

Abstract
The molecular crosstalk between the interkeukin-7 receptor (IL-7R) and pre-BCR in B lymphopoiesis has been enigmatic. We demonstrate that in pre-B cells, the IL-7R, but not the pre-BCR, was coupled to the phosphatidylinositol-3-OH kinase (PI(3)K)–Akt module, signaling by which prevents Rag expression. Attenuation of IL-7 signaling resulted in up-regulation of Foxo1 and Pax5, which co-activated many pre-B cell genes, including Rag1,2 and Blnk. Induction of the latter gene enabled pre-BCR signaling via the Syk-BLNK module and promoted immunoglobulin light chain rearrangement. BLNK signaling also antagonized Akt activation, thereby augmenting Foxo1 and Pax5 accumulation. This self-reinforcing molecular circuit appears to sense limiting concentrations of IL-7 and functions to control the expansion and differentiation of pre-B cells.
INTRODUCTION
During B cell development in-frame DNA rearrangement of an immunoglobulin heavy chain allele (Igh) results in assembly of a pre-BCR that, in concert with the IL-7R, promotes limited clonal expansion of pre-B cells followed by their cell-cycle arrest and Ig light (IgL) chain recombination1,2. Attenuation of IL-7 signaling3 or a qualitative change in the signaling state of the pre-BCR1 have been proposed as alternative means for inducing the switch from pre-B cell proliferation to differentiation. According to the former model attenuation of IL-7 signaling is necessary for inducing cell-cycle arrest and the induction of Rag gene expression. This model also proposes that pre-BCR signaling upregulates the transcription factor IRF-4 that in turn promotes increased expression of the chemokine receptor CXCR4 on pre-B cells and their CXCL12 directed movement away from stromal cells expressing IL-7, thereby attenuating IL-7 signaling3. Importantly, IRF-4 also induces germline transcription of Ig light chain loci and consequently their accessibility to recombination3, 4.
An alternate model1 posits that the pre-BCR exists in two distinct signaling states, one in which it stimulates pre-B cell proliferation and the inhibition of Rag gene expression and an altered state in which it promotes cell-cycle arrest, the induction of Rag gene expression as well as Ig light chain gene rearrangement. The differentiation inducing signaling state of the pre-BCR has been proposed to be dependent on the Ras-MEK-Erk pathway5 and also the signaling adapter protein BLNK (also known as SLP-65, ref. 6). The latter protein functions as a molecular scaffold for the pre-BCR and enables docking and activation of kinases such as Btk and PLC-γ2. The upregulation of the transcription factor IRF-4 by pre-BCR signaling is mediated via BLNK as restoration of its expression in Blnk−/− cells induces expression of IRF-4 (ref. 7). BLNK is also required for the activation of p38MAPK8 and the activation of the latter has been implicated in the induction Ig light chain gene rearrangement in pre-B cells3,9. Consistent with this finding, p38MAPK has been shown to promote cell cycle arrest and induce differentiation in a variety of developmental contexts10. In addition to promoting differentiation, pre-BCR signaling via the Erk-MAPK pathway has also been suggested to synergize with IL-7 signaling in regulating pre-B cell expansion11. Therefore the decision to undergo cellular expansion versus cell cycle arrest and differentiation involves a complex interplay between the IL-7R and pre-BCR signaling systems. However, each of the above models that focus primarily on one or the other receptor system lack completeness in that it they do not fully account for the signaling functions of the alternate receptor system. Furthermore each model fails to specify the molecular circuitry that controls the regulatory interplay between the IL-7R and pre-BCR.
Recently, the transcription factors Foxo1 and Foxo3a have been shown to directly target and activate Rag gene expression in pre-B cells6,9,12. Foxo protein stability and nuclear accumulation is in turn negatively regulated by PI(3)K-Akt signaling9. Thus, pre-B cell differentiation is dependent on the attenuation of PI(3)K-Akt signaling and the induction of Foxo transcription factors1. Attenuation of IL-7 signaling results in the robust induction of Rag1/2 gene expression in pre-B cells3,9, however it remains to be determined if this is associated with reduced PI(3)K-Akt activity and the induction of Foxo factors given that these cells continue to express a pre-BCR, which has been suggested to activate the former signaling module. Furthermore, if the pre-BCR activates PI(3)K-Akt signaling then the question remains how is its signaling state altered so as to enable the activation of Foxo factors and Rag gene expression1. We set out to explore these fundamental questions and in so doing to elucidate the nature of the regulatory interplay between the IL-7R and the pre-BCR in orchestrating the pre-B cell developmental checkpoint.
RESULTS
IL-7 signaling negatively regulates FoxO’s via PI(3)K-Akt
We utilized Irf4−/−Irf8−/− (called Irf4,8 −/− hereafter) pre-B cells3, 4, a model system for analyzing large cycling pre-B cells that express both the IL-7R and the pre-BCR, to determine if IL-7 signaling in these cells negatively regulates the expression of Foxo family transcription factors via the activation of the PI(3)K-Akt module. Lowering of IL-7 concentration in the culture medium substantially induced the amounts of Foxo1 and Foxo3a proteins but not that of Foxo4 (Fig. 1a). Increased Foxo1 and Foxo3a protein abundance was not accompanied by corresponding changes in the expression of their transcripts (data not shown), suggestive of a post-transcriptional mechanism of control. Reduced IL-7 signaling resulted in lower amounts of p-Akt and diminished phosphorylation of Foxo1 and Foxo3a (Fig. 1b) that was inversely correlated with their overall protein abundance (Fig. 1a). Given that Akt has been shown to phosphorylate Foxo proteins and promote their degradation via ubiquitination13–15, these results suggest that IL-7R signaling promotes Foxo protein degradation in pre-B cells via the PI(3)K-Akt pathway. Consistent with this inhibitory mechanism, attenuation of IL-7 signaling resulted in increased Foxo1 DNA binding activity, its nuclear accumulation (Supplementary Fig. 1a,b) and binding in vivo to enhancers (Erag1 and Erag2)12, 16 in the Rag locus (Fig. 1c). As shown previously, attenuation of IL-7 signaling in Irf4,8−/−pre-B cells resulted in the robust induction of Rag gene expression (ref. 3 and Fig. 1d) and importantly this was dependent on Foxo1 and Foxo3a as their knockdown in these cells via shRNAs impaired Rag gene activation (Supplementary Fig. 2a). We note that knockdown of Foxo1 or Foxo3a also resulted in a defect in the ability of Irf4,8−/− pre-B cells to undergo cell-cycle arrest upon the attenuation of IL-7 signaling (Supplementary Fig. 2b) and this was correlated with impaired induction of the gene encoding the CDK inhibitor p27 (ref. 3, 13, 14 and data not shown). Gain-of-function experiments with Foxo1 and Foxo3a reinforced the conclusion that they are potent activators of Rag gene expression in pre-B cells (Supplementary Fig. 2c). It should be noted that increased expression of Foxo1 and Foxo3a resulted in the selective induction of Rag gene expression but not kappa germline transcription whereas the restoration of IRF-4 expression in these cells primarily induced kappa germline transcription (Supplementary Fig. 2c). Thus we propose that efficient pre-B cell differentiation is contingent on induction of Foxo transcription factors via attenuation of IL-7R and PI(3)K-Akt signaling and the upregulation of IRF-4 via the pre-BCR.
Figure 1. The IL-7R/PI3K/Akt pathway negatively regulates FoxO activity and Rag gene expression in pre-B cells.

IL-7 signaling was attenuated in IRF-4,8−/− pre-B cells by reducing the concentration of IL-7 in the culture medium from 5.0 ng/mL (IL-7 Hi) to 0.1 ng/mL (IL-7 Lo) for a period of 48 h (a through d). (a) Immunoblot analysis of Foxo1, FoxO3a and FoxO4. Expression of these proteins was normalized to HPRT. (b) Immunoblot analysis of phosphorylated forms of Foxo1 (p-Foxo1), FoxO3a (p-FoxO3a) and Akt (p-Akt) in relation to total Akt (Akt). (c) ChIP analysis of Foxo1 binding to regulatory elements (Erag1,2,3) in the Rag locus1, 3. ChIP was performed with control IgG or αFoxo1 antibodies. Binding enrichment at the indicated Erag region is displayed as a percentage of input DNA and was calculated using quantitative PCR. (d) RT-PCR analysis of Rag1,2 transcripts normalized to those for β-2 microglobulin (β-2mi). For a–d, data are representative of three independent experiments, and error bars show the standard deviation from the mean value. (e) Flow cytometric analysis of IL-7Rα (surface) as well as p-Akt and Foxo1 (intracellular) expression in large cycling and small resting pre-B cells. Bone marrow pre-B cells were isolated from wild type mice (n=3) as detailed in Methods. Flow cytometry profiles of large (red) versus small pre-B cells (green) with the indicated antibodies and an isotype control (filled gray histogram) are shown. Data is representative of analysis of pre-B cells from three individual mice. (f) Effect of pre-BCR expression on Akt activity in pro-B cells. Rag2−/− pro-B cells were transduced with either MigR (Control) or MigR-μH chain (WT383) retroviral vectors and the infected cells were sorted based on expression of GFP. Flow cytometry analysis was used to confirm the surface expression of the pre-BCR (left panel). Phospho- and total Akt levels were monitored by immunoblotting in the indicated cells under IL-7 Hi or Lo conditions as detailed above. Data is representative of two independent experiments.
IL-7R but not pre-BCR couples to PI(3)K-Akt module
To determine if the inverse relationship between IL-7R mediated activation of the PI(3)K-Akt module and Foxo protein accumulation was also operative in vivo, we analyzed IL-7R, p-Akt and Foxo1 protein abundance in large versus small pre-B cells isolated from wild-type mice. Rag gene expression, Ig kappa germline transcription and Ig kappa rearrangement are highly induced in small pre-B cells (ref. 1, 3 and data not shown). As expected, the cell surface expression of IL-7Rα was reduced in small resting pre-B cells compared to their large cycling counterparts (ref. 17 and Fig. 1e) and this was also reflected in reduced amounts of intracellular IL-7Rα protein (data not shown). We note that attenuation of IL-7 signaling in Irf4,8−/− pre-B cells, which results in cell-cycle arrest and reduced size3, was also accompanied by the down regulation of cell surface and intracellular expression of IL-7Rα protein (Supplementary Fig. 3). Importantly, whereas p-Akt abundance was positively correlated with that of the IL-7R in both wild-type and Irf4,8−/− pre-B cells, the abundance of Foxo1 showed the reciprocal behavior (compare Fig. 1e with Fig. 1a,b). Thus, IL-7R signaling via p-Akt appears to negatively regulate Foxo1 accumulation in wild-type pre-B cells. Furthermore since the Irf4,8−/− pre-B cells manifest this regulatory pathway, they represent a suitable cellular system for analyzing the underlying molecular mechanisms.
Based on the signaling properties of the BCR it has been proposed that the pre-BCR also activates PI(3)K-Akt signaling1. However, this proposition has not been directly tested. To test this possibility we expressed a rearranged Ig heavy chain gene (μH383) in Rag2−/− pro-B cells5. This heavy chain is assembled into a pre-BCR that activates ERK signaling5. Intriguingly, this signaling competent pre-BCR did not enhance the activity of Akt (Fig. 1f). The latter was regulated to a comparable degree in response to IL-7 signaling in the control as well as reconstituted Rag2−/− pro-B cells. These results suggest that the PI(3)K-Akt module is primarily coupled to the IL-7R rather than to the pre-BCR in pre-B cells.
Foxo1 activates BLNK and Syk expression in pre-B cells
We have previously identified a large set of genes in Irf4,8−/− pre-B cells that are either induced or repressed upon the attenuation of IL-7 signaling3. Therefore, we performed ChIPseq analysis with Foxo1-specific antibodies under these conditions to determine which of these genes are direct targets. Deep sequencing of Foxo1 targeted regions identified 11,772 unique sequences. Analysis of the top 500 targeted regions using MEME revealed the Foxo binding site18 as the most frequently occurring motif (Fig. 2a; left). Within the Foxo1 cistrome, the majority of the binding sites resided in intergenic (52.4 %) or intronic (35.5 %) regions (Fig. 2a; right). We also performed genome-wide ChIPseq analysis with an epitope tagged Foxo3a protein and found its cistrome to substantially overlap with that of Foxo1 in Irf4,8−/− pre-B cells (data not shown). Mapping of the Foxo1 cistrome with respect to the IL-7 regulated genes showed that a majority of the induced genes (150/197; 76%) contained one or more Foxo1 binding sites within or in their vicinity (Fig. 2b). Selected genes that were of biological interest included those involved in V(D)J recombination (Rag1,2, Dntt), cell-cycle arrest (p21, p27) and pre-BCR signaling (Blnk, Syk) (Supplementary Fig. 4). These results suggested that in addition to coordinating cell-cycle arrest with the induction of the Rag genes, Foxo1 has a novel function in regulating pre-BCR signaling.
Figure 2. ChIPseq analysis of the Foxo1 cistrome in IRF-4,8−/− pre-B cells.

IRF-4,8−/− pre-B cells were cultured under IL-7 Lo conditions. Cross-linked and sheared chromatin was immunoprecipitated with αFoxo1 antibodies and then the eluted DNA was processed for massively parallel sequencing. (a) Left; De novo motif analysis of Foxo1 target sequences. 11,772 Foxo1 target sites were identified using QuEST. The target sequences associated with the top 500 Foxo1 peaks were analyzed using MEME to identify overrepresented motifs within +/− 100 bp from the peak maxima. Sequence logo depicts the Foxo1 motif (P-value for motif significance; P = 1.75E-6) that was present in all 500 sequences analyzed. Right; Genomic distribution of Foxo1 target sites was analyzed using Cis-regulatory Element Annotation System (CEAS). (b) Union analysis was used to compare the Foxo1 cistrome with IL-7 regulated genes identified by genome-wide expression analysis. The numbers within the circles indicate genes that are up regulated or down regulated by IL-7 signaling and also targeted by Foxo1. (c) and (d) Foxo1 binding peaks at the Rag1,2 and the Blnk, Dntt loci, respectively are depicted using Integrated Genome Browser (IGB). P and I denote Foxo1 binding peaks at promoter or intergenic regions, respectively. Pax5BS denotes a previously characterized Pax5 binding site19 that is also co-targeted by Foxo1. The y-axis shows the degree of target sequence enrichment in terms of normalized tag counts. Two independent ChIPseq experiments were performed.
The binding peaks of Foxo1 in the vicinity of the Rag1, 2 and Blnk, Dntt loci are shown in Fig. 2c,d and those for Syk in Supplementary Fig. 5c. These were validated as peaks that are induced upon attenuation of IL-7 signaling by ChIP and Q-PCR analysis (Supplementary Fig. 5a–c). Within the Rag locus, in addition to the known Erag region, two new intergenic regions were identified, Irag1 and Irag2. The former is located about 15 kb upstream of the Rag1 promoter and the latter is adjacent to Erag, approximately 25 kb upstream of the Rag2 promoter. As was the case with the Rag locus Foxo1 bound the Blnk, TdT locus at multiple sites with a prominent peak, IBlnk, positioned in the intergenic region between the divergent promoters of the two genes and a pair of sites near the promoter of the Blnk gene (PBlnk, Pax5BS19). The latter site is implicated in the regulation of the Blnk gene by the transcription factor Pax5. These findings raised the possibility that upon attenuation of IL-7 signaling, Foxo1 coordinately activates the Blnk gene in conjunction with Pax5 in pre-B cells and enables effective coupling of the pre-BCR with many to its downstream signaling components1,19,20. These results prompted us to explore if Foxo1, as is the case for Pax5, was functionally required for Blnk expression. Consistent with this possibility, BLNK mRNA and protein were substantially induced in Irf4,8−/− pre-B cells upon lowering IL-7 signaling (Fig. 3a). Importantly, BLNK protein expression was also induced during the transition from large cycling pre-B cells to the small resting stage in the bone marrow and was correlated with higher amounts of Foxo1 (Fig. 3b and Fig. 1e). Given that Foxo1 also targets the Syk gene we also sought to determine if Syk expression was correlated with and regulated by Foxo1. Although Syk protein is expressed at appreciable amounts in large cycling pre-B cells its expression was further induced upon transition to the small resting pre-B cell stage and correlated with increased Foxo1 protein (Fig. 3b and Fig. 1e). To demonstrate that Foxo1 regulates BLNK and Syk expression we carried out loss- and gain-of-function experiments. Knockdown of Foxo1 in Irf4,8−/− pre-B cells reduced BLNK and Syk expression (Fig. 3c). Conversely, ectopic expression of Foxo1 increased Syk and BLNK expression (Fig. 3d). As was the case with BLNK (Fig. 3a), Syk expression was also induced upon lowering of IL-7 signaling in Irf4,8−/− pre-B cells and this was accompanied by its association with BLNK in a signaling complex (ref. 20 and Fig. 3e,f). Thus, Foxo1 not only activates the expression of the Rag genes upon attenuation of IL-7 signaling but also that of the Blnk and Syk genes, thereby enabling the differentiation signaling functions of the pre-BCR.
Figure 3. Foxo1 is necessary for inducing two key components of pre-BCR signaling.

IL-7 signaling was attenuated in IRF-4,8−/− pre-B cells as described in Fig. 1. (a) RT-PCR and immunoblot analysis of Blnk expression. (b) Flow cytometric analysis of intracellular Blnk and Syk expression in large cycling and small resting pre-B cells. Bone marrow pre-B cells were isolated from wild-type mice (n=3) as detailed in Methods. Flow cytometry profiles of large (red) versus small pre-B cells (green) with Blnk and isotype control antibodies (filled gray histogram) are shown. (c) Immunoblot analysis of indicated proteins in IRF-4,8−/− pre-B cells transduced with a control or Foxo1 knock down vector encoding a hygromycin resistance gene as a selectable marker. Cells were transduced with retroviral vectors encoding short hairpin (sh) sequences targeting Foxo1 or Luciferase (control) transcripts. Infected cells were sorted based on hygromycin resistance under IL-7 Hi conditions, and plated in IL-7 Lo conditions for 48 h before generating lysates for immunoblotting. (d) Immunoblot analysis of Syk, Blnk, Pax5 and HPRT in IRF-4,8−/− pre-B cells transduced with a control or Foxo1 retrovirus vector. Cells were maintained in IL-7 Hi conditions and sorted based on GFP expression after transduction as described in Fig. 1f. MigR, MigR-Foxo1 denote control and Foxo1, respectively. (e) Immunoblot analysis of Syk and HPRT upon attenuation of IL-7 signaling in IRF-4,8−/− pre-B cells (see Fig. 1 for details). (f) Immunoprecipitation of Blnk complexes. Whole cell extracts were prepared from IRF-4,8−/− pre-B cells cultured in IL-7 Lo conditions for 48 h. Immunoprecipitations were performed using either control IgG or αBlnk antibodies followed by immunoblot analysis with αSyk or αBlnk. 2% of the pre-immunoprecipitated samples were used for input. All data are representative of three independent experiments except for 3c which is representative of two independent experiments.
Regulation of BLNK expression by Foxo1 and Pax5
Given that Pax5 has been shown to directly activate the Blnk gene in part by binding to the Pax5BS19, ChIP analysis was used to determine if Foxo1 co-bound this site along with Pax5. Foxo1 and Pax5 not only co-targeted the Pax5BS region but also a nearby segment, PBlnk (Fig. 4a and Supplementary Fig. 5b). Intriguingly, as was the case with Foxo1, Pax5 binding to both of these presumptive regulatory regions was induced upon the attenuation of IL-7 signaling. Immunoblotting revealed that Pax5 protein abundance was also upregulated by the lowering of IL-7 signaling and/or the inhibition of PI(3)K activity using LY294002 (Fig. 4b). Thus like Foxo1, Pax5 protein abundance was negatively regulated by IL-7 signaling in pre-B cells, likely via the PI(3)K-Akt module. The inducible co-binding of Pax5 and Foxo1 to promoter elements in the Blnk gene raised the possibility that the two transcription factors might physically interact. Consistent with this possibility, immunoprecipitation of Pax5 demonstrated its association with Foxo1 in Irf4,8−/− pre-B cells (Fig. 4c). Finally, ectopic expression of Foxo1 in Irf4,8−/− pre-B cells promoted the binding of Pax5 to the Blnk promoter region (Fig. 4d). Thus attenuation of IL-7 signaling in pre-B cells leads to the stabilization of Foxo1 and Pax5 transcription factors that co-bind the Blnk promoter region to induce transcription.
Figure 4. Foxo1 and Pax5 cooperate to induce Blnk expression upon attenuation of IL-7 signaling.

(a) ChIP analysis of Foxo1 and Pax5 binding to the denoted regulatory elements in the Blnk locus. ChIP was performed as in Fig. 1c. (b) Immunoblot analysis of Pax5. Cells were treated with vehicle or 53μg/mL of PI3K inhibitor LY294002 (LY). Protein expression was normalized with HPRT. (c) Immunoprecipitation of Pax5 complexes. Whole cell extracts were prepared from IRF-4,8−/− pre-B cells cultured under IL-7 Lo conditions for 48 h. Immunoprecipitation was performed using either control IgG or αPax5 antibodies, followed by immunoblot analysis with αFoxo1 or αPax5 antibodies. 5% of pre-immunoprecipitated samples were used for input. Data in a–c are represetative of three independent experiments. (d) Pax5 binding to the regulatory element in the Blnk locus in IRF-4,8−/− pre-B cells after transduction with Foxo1 retroviral vector as described above. ChIP assay was performed using control IgG or αPax5 antibodies. The error bars correspond to the standard deviation from the mean for three independent experiments. The κ3′E region is used as a negative control. (e) Comparative analysis of Foxo1 and Pax5 cistromes in IRF-4,8−/− pre-B cells. ChIPseq analysis of Pax5 binding was performed as in Fig. 2a. Left; A venn diagram comparing Foxo1 cistrome (red) with the Pax5 cistrome (blue). Based on 17,639 Pax5 binding peaks, de novo motif analysis was performed with the top 500 peaks as in Fig. 2a. Sequence logo depicts the Pax5 motif (P-value for motif significance; P = 6.3E-05) that was present in 341 of the 500 sequences analyzed. Right; Union analysis comparing the Foxo1 and Pax5 cistromes with IL-7 regulated genes as described in Fig. 2b. The number of genes in each category is shown. Numbers in parentheses indicate DNA sequences that display coincident binding of Foxo1 and Pax5 in IL-7 regulated genes that are co-targeted by both transcription factors.
The above results suggested that Foxo1 and Pax5 may co-bind regulatory regions in many other genes that are induced in pre-B cells upon attenuation of IL-7 signaling. Therefore we performed ChIPseq analysis with Pax5 antibodies in Irf4,8−/−pre-B cells under conditions of low IL-7 signaling. Comparison of the Pax5 cistrome with that of Foxo1 showed that about half of Foxo1 binding peaks (6,035/11,772; 51%) were coincident with those of Pax5 (Fig. 4e; left). Mapping of the Pax5 and Foxo1 cistromes with respect to the IL-7 regulated genes showed that a majority of those that were induced (121/197; 61%) were targeted by both Foxo1 and Pax5, with 350 coincident sites between these two factors (Fig. 4e; right). The Foxo1 and Pax5 binding landscapes on select genes that were of biological interest (Rag 1,2, Blnk, Syk and Irf4) are shown in Supplementary Fig. 5. Thus, upon attenuation of IL-7 signaling, Foxo1 and Pax-5 co-target and likely activate a large set of pre-B cell genes.
A positive feedback loop involving Foxo1-BLNK-p38
Our results suggested that a major consequence of lowering IL-7 signaling in pre-B cells is to strongly induce BLNK expression via the transcription factors Foxo1 and Pax5, thereby coupling the pre-BCR to multiple downstream effectors1, 8. To test this model we performed gain-of-function experiments with Foxo1 and BLNK. Expression of Foxo1 in Irf4,8−/− pre-B cells induced BLNK and the activation of p38MAPK (Fig. 5a). Secondly, expression of BLNK resulted in the activation of p38 (Fig. 5b). Importantly, BLNK expression under conditions of high IL-7 signaling also led to inhibition of Akt activity and the consequent elevation of Foxo1 and Pax5 proteins (Fig. 5b). These results are consistent with an earlier report utilizing Blnk−/− pre-B cells in which restoration of BLNK expression resulted in the inhibition of Akt activity6. Thus BLNK functions in a negative feedback loop to inhibit IL-7 signaling thereby promoting its own expression via Foxo1 and Pax5 and then coupling to the pre-BCR and downstream effectors such as p38 (ref. 8). Foxo1 transcriptional activity is positively regulated through its phosphorylation by p38 (ref. 9, 21). This finding raised the possibility that p38 activation may result in a positive feedback loop via Foxo1 that further induces BLNK and augments the inhibition of IL-7 signaling as well as promotes signaling by the pre-BCR. Therefore we tested if inhibition of p38 activity would impair the functioning of Foxo1 under conditions of low IL-7 signaling. Attenuation of IL-7 signaling in Irf4,8−/− pre-B cells resulted in the activation of p38 that was blocked by the inhibitor SB203580 (Fig. 5c). Inhibition of p38 activity under these conditions lowered the activation of Rag and Blnk gene expression (Supplementary Fig. 6a and data not shown) and reduced the binding of Foxo1 to multiple regulatory sequences in the Rag as well as the Blnk loci (Fig. 5d). We note that Irf4,8−/− pre-B cells expressed p38α and p38β MAPK isoforms (Supplementary Fig. 6b) and the p38 inhibitor SB203580 inhibited the activity of both. Finally, inhibition of p38 activity did not appreciably affect Pax5 binding to these sites in the Rag locus sites (data not shown), thereby highlighting the requirement of Foxo1 for efficient transcriptional activation of these genes. Thus we propose that p38 functions in a positive feedback loop in pre-B cells to augment Foxo1 binding to its target genes thereby further inducing Rag and Blnk gene expression. To examine the kinetics of activation of the Foxo1-BLNK-p38 positive feedback loop we analyzed the expression and/or activation of each of these regulatory components upon attenuation of IL-7 signaling in Irf4,8−/− pre-B cells (Fig. 5e). Foxo1 accumulation was initially discernable at 24 h, after the lowering of IL-7 signaling, and continued to increase up to 48 h. BLNK protein abundance paralleled that of Foxo1. p38 activation was observed at 36 h after substantial accumulation of Foxo1 and BLNK. Thus the positive feedback loop comprised of Foxo1-BLNK-p38 is gradually activated upon lowering of IL-7 signaling in pre-B cells as a consequence of stabilization of Foxo1 protein.
Figure 5. A positive feedback loop comprised of Foxo1-BLNK-p38 that regulates pre-B cell differentiation.
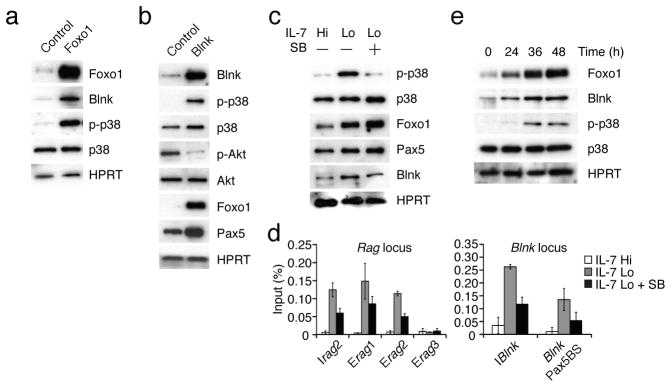
(a) and (b) Immunoblot analysis of indicated proteins in IRF-4,8−/− pre-B cells after transduction with Foxo1 or Blnk retroviral vectors. Cells were maintained in IL-7 Hi conditions and sorted based on GFP expression after transduction as described in Fig. 1f. MigR, MigR-Foxo1 and MigR-Blnk denote control and Foxo1 or Blnk encoding retroviruses, respectively. (c) Immunoblot analysis of phospho-p38 (p-p38), total p38 (p38), Foxo1 and HPRT in the absence or presence of the p38 inhibitor SB203580 (SB, 0.1 μg/mL) and the indicated IL-7 culture conditions. (d) Foxo1 binding to regulatory elements in the Rag and Blnk loci under the conditions described above. ChIP assay was performed using αFoxo1 antibodies as in Fig. 1c. (e) Immunoblot analysis of Foxo1, Blnk, p-p38, p38 and HPRT upon attenuation of IL-7 signaling. IRF-4,8−/− pre-B cells were cultured in IL-7 Lo conditions and whole cell lysates were prepared and analyzed by immunoblotting at the indicated time points. Data are representative of three independent experiments.
Given that p38 activation is dependent on pre-BCR signaling these results imply that attenuation of IL-7 signaling should lead to p38 activation, dependent on Foxo1 and BLNK, in pre-B but not in pro-B cells as the latter cells lack the pre-BCR. This result was indeed the case as lowering of IL-7 signaling in Rag2−/− pro-B cells did not result in activation of p38 (Fig. 6a). We note that although Rag2−/− pro-B cells expressed comparable amounts of Foxo1 upon attenuation of IL-7 signaling as Irf4,8−/− pre-B cells (data not shown), the former did not substantially induce BLNK expression (Fig. 6a). Consistent with these findings Rag gene expression was more highly induced in Irf4,8−/−pre-B cells than in Rag2−/− pro-B cells (Supplementary Fig. 6c). Thus we suggest that the acquisition of a pre-B cell receptor enables pre-B cells to uniquely exploit a self-reinforcing regulatory loop involving the Foxo1-BLNK-p38 module to antagonize IL-7 signaling and to activate BLNK-dependent pre-BCR signaling.
Figure 6. Activation of the Foxo1-BLNK-p38 module is specific to pre-B cells.
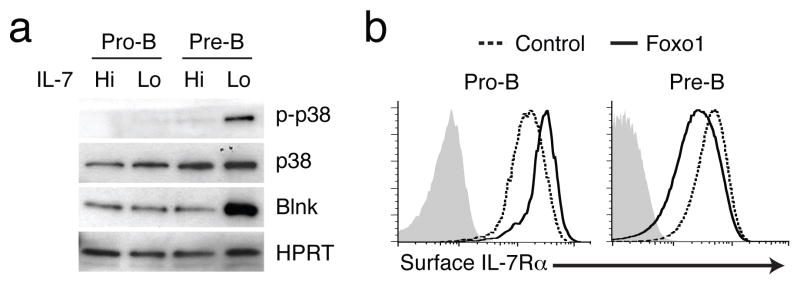
(a) Immunoblot analysis of p38 and Blnk in Rag2−/− pro-B (Pro-B) or IRF-4,8−/− pre-B (Pre-B) cells cultured in IL-7 Hi medium or shifted to IL-7 Lo conditions for 48 h. p-p38, p38, Blnk and HPRT levels were analyzed as in Fig. 5a. (b) Flow cytometric analysis of IL-7Rα expression upon transduction of Foxo1 in Rag2−/− pro-B (Pro-B) or IRF-4,8−/− pre-B (Pre-B) cells. Indicated cells were transduced with control (red) or Foxo1 (green) retrovirus as described in Fig. 5a. Cells were maintained in IL-7 Hi conditions after retroviral transduction and gated on GFP+ cells for the analysis of IL-7Rα levels. Data are representative of three independent experiments.
A key function of pre-BCR signaling is to up-regulate the expression of IRF-4 that is required for efficient activation of germline kappa transcription and rearrangement (ref. 3, 22 and Supplementary Fig. 2c). ChIPseq analyses demonstrate that Foxo1 binds to multiple presumptive regulatory sequences in the Irf4 gene along with Pax5, the latter is required for IRF-4 expression (ref. 19 and Supplementary Fig. 5d). Knockdown of Foxo1 in a v-abl transformed B-lineage cell line resulted in diminished expression of IRF-4 (Supplementary Fig. 7). Thus the selective activation of the Foxo1-BLNK module in pre-B cells ensures effective coupling of Rag gene expression with the induction of IRF-4 expression and Ig light chain rearrangement. Finally we note that IRF-4 along with Foxo1 and Pax5 targets the BLNK and Cxcr4 loci (Supplementary Fig. 8). Thus IRF-4 also functions in a positive feedback loop to augment BLNK expression and attenuate IL-7 signaling, the latter in part by inducing expression of Cxcr4 and promoting the migration of pre-B cells away from stromal cells expressing IL-7 (ref. 3).
Down regulation of the IL-7R by Foxo1 in pre-B cells
Paradoxically, although Foxo1 targets and activates transcription of the Il7ra gene in Irf4,8−/− pre-B cells (ref. 12, 23 and Supplementary Fig. 4), the lowering of IL-7 signaling actually results in the down regulation of the IL-7Rα (Supplementary Fig. 3). Therefore we tested the consequences of increased Foxo1 expression in these cells. Elevated amounts of Foxo1 resulted in the down regulation of surface bound and intracellular IL-7Rα (Fig. 6b, data not shown). It should be noted that over expression of Foxo1 in Rag2−/− pro-B cells did not lead to down regulation of IL-7Rα (Fig. 6b). Thus in pre-B cells the induction of Il7ra gene activity by Foxo1 (Fig. S4) appears to be selectively overridden by a post-translational regulatory mechanism that likely promotes degradation of IL-7Rα protein in a Foxo1 dependent manner. We propose that at the pre-B cell developmental stage, Foxo1 also functions in a negative feedback loop to diminish IL-7 signaling via down regulating IL-7Rα protein while promoting the coupling of the pre-BCR to its downstream effectors.
A self-reinforcing differentiation circuit
Based on the experimental evidence presented herein, Fig. 7 depicts the components and connections that comprise the gene regulatory network which orchestrates the interplay between IL-7R and pre-BCR signaling. Attenuation of IL-7 signaling and the consequent diminution of PI(3)K–Akt activity results in stabilization of the Foxo1 and Pax5 proteins. These transcription factors mediate the activation of the Rag and Dntt genes as well as that of Syk and Blnk. They also co-target a large set of genes that are important in BCR and cytokine signaling (Supplementary Fig. 9). Importantly, BLNK inhibits the activity of the PI(3)K–Akt module thereby further enhancing Foxo1 and Pax5 protein abundance. Induction of BLNK expression by Foxo1 also sets in motion a positive auto-regulatory loop that involves the activation of p38 and in turn its stimulation of Foxo1 activity. Finally, BLNK dependent pre-BCR signaling induces the expression of IRF-4 which targets Ig light chain enhancers to drive chromatin modifications and germ line transcription3, 5, 22. Thus, the architecture of the pre-B cell gene regulatory network involves antagonistic interplay between a growth factor and a differentiation inducing receptor through a series of self-reinforcing positive and negative feedback loops.
Figure 7. A gene regulatory network triggered by limiting IL-7 activates pre-BCR signaling and differentiation.

The schematic depicts the organization of components and connections of a signaling and gene regulatory network that orchestrates the pre-B cell developmental checkpoint. Within the network, proliferation is primarily driven by IL-7R activated signaling pathways (blue lines). Under these conditions, the activity of the PI3K/Akt module inhibits the accumulation of Foxo1 and Pax5 that are needed to induce BLNK expression and it’s coupling to the pre-BCR via Syk. A feed forward loop comprising of Foxo1, Blnk and p38 is activated in cells sensing a lower concentration of IL-7 (bold green lines). This module triggers pre-BCR signaling and down regulates the IL-7R as well as its signaling activity via inhibition of PI3K/Akt. Thus pre-B cells undergo cell-cycle arrest, activate Rag gene expression and immunoglobulin kappa light chain locus transcription and rearrangement to generate naïve B cells (green lines).
Discussion
Our findings reconcile alternate models that either invoke the attenuation of IL-7 signaling3 or a qualitative change in the signaling state of the pre-BCR1 as a means of regulating the pre-B cell developmental checkpoint. We demonstrate that attenuation of IL-7 signaling is not only required for cell cycle arrest and the induction of Rag gene expression via the stabilization of Foxo1 but that this transcription factor along with Pax5 is needed to activate the Blnk gene which encodes a critical signaling adapter for the pre-BCR. Thus in pre-B cells the pre-BCR indeed manifests two distinct signaling states involving low versus high amounts of BLNK expression. Importantly IL-7 signaling negatively regulates the BLNK dependent signaling state of the pre-BCR. Therefore we propose that attenuation of IL-7 signaling induces a qualitative change in the signaling functions of the pre-BCR by enabling its coupling to the adapter BLNK and the activation of downstream effectors such as Btk, PLC-γ2 and p38MAPK.
The conclusion that Foxo1 and Pax5 cooperate in regulating the Blnk gene is based on the following lines of evidence. Foxo1 and Pax5 co-bind to several regulatory regions in the Blnk gene in an inducible manner requiring attenuation of IL-7 signaling. Foxo1 and Pax5 associate with one another and increased Foxo1 expression that results in Blnk activation also leads to increased binding of Pax5 to a shared regulatory element in the Blnk gene promoter. It remains to be determined if Foxo1 and Pax5 directly interact with one another and cooperatively bind in vitro to their closely spaced sites in co-targeted genes.
Our results suggest that Foxo1-BLNK-p38 function as components of a positive feedback loop to augment Foxo1 activity. p38 MAPK has been previously implicated in the induction of Rag gene expression in pre-B cells but the mechanism has remained obscure9. Importantly p38α has been shown to phosphorylate multiple sites on Foxo1 in vitro and in vivo, the latter in NIH3T3 cells, and these sites are implicated in transcriptional activation21. These results, along with our demonstration that inhibition of p38 activity results in reduced binding of Foxo1 to regulatory elements in the Rag and BLNK genes, strongly suggest that p38 positively regulates Foxo1 activity. We note that disruption of the MAPK p38α gene has not been associated with a defect in B cell development at the pre-B cell stage24. However pre-B cells express both p38α and p38β MAPK isoforms (Fig. S6b). Given the relatedness of these two MAPK isoforms and several shared substrates it is likely that they perform overlapping functions. Finally we note that the p38 inhibitor SB203580 inhibits the activity of both isoforms. It will be important to determine the sites of p38 mediated phosphorylation of Foxo1 in pre-B cells and their molecular consequences on Foxo1 DNA binding and transcriptional activating functions.
We have shown that in cycling pre-B cells that are dependent on IL-7 signaling, the pre-BCR is not efficiently coupled to the PI(3)K-Akt or Syk-BLNK modules. However, in this context the pre-BCR can function to augment proliferation driven by the IL-7R11, 17. We propose that following Ig light chain gene rearrangement, the expression of a non-self reactive BCR terminates the Foxo1-BLNK-p38 feed forward by coupling to the PI(3)K-Akt module. Basal or “tonic” signaling25 through the BCR leads to inactivation of the Foxo transcription factors26. The down regulation of the IL-7R at this developmental stage would also favor the coupling of the PI(3)K-Akt module to the BCR. Our results imply a fundamental structural difference between pre-BCR and BCR signal transduction complexes in terms of their coupling with the PI(3)K-Akt module. The structural basis of the difference between these two signal transduction complexes remains to be elucidated and may partly depend on differing co-receptors.
If light chain rearrangement results in the generation of an autoreactive BCR that encounters self-antigen, it will undergo endocytosis upon binding antigen thereby resulting in a lowering of basal PI(3)K-Akt signaling and therefore the re-induction of Rag gene expression and receptor editing. Consistent with our model either loss of the BCR or inhibition of PI3K activity in immature B cells results in the induction of Rag gene expression and light chain gene rearrangement25. We note that expression of an autoreactive BCR that resembles the pre-BCR in its structure and signaling properties27 would not be predicted to terminate the feed forward loop. In this latter circumstance Rag gene expression would be sustained so to enable receptor editing. Thus our regulatory model provides an attractive molecular logic for the control of receptor editing in response to autoreactive BCRs.
In our regulatory model the attenuation of IL-7 signaling is critical for initiating pre-B cell differentiation. How could IL-7 signaling be attenuated in vivo? The survival and proliferation of pro-B cells is dependent on interactions with IL-7 expressing stromal cells28. Following productive heavy chain gene rearrangement and the ensuing assembly of the pre-BCR, pre-B cells undergo a proliferative burst. Assuming a limiting niche of IL-7 expressing stromal cells29, dividing pre-B cells that undergo cytokinesis orthogonally to the stromal cell layer would generate daughter cells that sense a somewhat lower concentration of IL-7. Such environmental asymmetry29,30 may be sufficient to initiate activation of the Foxo1-BLNK-p38 feed forward loop in daughter cells that are positioned away from the stroma, thereby down regulating the IL-7 receptor. As Foxo1 and IRF-4 co-target the Cxcr4 gene, their accumulation would promote the movement of pre-B cells away from IL-7 expressing stromal cells3, 31. Consequently such pre-B cells would undergo cell-cycle arrest and induce light chain recombination.
METHODS
Cells and culture conditions
Irf4,8−/− pre-B cells and Rag2−/− pro-B cells were cultured in Opti-MEM (Invitrogen) media supplemented with 5% FCS and 5 ng/ml of IL-7 (IL-7 Hi conditions)4, 33. IL-7 signaling was attenuated in B lineage cells by lowering the IL-7 concentration to 0.1 ng/ml (IL-7 Lo conditions)3. Pharmacological inhibitors were from Cell Signaling (PI(3)K inhibitor, LY294002) and Promega (p38 inhibitor, SB203580).
Retroviral vectors and transduction of B lineage cells
Retroviruses encoding murine Foxo1 and Foxo3a were constructed by sub-cloning amino-terminal FLAG-tagged cDNA segments into the MigR retrovirus34. Foxo1 and Foxo3a coding sequences were amplified by PCR using cDNA prepared from B220+ B lineage cells isolated from mouse bone marrow. The murine Blnk cDNA was a kind gift of M. Veselitis (University of Chicago). Plasmids encoding shRNA oligonucleotides were constructed as previously described35. Oligonucleotide sequences of the shRNAs directed against Foxo1 or Foxo3a mRNAs were: Foxo1, 5′-TGCCCAGTCTGTCTGAAATCAG-3′; Foxo3a, 5′-CCTTTCCCTAGCACACTTAAA-3′. To generate retroviral stocks, PlatE packaging cells were transiently transfected with retroviral constructs using FuGENE reagent (Roche). Viral supernatants were collected at 48 and 72 h after transfection and pooled. Irf4,8−/− pre-B or Rag2−/− pro-B cells were suspended in retroviral supernatant, generated as described above, with 12.5 μg/ml polybrene and centrifuged for 90 min at 1,500 g at 32 °C. After centrifugation, cells were incubated at 32 °C for 3 h and then washed and plated on OP9 cells in Opti-MEM supplemented with 5% FCS and 5 ng/ml of IL-7 (IL-7 Hi). After 48 h, transduced cells were sorted on the basis of GFP expression and were used for generating protein lystes or the isolation of RNA. For experiments requiring analysis of the effects of attenuation of IL-7 signaling, these cells were re-plated on OP9 cells in IL-7 Hi or IL-7 Lo medium for 48 h.
Immunoblotting, immunoprecipitation and immunohistochemical analyses
Protein extracts were prepared as previously described36. Protein lysates were separated by SDS-PAGE and transferred to Immobilon-P membrane (Millipore). Antibodies used to probe the protein blots were; anti-Foxo1 (Cell Signaling, #9462), anti-Foxo3a (Cell Signaling, #9467), anti-Foxo4 (Cell Signaling, #9472), anti-p-Foxo1 (Cell Signaling, #9461), anti-p-Foxo3a (Cell Signaling, #9466), anti-p-Akt (Cell Signaling, #4058), anti-Akt (Cell Signaling, #9272), anti-p-p38 (Cell Signaling, #9211), anti-p-38 (Cell Signaling, #9212), anti-Pax5 (Santa Cruz, sc-1974), anti-BLNK (Santa Cruz, sc-15345), anti-Syk (Santa Cruz, sc-1077), anti-IRF-4 (Santa Cruz, sc-6059), anti-IRF-8 (sc-6058), anti-HPRT (Santa Cruz, sc-20975). Specifications of antibody reagents are provided in Supplementary Table 2. For immunoprecipitation, whole cell lysates were incubated with specific antibodies and processed as previously described36. Immunohistochemistry was performed as described in Reddy et al.,37 using anti-Foxo1 (noted above) and anti-laminB37 antibodies.
Flow cytometric analyses
Procedures for surface and intracellular staining have been described previously34. Antibodies used for staining were anti-IL-7Rα (eBioscience, 25-1271-82) and anti-pre-BCR (BD Biosciences, 551863). For cell-cycles analysis, cells were stained with 10 μg/ml Hoechst 33342 (Molecular Probes) in the Hoechst buffer at 37 °C for 90 min. Data were collected with an LSR II and analyzed with Flow Jo software (Tree Star).
Isolation and flow cytometric analysis of bone marrow B-lineage progenitors
Bone marrow was collected from C57BL/6 mice (JAX, 8–12 weeks of age) according to protocol approved by the University of Chicago Institutional Animal Use and Care Committee. Cells were resuspended in PBS containing 1 % FCS. Erythrocytes were lysed and cells were stained with anti-B220, anti-CD43, anti-IgM and anti-IgD. Pre-B cell populations were gated on the basis of the following combination of cell surface markers B220+CD43loIgM−IgD−. Large pre-B and small pre-B cells were distinguished by cell size; large pre-B (FSChi SSChi) and small pre-B (FSCloSSClo)..
RT-PCR
Total RNA was isolated with Trizol (Invitrogen) and cDNA was made with SuperScript II reverse transcriptase (Invitrogen). Quantitative PCR was performed with a SYBR qPCR Master Mix (Clontech) by using a Bio-Rad CFX96 Real-Time PCR Detection System. Primers used are listed in Supplementary Table 1.
ChIP and ChIPseq analyses
ChIP assay was performed as previously described34. Chromatin from Irf4,8−/− pre-B cells was isolated, sonicated to obtain DNA fragments ranging in size from 100 to 500 bp. Chromatin fragments were immunoprecipitated using control IgG (Santa Cruz, sc-2027), anti-Foxo1 (Abcam, ab70382) or anti-Pax5 (Santa Cruz, sc-1974). After reversal of formaldehyde crosslinks, specific DNA sequences were assessed by quantitative real-time PCR. Primers used for PCR are listed in Supplementary Table 1. For Foxo1 and Pax5 ChIPseq, Irf4,8−/− pre-B cells were cultured under IL-7 Lo conditions for 48 h. For IRF-4 ChIPseq, splenic B cells purified from B1-8i BCR knock-in mice were stimulated and cultured for 3 days as previously described38. DNA libraries were then prepared using sheared chromatin that was immunoprecipitated with anti-Foxo1, anti-Pax5 or anti-IRF-4 (refs.34, 39). Genomic DNA was sequenced using a Illumina Genome Analyzer II, aligned using ELAND software, allowing for no more than two mismatches per sequence. Foxo1 or Pax5 binding peaks were identified using Quantitative Enrichment of Sequence Tags (QuEST) v2.4 with input chromatin as control using a 1% false discovery rate (FDR). The MEME algorithm (v4.4.0) was used to identify overrepresented motifs within +/− 100bp from peak maxima. Genomic localization and distance of binding sites from nearest transcriptional start sites were analyzed with software programs created by the Dinner Lab. Scripts used for QuEST 1% FDR, peak annotation and coincidence are described below. Schematic representations of peaks in relation to genome sequence were generated using the Integrated Genome Browser (IGB) v6.5. Microarray data have been described previously3.
Computational Methods for ChIPseq
QuEST 1% FDR
QuEST was first run using a low threshold, which generally resulted in an FDR of >1%. A 1% FDR was then achieved post-QuEST: the lowest ChIP regions and pseudo-ChIP (input) regions were systematically eliminated by raising the threshold until the FDR (ratio of the number of pseudo-ChIP regions to the number of ChIP regions) was 1%.
Annotation of peaks in relation to genes
Peaks were annotated with UCSC genes. Peaks were annotated for the closest upstream and downstream gene (judged by distance to gene TSS), plus any genes that the peak happened to lie within. For peaks within the coordinates of a gene, the exon/intron position of the location was noted. Distance to TSS was recorded for each gene, as was the closest gene.
Coincident peaks
Peaks were called as coincident if they were less than 200 bp apart.
Electrophoretic mobility shift assays (EMSAs)
Nuclear extracts were prepared from Irf4,8−/− pre-B cells and assayed with a Foxo binding site double-stranded oligonucleotide as described previously3. The oligonucleotide sequence derived from the Erag1 region and its Foxo binding motif (underlined) is shown below:
5′-AAGAGTATCAAAACAATGCTAAGCCCTCGG-3′.
Supplementary Material
Acknowledgments
We are grateful to S. Heinz (Univ. California-San Diego) for advice concerning preparation of DNA libraries for ChIPseq, M. Veselitis (Univ. of Chicago) for providing murine Blnk cDNA and S. E. Powers (Univ. of Chicago) for helpful discussions. We thank the Bendelac lab for assistance with flow cytometry, R. Duggan, D. Leclerc, M. Olson and J. Cao for assistance with cell sorting and K. Michelini and J. Zekos for operating the Illumina Genome Analyzer II at University of Chicago supported by the Howard Hughes Medical Institute.
Footnotes
AUTHOR CONTRIBUTIONS
K.O. and H.S. designed the experiments. K.O. performed majority of the experiments. M.M.C. created the program for ChIPseq data analysis; M.M. analyzed p-Akt abundance in pre-B cells and in IgμH transduced Rag2−/− pro-B cells; J.R.T. and E.B. assisted with ChIPseq data analysis and EMSAs respectively. M.R.C, A.R.D. and R.S. provided experimental advice and input on the manuscript. K.O. and H.S. wrote the manuscript.
References
- 1.Herzog S, Reth M, Jumaa H. Regulation of B-cell proliferation and differentiation by pre-B-cell receptor signalling. Nat Rev Immunol. 2009;9:195–205. doi: 10.1038/nri2491. [DOI] [PubMed] [Google Scholar]
- 2.Geier JK, Schlissel MS. Pre-BCR signals and the control of Ig gene rearrangements. Semin Immunol. 2006;18:31–39. doi: 10.1016/j.smim.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 3.Johnson K, et al. Regulation of immunoglobulin light-chain recombination by the transcription factor IRF-4 and the attenuation of interleukin-7 signaling. Immunity. 2008;28:335–345. doi: 10.1016/j.immuni.2007.12.019. [DOI] [PubMed] [Google Scholar]
- 4.Lu R, Medina KL, Lancki DW, Singh H. IRF-4,8 orchestrate the pre-B-to-B transition in lymphocyte development. Genes Dev. 2003;17:1703–1708. doi: 10.1101/gad.1104803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mandal M, et al. Ras orchestrates exit from the cell cycle and light-chain recombination during early B cell development. Nat Immunol. 2009;10:1110–1117. doi: 10.1038/ni.1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herzog S, et al. SLP-65 regulates immunoglobulin light chain gene recombination through the PI(3)K-PKB-Foxo pathway. Nat Immunol. 2008;9:623–631. doi: 10.1038/ni.1616. [DOI] [PubMed] [Google Scholar]
- 7.Thompson EC, et al. Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity. 2007;26:335–344. doi: 10.1016/j.immuni.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 8.Ishiai M, et al. BLNK required for coupling Syk to PLC gamma 2 and Rac1-JNK in B cells. Immunity. 1999;10:117–125. doi: 10.1016/s1074-7613(00)80012-6. [DOI] [PubMed] [Google Scholar]
- 9.Amin RH, Schlissel MS. Foxo1 directly regulates the transcription of recombination-activating genes during B cell development. Nat Immunol. 2008;9:613–622. doi: 10.1038/ni.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Han J, Sun P. The pathways to tumor suppression via route p38. Trends Biochem Sci. 2007;32:364–371. doi: 10.1016/j.tibs.2007.06.007. [DOI] [PubMed] [Google Scholar]
- 11.Fleming HE, Paige CJ. Pre-B cell receptor signaling mediates selective response to IL-7 at the pro-B to pre-B cell transition via an ERK/MAP kinase-dependent pathway. Immunity. 2001;15:521–531. doi: 10.1016/s1074-7613(01)00216-3. [DOI] [PubMed] [Google Scholar]
- 12.Dengler HS, et al. Distinct functions for the transcription factor Foxo1 at various stages of B cell differentiation. Nat Immunol. 2008;9:1388–1398. doi: 10.1038/ni.1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci USA. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plas DR, Thompson CB. Akt activation promotes degradation of tuberin and FOXO3a via the proteasome. J Biol Chem. 2003;278:12361–12366. doi: 10.1074/jbc.M213069200. [DOI] [PubMed] [Google Scholar]
- 15.Huang H, Tindall DJ. Regulation of FOXO protein stability via ubiquitination and proteasome degradation. Biochim Biophys Acta. 2011;1813:1961–1964. doi: 10.1016/j.bbamcr.2011.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hu H, et al. Foxp1 is an essential transcriptional regulator of B cell development. Nat Immunol. 2006;7:819–826. doi: 10.1038/ni1358. [DOI] [PubMed] [Google Scholar]
- 17.Erlandsson L, et al. Both the pre-BCR and the IL-7Ralpha are essential for expansion at the pre-BII cell stage in vivo. Eur J Immunol. 2005;35:1969–1976. doi: 10.1002/eji.200425821. [DOI] [PubMed] [Google Scholar]
- 18.Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J. 2003;375:365–371. doi: 10.1042/BJ20030022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schebesta M, Pfeffer PL, Busslinger M. Control of pre-BCR signaling by Pax5-dependent activation of the BLNK gene. Immunity. 2002;17:473–485. doi: 10.1016/s1074-7613(02)00418-1. [DOI] [PubMed] [Google Scholar]
- 20.Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol. 2010;10:387–402. doi: 10.1038/nri2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Asada S, et al. Mitogen-activated protein kinases, Erk and p38, phosphorylate and regulate Foxo1. Cell Signal. 2007;19:519–527. doi: 10.1016/j.cellsig.2006.08.015. [DOI] [PubMed] [Google Scholar]
- 22.Oda A, Ono T, Yamamoto M, Goitsuka R, Kitamura D. PKC eta directs induction of IRF-4 expression and Ig kappa gene rearrangement in pre-BCR signaling pathway. Int Immunol. 2008;20:1417–1426. doi: 10.1093/intimm/dxn101. [DOI] [PubMed] [Google Scholar]
- 23.Kerdiles YM, et al. Foxo1 links homing and survival of naive T cells by regulating L-selectin, CCR7 and interleukin 7 receptor. Nat Immunol. 2009;10:176–184. doi: 10.1038/ni.1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JM, White JM, Shaw AS, Sleckman BP. MAPK p38 alpha is dispensable for lymphocyte development and proliferation. J Immunol. 2005;174:1239–1244. doi: 10.4049/jimmunol.174.3.1239. [DOI] [PubMed] [Google Scholar]
- 25.Tze LE, et al. Basal immunoglobulin signaling actively maintains developmental stage in immature B cells. PLoS Biol. 2005;3:463–475. doi: 10.1371/journal.pbio.0030082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verkoczy L, et al. Basal B cell receptor-directed phosphatidylinositol 3-kinase signaling turns off RAGs and promotes B cell-positive selection. J Immunol. 2007;178:6332–6341. doi: 10.4049/jimmunol.178.10.6332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kohler F, et al. Autoreactive B cell receptors mimic autonomous pre-B cell receptor signaling and induce proliferation of early B cells. Immunity. 2008;29:912–921. doi: 10.1016/j.immuni.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 28.Bertolino E, et al. Regulation of interleukin 7-dependent immunoglobulin heavy-chain variable gene rearrangements by transcription factor STAT5. Nat Immunol. 2005;6:836–843. doi: 10.1038/ni1226. [DOI] [PubMed] [Google Scholar]
- 29.Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nat Rev Immunol. 2006;6:107–116. doi: 10.1038/nri1780. [DOI] [PubMed] [Google Scholar]
- 30.Wilson A, Trumpp A. Bone-marrow haematopoietic-stem-cell niches. Nat Rev Immunol. 2006;6:93–106. doi: 10.1038/nri1779. [DOI] [PubMed] [Google Scholar]
- 31.Lin YC, et al. A global network of transcription factors, involving E2A, EBF1 and Foxo1, that orchestrates B cell fate. Nat Immunol. 2010;11:635–643. doi: 10.1038/ni.1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hedrick SM. The cunning little vixen: Foxo and the cycle of life and death. Nat Immunol. 2009;10:1057–1063. doi: 10.1038/ni.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reynaud D, et al. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol. 2008;9:927–936. doi: 10.1038/ni.1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sciammas R, et al. Graded expression of interferon regulatory factor-4 coordinates isotype switching with plasma cell differentiation. Immunity. 2006;25:225–236. doi: 10.1016/j.immuni.2006.07.009. [DOI] [PubMed] [Google Scholar]
- 35.Laslo P, et al. Multilineage transcriptional priming and determination of alternate hematopoietic cell fates. Cell. 2006;126:755–766. doi: 10.1016/j.cell.2006.06.052. [DOI] [PubMed] [Google Scholar]
- 36.Ochiai K, Muto A, Tanaka H, Takahashi S, Igarashi K. Regulation of the plasma cell transcription factor Blimp-1 gene by Bach2 and Bcl6. Int Immunol. 2008;20:453–460. doi: 10.1093/intimm/dxn005. [DOI] [PubMed] [Google Scholar]
- 37.Reddy KL, Zullo JM, Bertolino E, Singh H. Transcriptional repression mediated by repositioning of genes to the nuclear lamina. Nature. 2008;452:243–247. doi: 10.1038/nature06727. [DOI] [PubMed] [Google Scholar]
- 38.Sciammas R, et al. An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol Syst Biol. 2011;7:1–15. doi: 10.1038/msb.2011.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heinz S, et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.