

Abstract
The identification of potent, highly selective orally bioavailable ghrelin receptor inverse agonists from a spiro-azetidino-piperidine series is described. Examples from this series have promising in vivo pharmacokinetics and increase glucose-stimulated insulin secretion in human whole and dispersed islets. A physicochemistry-based strategy to increase lipophilic efficiency for ghrelin receptor potency and retain low clearance and satisfactory permeability while reducing off-target pharmacology led to the discovery of 16h. Compound 16h has a superior balance of ghrelin receptor pharmacology and off-target selectivity. On the basis of its promising pharmacological and safety profile, 16h was advanced to human clinical trials.
Keywords: Ghrelin, ghrelin receptor inverse agonist, ghrelin receptor antagonist, diabetes, PF-5190457
Type 2 diabetes mellitus (T2DM) is a rapidly expanding public health problem affecting over 285 million people worldwide.1 The disease is characterized by elevated fasting plasma glucose, insulin resistance, abnormally elevated hepatic glucose production, and reduced glucose-stimulated insulin secretion.2 Uncontrolled glucose levels can lead to severe downstream complications, such as elevated risks of cardiovascular disease, retinopathy, and nephropathy. While several classes of antidiabetic therapy are available for clinical use, there still remains a significant need for new therapies with improved efficacy, safety, and tolerability to help diabetes patients achieve their treatment goals and avoid long-term complications.3
Ghrelin, an agonist of the ghrelin receptor (earlier names for the receptor that have been used are GHS-R1a, GRLN, etc.), stimulates release of growth hormone (GH) from the pituitary gland and increases food intake.4−8 Through infusion studies in man, ghrelin has been shown to suppress both glucose-dependent insulin secretion and insulin sensitivity.9−13 The ghrelin receptor is expressed in pancreatic islets, and ghrelin is released into the pancreatic microcirculation. Islet-derived ghrelin has been reported to play an important role in the regulation of insulin release in rodents.14 Although ghrelin has been shown to cross the blood–brain barrier,15 several effects of ghrelin appear to be peripherally mediated as shown by the clinical results from the brain-impaired ghrelin receptor agonist capromorelin (CP-424391).16,17 Treatment with capromorelin led to increases in appetite, fasting glucose, HbA1c levels, and insulin resistance. Thus, ghrelin receptor antagonists/inverse agonists are anticipated to improve glucose homeostasis and insulin sensitivity. Described herein is the discovery of PF-5190457, a potent and selective ghrelin receptor inverse agonist that increases glucose-stimulated insulin secretion (GSIS) in human whole and dispersed islets.
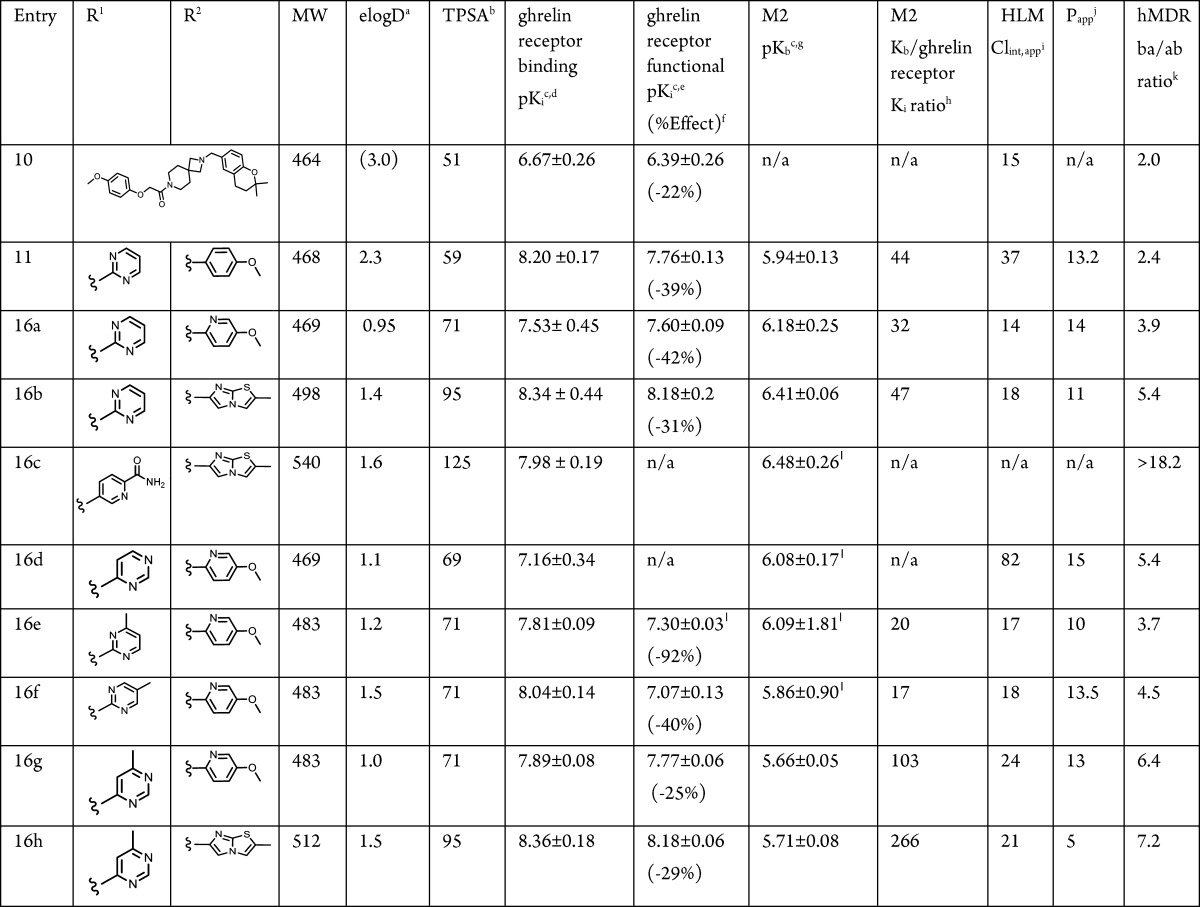
A number of small molecule ghrelin receptor antagonists have been described in the literature (see Figure 1).18−26 The vast majority of these efforts have focused on centrally acting agents targeted toward the treatment of obesity. We previously reported on a spiro-azetidino-piperidine series that was identified from high-throughput screening (HTS) of our corporate file.27 In a later report, we described efforts to optimize this series to achieve high central receptor occupancy targeting an obesity indication. Thus, the HTS-hit (10) in the aforesaid series was transformed to a centrally acting ghrelin receptor inverse agonist lead (11, Table 1).28
Figure 1.

Examples of ghrelin receptor antagonists.
Table 1. Data for Spiro-azetidino-piperidine Analogues 10, 11, and 16a–h.
elogD = measured logD at pH 7.4 [calculated logD (ACD Laboratories program v12) in parentheses].
TPSA = topological polar surface area.
Geometric mean ± standard error of ≥3 measurements (unless noted otherwise).
Human ghrelin receptor SPA receptor binding assay as published.27
Human ghrelin receptor agonist/antagonist/inverse agonist GTP-γ-S functional assay as published.27
Minimum percent effect in parentheses; a negative value indicates an inverse agonist.
Muscarinic M2 β-Arrestin PathHunter assay from Discoverx. Kb for antagonists determined in the presence of EC80 concentration of agonist, oxotremorine.39
Ratio of M2 Kb over human ghrelin receptor functional Ki values.
Human liver microsome (HLM) apparent intrinsic clearance, mL/min/kg, uncorrected for fraction unbound.
Passive permeability (Papp in 10–6 cm/s) measured using low-efflux MDCKII cells as described.40
MDR BA/AB efflux ratio using MDCK cell line transfected with human MDR1.41
Less than three independent replicates.
Emerging ghrelin biology suggests that blocking ghrelin receptor signaling has the potential to ameliorate diabetes through direct action on the pancreas.29 To test this hypothesis, lead 11 was assessed in a human dispersed islet cell assay. As anticipated, 11 induced insulin secretion in a glucose-dependent manner (Supporting Information, Figure S1). These data, in conjunction with other published work linking ghrelin receptor inhibition to improved insulin sensitivity, caused us to redirect our research efforts toward diabetes (T2DM) as the primary indication, and we targeted the peripheral compartment as the key site of action.
The spiro-azetidino-piperidine series was an attractive starting point for optimization for this indication. It provided a rigid core scaffold with high fractional sp3 content (for leads in series, Fsp3 = 0.41–0.53)30 and two point vectors for introducing orthogonal diversity using simple coupling methodologies. A significant factor in the selection of this series was consistent inverse agonism of ghrelin receptor. Functional switching (between antagonism and partial agonism) has been observed by us and others and has demonstrated impact on the in vivo performance of compounds.26,31 We believed that by focusing on a series where receptor functionality was not an issue, we could accelerate our drug discovery program by eliminating one dimension from our lead optimization efforts and, at the same time, streamline our in vitro pharmacology testing funnel. Furthermore, because of the high constitutive activity of the ghrelin receptor, inverse agonism of ghrelin receptor may provide a greater opportunity for in vivo efficacy through reductions in basal receptor firing.32
Although lead 11 showed encouraging insulin secretion effects in islet studies, more extensive profiling highlighted issues that prevented its further development. In preclinical CV safety testing, 11 showed inadequate safety margins to allow progression. The discovery of the indane amine moiety in 11 (built as a less lipophilic surrogate of an ortho-chloro benzylic amine) had allowed improvements in selectivity within the spiro-azetidino-piperidine series.28 However, while selectivity over muscarinic receptors (in particular M2) was improved with the desired chiral indane, in wide-ligand promiscuity profiling (CEREP; tested at a dose of 10 μM, Figure 2) 11 was active at a number of targets including adrenergic α2c and α2a, dopamine D2s, D3, and D4, and histamine H1. Compound 11 also showed human ether-à-go-go-related gene (hERG) activity (IC50 = 2.9 μM). Thus, the overall off-target pharmacology profile, coupled with the physicochemical properties of 11 (Table 1), was of concern. An increased risk of adverse safety findings for compounds with higher clogP and lower TPSA has been described.33 In addition, several groups report that off-target promiscuity leads to elevated safety risk.34,35 Therefore, our primary objective was to identify a compound in this series with reduced off-target pharmacology in order to achieve broad safety margins to allow advancement to the clinic. Our strategy to reduce off-target pharmacology was to increase the lipophilic efficiency (LipE)36,37 and retain or improve ghrelin receptor potency while increasing the overall polarity of the inverse agonists. This physicochemistry-based strategy should provide the added benefit of decreasing CNS penetration, leading to a reduction in CNS-based side effects.
Figure 2.
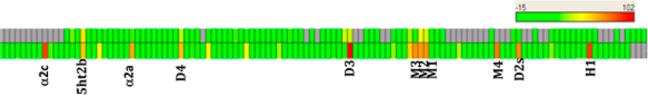
Comparative CEREP profile of 11 (bottom) and 16h (top).
Scaffold 13 provided a modular framework for exploring structure–activity relationship (SAR), and earlier work established the preference for the R-enantiomer at C(5) as providing robust ghrelin receptor activity. A one-pot sequence reacting chiral amine 12a with chloroaldehyde 12b in the presence of NaCNBH3 was developed and used to deliver bulk quantities of 13 (with retention of stereochemistry at the C(5) position), a key chiral intermediate for flexible incorporation of a variety of R1 and R2 groups onto scaffold 16.28 Suzuki coupling to incorporate R1 followed by in situ deprotection (Scheme 1) under acidic conditions unmasked the piperidine nitrogen (15b) for incorporation of the R2 amide. Amide coupling with a variety of carboxylic acids was accomplished with HBTU or CDI as the preferred coupling agent.
Scheme 1. Synthesis of Analogues 16a–h.

Reagents and conditions: (a) (i) HOAc, MeOH, 50 °C, 2 h; (ii) NaCNBH3, 70 °C; 99%; (b) bis(pinacolato)diboron, Pd(dppf)Cl2 (1.6 mol %), KOAc, dioxane, 110 °C, 1 h, >99% conversion; (c) R1-Cl or R1-Br, Pd(dppf)Cl2 (2.5 mol %), K2CO3 (aq), dioxane, 110 °C, 3 h, 90% conversion; (d) HCl; (e) TFA; (f) R2CH2CO2H (14), HBTU or CDI, DIEA or TEA, 50–99% (over 2 steps).
We identified two R2 moieties, pyridyl-3-methoxy (16a) and the bicyclic imidazothiazole (16b),27 that maintained ghrelin receptor potency at lower logD with retention of other properties, such as low clearance and satisfactory permeability. Unfortunately, when combined with 2-pyrimidinyl at R1, neither compound had a positive effect on selectivity over M2 (Table 1). As anticipated, these changes, which were designed to lower lipophilicity, had the effect of increasing efflux ratios in an in vitro MDR assay (b-a/a-b efflux ratio of >2.5 being generally indicative of reduced CNS exposure; Table 1).38
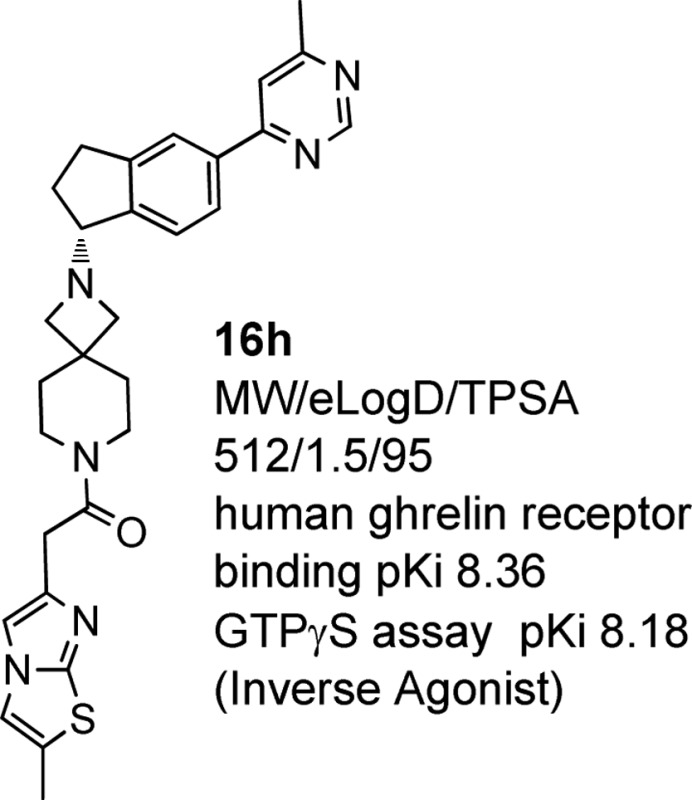
In an attempt to improve M2 selectivity, we switched attention to modification of R1. Attempts to further increase polarity, as exemplified by carboxamide 16c, had no effect on selectivity (Table 1). Thus, to complement the property-based design strategy, we investigated the effect of more subtle structural changes on M2 potency. Small changes around the pyrimidinyl group (as exemplified by 16d, 16e, and 16f) did not provide significant improvement with the exception of the 6-methyl-4-pyrimidinyl group (as in 16g), which showed improved ghrelin receptor potency, as compared to 16a, and a marked enhancement in M2 selectivity. Gratifyingly, combination of the 6-methyl-4-pyrimidinyl group at R1 with the more polar imidazothiazole moiety at R2, provided 16h, which demonstrated significant improvements in both functional potency and selectivity against M2 relative to 11 (Table 1). Indeed, compound 16h has one of the highest lipophilic efficiencies (LipEelogD = 6.9) in this series, which is a desirable predictor for lowered promiscuity-related developmental attrition risk.34,36 Compound 16h maintained a moderate clearance in human liver microsomes (HLM). In addition, the reductions in logP achieved in moving to 16h had the anticipated and desired effects on MDR efflux ratio, suggesting a high probability of reduced CNS exposure. (Incidentally, 16h was 11-fold impaired in rat brain after chronic dosing for 14 days.)
Given its promising in vitro pharmacology and ADMET parameters, compound 16h was profiled in more detail. Compound 16h showed reduced off-target activity, as assessed by the CEREP panel (screened at 10 μM, Figure 2), with serotonin 5-HT2B (IC50 = 3700 nM) being the only target inhibited to >50%. In follow-up screening, 16h did not demonstrate any agonist (or antagonist) functional effects at this receptor.
Compound 16h was advanced to a number of ex vivo and in vivo studies. The full pharmacology profile for 16h (GTP-γ-S functional profile in Supporting Information, Figure S2), including a biomarker for target engagement, will be described separately.42 A human islet assay was used in order to gain confidence in the ability of 16h to increase insulin secretion in humans (Figure 3). Human whole islets in static culture were incubated at both low (2.8 mM) and high (11.2 mM) glucose concentrations and demonstrated that the islets were glucose responsive.43 The sulfonylurea, glibenclamide (glyburide), was tested as a positive control. Compound 16h (1 μM) significantly increased insulin secretion above the 11.2 mM glucose control.
Figure 3.
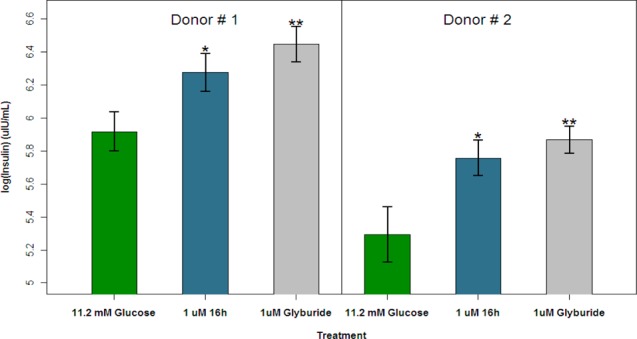
Glucose-stimulated insulin secretion in human whole islet static culture following incubation with 16h at 1 μM. **p < 0.001; *p < 0.05. Measurement data are expressed as the arithmetic mean ± standard error.
To predict human pharmacokinetics and enable clinical dose setting, the pharmacokinetics of 16h were evaluated preclinically in three species (Supporting Information, Table S1). Rat pharmacokinetics revealed both high plasma clearance and volume of distribution (consistent with the high clearance value derived from rat liver microsomal in vitro assay). Dog and monkey plasma clearance and volume of distribution were moderate. Urinary and biliary elimination of 16h contributed minimally to the overall clearance in the species investigated. Because of its high in vivo rat clearance, 16h was tested in portal vein cannulated rats and demonstrated excellent absorption (Fa = 100%) as anticipated given its high solubility and moderate passive permeability.44 The human plasma clearance of 16h was predicted by scaling human liver microsomal data (human hepatocyte data was consistent with microsomal data). When corrected for microsomal (fu,mic = 0.8) and plasma binding (fu = 0.15), human plasma clearance was predicted to be low (3.3 mL/min/kg). Using preclinical data and physiologically based pharmacokinetic (PBPK) models,4516h was predicted to have moderate to high human absorption (86%) along with moderate measures of bioavailability (67%), Vdss (1.79 L/kg), and half-life (6.3 h). Human free drug concentrations (Ceff,u) needed to achieve efficacy were based on the equilibrium binding constant (Kd) of 16h, which was determined using the Motulsky kinetics procedure (3.04 ± 0.91 nM).42,46 We sought to maintain minimal free drug concentrations of 10 × Kd for the full dosing period (∼30 nM). Combining this Ceff,u value with predicted human PK parameters provided a projected human oral dose of 35 mg bid. The corresponding predicted Cmax,u and AUCu values are 52.4 nM and 1010 nM·h, respectively.
Compound 16h was evaluated in a variety of assays to ensure robust safety (see Supporting Information). While 16h showed moderate potential for inhibition of hERG current, the IC50 (6.9 μM) represents a significant multiple (>100×) over the predicted human Cmax required to maintain Ceff,u for the full dosing interval (52.4 nM free). Thus, the likelihood of achieving drug exposures in the clinic that would effect QTc prolongation was considered low.
With the primary design objectives of delivering a peripherally acting ghrelin receptor inverse agonist with improved selectivity satisfied, the nonclinical safety profile of 16h was assessed in both rats and dogs in 1-month toxicology studies and in safety pharmacology studies. Gratifyingly, the safety profile of 16h demonstrated sufficient safety margins above the projected Ceff in humans and supported continued development of the compound.
In summary, we set out to transform a centrally acting ghrelin receptor inverse agonist indane lead (11) to a peripherally restricted one, by pursuing a physicochemistry-based strategy to increase LipE for ghrelin receptor potency and reduce off-target pharmacology while retaining low clearance and satisfactory permeability. The addition of the 6-methyl-4-pyrimidinyl indane in the R configuration and the imidazothiazole group to the spiro-azetidino-piperidine core led to the discovery of 16h. Compound 16h is a potent inverse agonist with excellent selectivity and demonstrated robust increases in glucose-stimulated insulin secretion in human islets. Human pharmacokinetic predictions project a dose of 35 mg bid to achieve 10 × Kd at trough concentrations in the clinic. On the basis of the pharmacological profile and safety results, 16h (PF-5190457) was advanced to human clinical trials, and results from these trials will be reported in due course.
Glossary
ABBREVIATIONS
- TPSA
topological polar surface area
- dppf
1,1′-bis(diphenylphosphino)ferrocene
- HBTU
O-(benzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate
- CDI
1,1′-carbonyldiimidazole
- GHS-R1a
growth hormone secretagogue receptor
- GSIS
glucose stimulated insulin secretion
- QTc
heart rate-corrected QT interval
- PK
pharmacokinetic
Supporting Information Available
Experimental procedures of analogue preparation and description of biological assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- IDF Diabetes Atlas. http://www.diabetesatlas.org.
- Saltiel A. R. New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 2001, 104, 517–29. [DOI] [PubMed] [Google Scholar]
- Xu J. Current and emerging therapies for type 2 diabetes. IDrugs 2004, 7, 249–56. [PubMed] [Google Scholar]
- Ghrelin receptor. http://www.iuphar-db.org/DATABASE/ObjectDisplayForward?familyId=28&objectId=246&familyType=RECEPTOR.
- Tong J.; Prigeon R. L.; Davis H. W.; Bidlingmaier M.; Kahn S. E.; Cummings D. E.; Tschop M. H.; D’Alessio D. Ghrelin suppresses glucose-stimulated insulin secretion and deteriorates glucose tolerance in healthy humans. Diabetes 2010, 59, 2145–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucidi P.; Murdolo G.; Di Loreto C.; Parlanti N.; De Cicco A.; Fatone C.; Taglioni C.; Fanelli C.; Broglio F.; Ghigo E.; Bolli G. B.; Santeusanio F.; De Feo P. Metabolic and endocrine effects of physiological increments in plasma ghrelin concentrations. Nutr. Metab. Cardiovasc. Dis. 2005, 15, 410–7. [DOI] [PubMed] [Google Scholar]
- Reimer M. K.; Pacini G.; Ahren B. Dose-dependent inhibition by ghrelin of insulin secretion in the mouse. Endocrinology 2003, 144, 916–21. [DOI] [PubMed] [Google Scholar]
- Dezaki K.; Kakei M.; Yada T. Ghrelin uses Galphai2 and activates voltage-dependent K+ channels to attenuate glucose-induced Ca2+ signaling and insulin release in islet beta-cells: novel signal transduction of ghrelin. Diabetes 2007, 56, 2319–27. [DOI] [PubMed] [Google Scholar]
- Gauna C.; Meyler F. M.; Janssen J. A.; Delhanty P. J.; Abribat T.; van Koetsveld P.; Hofland L. J.; Broglio F.; Ghigo E.; van der Lely A. J. Administration of acylated ghrelin reduces insulin sensitivity, whereas the combination of acylated plus unacylated ghrelin strongly improves insulin sensitivity. J. Clin. Endocrinol. Metab. 2004, 89, 5035–42. [DOI] [PubMed] [Google Scholar]
- Damjanovic S. S.; Lalic N. M.; Pesko P. M.; Petakov M. S.; Jotic A.; Miljic D.; Lalic K. S.; Lukic L.; Djurovic M.; Djukic V. B. Acute effects of ghrelin on insulin secretion and glucose disposal rate in gastrectomized patients. J. Clin. Endocrinol. Metab. 2006, 91, 2574–81. [DOI] [PubMed] [Google Scholar]
- Vestergaard E. T.; Hansen T. K.; Gormsen L. C.; Jakobsen P.; Moller N.; Christiansen J. S.; Jorgensen J. O. Constant intravenous ghrelin infusion in healthy young men: clinical pharmacokinetics and metabolic effects. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1829–36. [DOI] [PubMed] [Google Scholar]
- Vestergaard E. T.; Djurhuus C. B.; Gjedsted J.; Nielsen S.; Moller N.; Holst J. J.; Jorgensen J. O.; Schmitz O. Acute effects of ghrelin administration on glucose and lipid metabolism. J. Clin. Endocrinol. Metab. 2008, 93, 438–44. [DOI] [PubMed] [Google Scholar]
- Vestergaard E. T.; Gormsen L. C.; Jessen N.; Lund S.; Hansen T. K.; Moller N.; Jorgensen J. O. Ghrelin infusion in humans induces acute insulin resistance and lipolysis independent of growth hormone signaling. Diabetes 2008, 57, 3205–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dezaki K.; Sone H.; Yada T. Ghrelin is a physiological regulator of insulin release in pancreatic islets and glucose homeostasis. Pharmacol. Ther. 2008, 118, 239–49. [DOI] [PubMed] [Google Scholar]
- Banks W. A.; Tschop M.; Robinson S. M.; Heiman M. L. Extent and direction of ghrelin transport across the blood–brain barrier is determined by its unique primary structure. J. Pharmacol. Exp. Ther. 2002, 302, 822–7. [DOI] [PubMed] [Google Scholar]
- White H. K.; Petrie C. D.; Landschulz W.; MacLean D.; Taylor A.; Lyles K.; Wei J. Y.; Hoffman A. R.; Salvatori R.; Ettinger M. P.; Morey M. C.; Blackman M. R.; Merriam G. R.; Effects of an oral growth hormone secretagogue in older adults. J. Clin. Endocrinol. Metab. 2009, 94, 1198–206. [DOI] [PubMed] [Google Scholar]
- Khojasteh-Bakht S. C.; O’Donnell J. P.; Fouda H. G.; Potchoiba M. J. Metabolism, pharmacokinetics, tissue distribution, and excretion of [14C]CP-424391 in rats. Drug Metab. Dispos. 2005, 33, 190–9. [DOI] [PubMed] [Google Scholar]
- Costantino L.; Barlocco D. Ghrelin receptor modulators and their therapeutic potential. Future Med. Chem. 2009, 1, 157–77. [DOI] [PubMed] [Google Scholar]
- Longo K. A.; Govek E. K.; Nolan A.; McDonagh T.; Charoenthongtrakul S.; Giuliana D. J.; Morgan K.; Hixon J.; Zhou C.; Kelder B.; Kopchick J. J.; Saunders J. O.; Navia M. A.; Curtis R.; DiStefano P. S.; Geddes B. J. Pharmacologic inhibition of ghrelin receptor signaling is insulin sparing and promotes insulin sensitivity. J. Pharmacol. Exp. Ther. 2011, 339, 115–24. [DOI] [PubMed] [Google Scholar]
- Yu M.; Lizarzaburu M.; Beckmann H.; Connors R.; Dai K.; Haller K.; Li C.; Liang L.; Lindstrom M.; Ma J.; Motani A.; Wanska M.; Zhang A.; Li L.; Medina J. C. Identification of piperazine-bisamide GHSR antagonists for the treatment of obesity. Bioorg. Med. Chem. Lett. 2010, 20, 1758–62. [DOI] [PubMed] [Google Scholar]
- Pasternak A.; Goble S. D.; deJesus R. K.; Hreniuk D. L.; Chung C. C.; Tota M. R.; Mazur P.; Feighner S. D.; Howard A. D.; Mills S. G.; Yang L. Discovery and optimization of novel 4-[(aminocarbonyl)amino]-N-[4-(2-aminoethyl)phenyl]benzenesulfonamide ghrelin receptor antagonists. Bioorg. Med. Chem. Lett. 2009, 19, 6237–40. [DOI] [PubMed] [Google Scholar]
- Perdona E.; Faggioni F.; Buson A.; Sabbatini F. M.; Corti C.; Corsi M. Pharmacological characterization of the ghrelin receptor antagonist, GSK1614343 in rat RC-4B/C cells natively expressing GHS type 1a receptors. Eur. J. Pharmacol. 2011, 650, 178–83. [DOI] [PubMed] [Google Scholar]
- Puleo L.; Marini P.; Avallone R.; Zanchet M.; Bandiera S.; Baroni M.; Croci T. Synthesis and pharmacological evaluation of indolinone derivatives as novel ghrelin receptor antagonists. Bioorg. Med. Chem. 2012, 20, 5623–36. [DOI] [PubMed] [Google Scholar]
- Mihalic J. T.; Kim Y. J.; Lizarzaburu M.; Chen X.; Deignan J.; Wanska M.; Yu M.; Fu J.; Chen X.; Zhang A.; Connors R.; Liang L.; Lindstrom M.; Ma J.; Tang L.; Dai K.; Li L. Discovery of a new class of ghrelin receptor antagonists. Bioorg. Med. Chem. Lett. 2012, 22, 2046–51. [DOI] [PubMed] [Google Scholar]
- McCoull W.; Barton P.; Broo A.; Brown A. J. H.; Clarke D. S.; Coope G.; Davies R. D. M.; Dossetter A. G.; Kelly E. E.; Knerr L.; MacFaul P.; Holmes J. L.; Martin N.; Moore J. E.; Morgan D.; Newton C.; Osterlund K.; Robb G. R.; Rosevere E.; Selmi N.; Stokes S.; Svensson T. S.; Ullah V. B. K.; Williams E. J. Identification of pyrazolo-pyrimidinones as GHS-R1a antagonists and inverse agonists for the treatment of obesity. MedChemComm 2013, 4, 456–462. [Google Scholar]
- Sabbatini F. M.; Melotto S.; Bernasconi G.; Bromidge S. M.; D’Adamo L.; Rinaldi M.; Savoia C.; Mundi C.; Di Francesco C.; Zonzini L.; Costantini V. J.; Perini B.; Valerio E.; Pozzan A.; Perdona E.; Visentini F.; Corsi M.; Di Fabio R. Azabicyclo[3.1.0]hexane-1-carbohydrazides as potent and selective GHSR1a ligands presenting a specific in vivo behavior. ChemMedChem 2011, 6, 1981–5. [DOI] [PubMed] [Google Scholar]
- Kung D. W.; Coffey S. B.; Jones R. M.; Cabral S.; Jiao W.; Fichtner M.; Carpino P. A.; Rose C. R.; Hank R. F.; Lopaze M. G.; Swartz R.; Chen H. T.; Hendsch Z.; Posner B.; Wielis C. F.; Manning B.; Dubins J.; Stock I. A.; Varma S.; Campbell M.; DeBartola D.; Kosa-Maines R.; Steyn S. J.; McClure K. F. Identification of spirocyclic piperidine-azetidine inverse agonists of the ghrelin receptor. Bioorg. Med. Chem. Lett. 2012, 22, 4281–7. [DOI] [PubMed] [Google Scholar]
- McClure K. F.; Jackson M.; Cameron K. O.; Kung D. W.; Perry D. A.; Orr S. T.; Zhang Y.; Kohrt J.; Tu M.; Gao H.; Fernando D.; Jones R.; Erasga N.; Wang G.; Polivkova J.; Jiao W.; Swartz R.; Ueno H.; Bhattacharya S. K.; Stock I. A.; Varma S.; Bagdasarian V.; Perez S.; Kelly-Sullivan D.; Wang R.; Kong J.; Cornelius P.; Michael L.; Lee E.; Janssen A.; Steyn S. J.; Lapham K.; Goosen T. Identification of potent, selective, CNS-targeted inverse agonists of the ghrelin receptor. Bioorg. Med. Chem. Lett. 2013, 23, 5410–4. [DOI] [PubMed] [Google Scholar]
- Meyer C. Final answer: ghrelin can suppress insulin secretion in humans, but is it clinically relevant?. Diabetes 2010, 59, 2726–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovering F.; Bikker J.; Humblet C. Escape from flatland: increasing saturation as an approach to improving clinical success. J. Med. Chem. 2009, 52, 6752–6. [DOI] [PubMed] [Google Scholar]
- Costantini V. J.; Vicentini E.; Sabbatini F. M.; Valerio E.; Lepore S.; Tessari M.; Sartori M.; Michielin F.; Melotto S.; Bifone A.; Pich E. M.; Corsi M. GSK1614343, a novel ghrelin receptor antagonist, produces an unexpected increase of food intake and body weight in rodents and dogs. Neuroendocrinology 2011, 94, 158–68. [DOI] [PubMed] [Google Scholar]
- Els S.; Beck-Sickinger A. G.; Chollet C. Ghrelin receptor: high constitutive activity and methods for developing inverse agonists. Methods Enzymol. 2010, 485, 103–21. [DOI] [PubMed] [Google Scholar]
- Hughes J. D.; Blagg J.; Price D. A.; Bailey S.; Decrescenzo G. A.; Devraj R. V.; Ellsworth E.; Fobian Y. M.; Gibbs M. E.; Gilles R. W.; Greene N.; Huang E.; Krieger-Burke T.; Loesel J.; Wager T.; Whiteley L.; Zhang Y. Physiochemical drug properties associated with in vivo toxicological outcomes. Bioorg. Med. Chem. Lett. 2008, 18, 4872–5. [DOI] [PubMed] [Google Scholar]
- Leeson P. D.; Springthorpe B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discovery 2007, 6, 881–90. [DOI] [PubMed] [Google Scholar]
- Gleeson M. P.; Hersey A.; Montanari D.; Overington J. Probing the links between in vitro potency, ADMET and physicochemical parameters. Nat. Rev. Drug Discovery 2011, 10, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. P.; Price D. A.. Role of Physicochemical Properties and Ligand Lipophilicity Efficiency in Addressing Drug Safety Risks. In Annual Reports in Medicinal Chemistry; John E. M., Ed.; Academic Press: New York, 2010; Vol. 45, Chapter 23, pp 380–391LipE calculated using elogD as a measure of lipophilicity.. [Google Scholar]
- Shultz M. D. The thermodynamic basis for the use of lipophilic efficiency (LipE) in enthalpic optimizations. Bioorg. Med. Chem. Lett. 2013, 23, 5992–6000. [DOI] [PubMed] [Google Scholar]
- Hitchcock S. A. Structural modifications that alter the P-glycoprotein efflux properties of compounds. J. Med. Chem. 2012, 55, 4877–95. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for experimental conditions of the muscarinic M2 b-arrestin functional assay.
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y.; Federico J. J. III; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-Kattan A. F.; Liston T. E.; Troutman M. D. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100, 4974–85. [DOI] [PubMed] [Google Scholar]
- Feng B.; Mills J. B.; Davidson R. E.; Mireles R. J.; Janiszewski J. S.; Troutman M. D.; de Morais S. M. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab. Dispos. 2008, 36, 268–275. [DOI] [PubMed] [Google Scholar]
- Kong J. S.; Stock I.; Loria P. M.; Straub S. V.; Vage C.; Cameron K. O.; Bhattacharya S. K.; Zhang Y.; Jackson V. M.. Pharmacological and translational characterization of PF-5190457, a novel selective ghrelin receptor competitive antagonist with inverse agonism that increases vagal afferent firing and glucose-dependent insulin secretion ex vivo. Br. J. Pharmacol. Manuscript in preparation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- See Supporting Information for experimental conditions of the human islet assay.
- The measured solubility of crystalline 16h was determined to be 0.18 mg/mL in water (at pH 7.9); 4.57 mg/mL in phosphate buffered saline (PBS, pH 6.7); and >25 mg/mL in SGF (simulated gastric fluid, pH 1.2).
- Jones H. M.; Mayawala K.; Poulin P. Dose selection based on physiologically based pharmacokinetic (PBPK) approaches. AAPS J. 2013, 15, 377–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motulsky H. J.; Mahan L. C. The kinetics of competitive radioligand binding predicted by the law of mass action. Mol. Pharmacol. 1984, 25, 1–9. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.