chandramouli G. V. R.; Chuang Y. E.; Khan J.; Sotiriou C.; Aprelikova O.; Yee C. J.; Zorn K. K.; Birrer M. J.; Barrett J. C.; Boyd J. Gene expression profiles associated with response to chemotherapy in epithelial ovarian cancers. Clin. Cancer Res. 2005, 11176300–6310. [DOI] [PubMed] [Google Scholar]
- Vidot S.; Witham J.; Agarwal R.; Greenhough S.; Bamrah H. S.; Tigyi G. J.; Kaye S. B.; Richardson A. Autotaxin delays apoptosis induced by carboplatin in ovarian cancer cells. Cell. Signal. 2010, 226926–35. [DOI] [PubMed] [Google Scholar]
- King-Underwood J.; Allin S. M.; Redman C. W.; Richardson A.. Autotaxin—A Target for the Treatment of Drug-Resistant Ovarian Cancer? In Ovarian Cancer—Basic Science Perspective [Online on InTech]; Farghaly S. A., Ed.; 2012. http://www.intechopen.com/books/ovarian-cancer-basic-science-perspective/autotaxin-a-target-for-the-treatment-of-drug-resistant-ovarian-cancer- (accessed 06/11/13). [Google Scholar]
- van Meeteren L. A.; Ruurs P.; Christodoulou E.; Goding J. W.; Takakusa H.; Kikuchi K.; Perrakis A.; Nagano T.; Moolenaar W. H. Inhibition of autotaxin by lysophosphatidic acid and sphingosine 1-phosphate. J. Biol. Chem. 2005, 2802221155–21161. [DOI] [PubMed] [Google Scholar]
- Ferry G.; Moulharat N.; Pradere J. P.; Desos P.; Try A.; Genton A.; Giganti A.; Beucher-Gaudin M.; Lonchampt M.; Bertrand M.; Saulnier-Blache J. S.; Tucker G. C.; Cordi A.; Boutin J. A. S32826, a nanomolar inhibitor of autotaxin: discovery, synthesis and applications as a pharmacological tool. J. Pharmacol. Exp. Ther. 2008, 3273809–19. [DOI] [PubMed] [Google Scholar]
- Gierse J.; Thorarensen A.; Beltey K.; Bradshaw-Pierce E.; Cortes-Burgos L.; Hall T.; Johnston A.; Murphy M.; Nemirovskiy O.; Ogawa S.; Pegg L.; Pelc M.; Prinsen M.; Schnute M.; Wendling J.; Wene S.; Weinberg R.; Wittwer A.; Zweifel B.; Masferrer J. A novel autotaxin inhibitor reduces lysophosphatidic acid levels in plasma and the site of inflammation. J. Pharmacol. Exp. Ther. 2010, 3341310–7. [DOI] [PubMed] [Google Scholar]
- Albers H. M.; Dong A.; van Meeteren L. A.; Egan D. A.; Sunkara M.; van Tilburg E. W.; Schuurman K.; van Tellingen O.; Morris A. J.; Smyth S. S.; Moolenaar W. H.; Ovaa H. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc. Natl. Acad. Sci. U. S. A. 2010, 107167257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esfand R.; Tomalia D. A. Poly(amidoamine) (PAMAM) dendrimers: from biomimicry to drug delivery and biomedical applications. Drug Discovery Today 2001, 68427–436. [DOI] [PubMed] [Google Scholar]
- Malik N.; Wiwattanapatapee R.; Klopsch R.; Lorenz K.; Frey H.; Weenerc J. W.; Meijerc E. W.; Paulusd W.; Duncan R. Dendrimers: Relationship between structure and biocompatibility in vitro, and preliminary studies on the biodistribution of 125I-labelled polyamidoamine dendrimers in vivo. J. Controlled Release 2000, 63, 133–1448. [DOI] [PubMed] [Google Scholar]
- Seymour L. W.; Miyamoto Y.; Maeda H.; Brereton M.; Strohalm J.; Ulbrich K.; Duncan R. Influence of molecular-weight on passive tumor accumulation of a soluble macromolecular drug carrier. Eur. J. Cancer 1995, 31A5766–770. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Seymour L. W.; Miyamoto Y. Conjugates of anticancer agents and polymers: advantages of macromolecular therapeutics in vivo. Bioconjugate Chem. 1992, 35351–362. [DOI] [PubMed] [Google Scholar]
- Najlah M.; Freeman S.; Attwood D.; D’Emanuele A. Synthesis, characterization and stability of dendrimer prodrugs. Int. J. Pharm. 2006, 3081–2175–182. [DOI] [PubMed] [Google Scholar]
- Malik N.; Evagorou E. G.; Duncan R. Dendrimer-platinate: a novel approach to cancer chemotherapy. Anti-Cancer Drugs 1999, 108767–776. [PubMed] [Google Scholar]
- Furui T.; LaPushin R.; Mao M. Overexpression of Edg-2/vzg-1 Induces Apoptosis and Acid-independent Manner Anoikisin Ovarian Cancer Cells in a Lysophosphatidic. Clin. Cancer Res. 1999, 5, 4308–4318. [PubMed] [Google Scholar]
- Roberts J. C.; Bhalgat M. K.; Zera R. T. Preliminary biological evaluation of polyamidoamine (PAMAM) StarburstTM dendrimers. J. Biomed. Mater. Res. 1996, 30153–65. [DOI] [PubMed] [Google Scholar]
- Ferguson C. G.; Bigman C. S.; Richardson R. D.; van Meeteren L. A.; Moolenaar W. H.; Prestwich G. D. Fluorogenic phospholipid substrate to detect lysophospholipase D/autotaxin activity. Org. Lett. 2006, 8102023–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang G.; Madan D.; Prestwich G. D. Aromatic phosphonates inhibit the lysophospholipase D activity of autotaxin. Bioorg. Med. Chem. Lett. 2011, 21175098–5101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaetano C. G.; Samadi N.; Tomsig J. L.; Macdonald T. L.; Lynch K. R.; Brindley D. N. Inhibition of Autotaxin Production or Activity Blocks Lysophosphatidylcholine-Induced Migration of Human Breast Cancer and Melanoma Cells. Mol. Carcinog. 2009, 489801–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. L.; Xu X. Y.; Gajewiak J.; Tsukahara R.; Fujiwara Y.; Liu J. X.; Fells J. I.; Perygin D.; Parrill A. L.; Tigyi G.; Prestwich G. D. Dual Activity Lysophosphatidic Acid Receptor Pan-Antagonist/Autotaxin Inhibitor Reduces Breast Cancer Cell Migration In vitro and Causes Tumor Regression In vivo. Cancer Res. 2009, 69135441–5449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders L. P.; Ouellette A.; Bandle R.; Chang W. C.; Zhou H.; Misra R. N.; De La Cruz E. M.; Braddock D. T. Identification of small-molecule inhibitors of autotaxin that inhibit melanoma cell migration and invasion. Mol. Cancer Ther. 2008, 7103352–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frankel A.; Mills G. B. Peptide and Lipid growth factors decrease cis-diamminedichloroplatinum-induced cell death in Human Ovarian Cancer Cells. Clinical Cancer Res. 1996, 2, 1307–1313. [PubMed] [Google Scholar]
- Samadi N.; Gaetano C.; Goping I. S.; Brindley D. N. Autotaxin protects MCF-7 breast cancer and MDA-MB-435 melanoma cells against Taxol-induced apoptosis. Oncogene 2009, 2871028–1039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.