

Background: Mortalin was shown to contribute to removal of the complement membranolytic C5b-9 complex from the target cell surface.
Results: Modulations of mortalin expression and activity affect deposition of C5b-9 and cell death.
Conclusion: Mortalin, through its ATPase domain, regulates the C5b-9 deposition and confers resistance to complement-dependent cytotoxicity.
Significance: Mortalin is a potential therapeutic target in autoimmune diseases and in cancer immunotherapy.
Keywords: Complement System, Heat Shock Protein, Immunology, Molecular Chaperone, Necrosis (Necrotic Death), C5b-9, GRP75, Mortalin
Abstract
Mortalin/GRP75, the mitochondrial heat shock protein 70, plays a role in cell protection from complement-dependent cytotoxicity (CDC). As shown here, interference with mortalin synthesis enhances sensitivity of K562 erythroleukemia cells to CDC, whereas overexpression of mortalin leads to their resistance to CDC. Quantification of the binding of the C5b-9 membrane attack complex to cells during complement activation shows an inverse correlation between C5b-9 deposition and the level of mortalin in the cell. Following transfection, mortalin-enhanced GFP (EGFP) is located primarily in mitochondria, whereas mortalinΔ51-EGFP lacking the mitochondrial targeting sequence is distributed throughout the cytoplasm. Overexpressed cytosolic mortalinΔ51-EGFP has a reduced protective capacity against CDC relative to mitochondrial mortalin-EGFP. Mortalin was previously shown by us to bind to components of the C5b-9 complex. Two functional domains of mortalin, the N-terminal ATPase domain and the C-terminal substrate-binding domain, were purified after expression in bacteria. Similar to intact mortalin, the ATPase domain, but not the substrate-binding domain, was found to bind to complement proteins C8 and C9 and to inhibit zinc-induced polymerization of C9. Binding of mortalin to complement C9 and C8 occurs through an ionic interaction that is nucleotide-sensitive. We suggest that to express its full protective effect from CDC, mortalin must first reach the mitochondria. In addition, mortalin can potentially target the C8 and C9 complement components through its ATPase domain and inhibit C5b-9 assembly and stability.
Introduction
The humoral innate immune system exerts its cytotoxic activity through the action of the complement membrane attack complex (MAC),2 comprising the complement proteins C5b, C6, C7, C8, and C9 (1). Complement-dependent cytotoxicity (CDC) is inflicted following insertion of the MAC into the target cell membrane. CDC is essential for ridding the body of infective pathogenic organisms and for successful antibody-based cancer immunotherapy (2), yet accidental deposition of MAC is implicated in pathogenesis of numerous life-threatening ailments (3). To evade CDC, cells have developed multiple strategies that are targeted at the complement activation cascades and at the intracellular cell death signals (4). Cells can also evade CDC by elimination of the MAC from their surface by using outward and inward membrane vesicles release. Exovesiculation of the MAC was demonstrated in several normal and tumor cells (5–9). The presence of elevated levels of cholesterol and diacylglycerol in shed membrane vesicles containing the MAC suggests a selective sorting process (7). Endocytosis of the MAC is caveolin- and dynamin-dependent (10).
Mortalin/GRP75, the mitochondrial heat shock protein 70, assists in import and folding of mitochondrial proteins (11–13), protects cells from glucose and serum deprivation and reactive oxygen species accumulation (14, 15), and is involved in cell senescence (16). Expression of mortalin is elevated in cancer cells and correlates with cancer progression and poor patient survival (17, 18).
Mortalin plays also a role in cell resistance to CDC and in MAC elimination (19, 20). K562 cells that have been exposed to sublytic doses of complement shed membrane vesicles that contain the complement MAC as well as mortalin. This process of MAC exovesiculation depends to a large extent on mortalin. Blocking of mortalin with MKT-077, a cationic rhodacyanine dye analog that binds to mortalin and inhibits its activity, sensitizes K562 as well as HCT human colon carcinoma cells to CDC (20). An inverse correlation between mortalin level in cells and cell sensitivity to CDC implicates mortalin in protection from complement. Addition of anti-mortalin antibodies to cells under a complement attack reduces release of MAC and mortalin and augments cell death (19). Mortalin can bind directly to two components of the C5b-9 complex, complement C8 and C9 (19). Interestingly, exogenously added recombinant human mortalin significantly inhibits hemolysis of rabbit erythrocytes by complement as well as polymerization of purified C9 (20).
In this study we studied further the protective effect of mortalin. We found that overexpression of mortalin reduces MAC deposition and CDC, whereas inhibition of mortalin synthesis by RNAi promotes MAC binding to the cell surface. Mortalin has two principal domains, the N-terminal ATPase and C-terminal substrate-binding domains (SBDs). Unfolded proteins bind to the SBD, whereas the ATPase domain synchronizes the chaperone activity (21). We show here that the ATPase domain of mortalin, but not the SBD, binds and inhibits C9 polymerization, and that the binding is nucleotide-sensitive.
EXPERIMENTAL PROCEDURES
Sera and Reagents
Normal human serum (NHS), used as a source for complement proteins, was prepared from healthy individuals after obtaining approval of the Ethical Committee of Tel Aviv University. Heat inactivation of NHS (HIS) was performed at 56 °C for 30 min. Purified human C7, C8, and C9 proteins and complement C9-depleted human serum (C9D-NHS) were purchased from Complement Technology Inc. (Tyler, TX). A polyclonal antiserum directed to K562 cells was prepared in rabbits and mice. Anti-human C9 antibodies were prepared in goats. Mouse anti-GRP75 (mortalin) (clone S52A-42) antibodies were purchased from StressMarq (Victoria, BC, Canada), mouse anti-actin (clone C4) antibodies from Chemicon International (Temecula, CA), mouse anti-His antibodies from Clontech (Mountain View, CA), mouse anti-GFP antibodies from Santa Cruz Biotechnololgy (Santa Cruz, CA), and peroxidase-conjugated goat anti-mouse IgG and Cy3-conjugated goat anti-mouse IgG (H+L) from Jackson ImmunoResearch (West Grove, PA). Alexa Fluor protein labeling kits (AF555 and AF488) and MitoTracker were purchased from Molecular Probes (Eugene, OR). ATP, AMP-PNP, and propidium iodide were purchased from Sigma and ADP from Fluka (Buchs, Switzerland).
Cell Culture and Cell Lysis
K562, a human erythroleukemic cell line, was cultured in RPMI 1640 medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Invitrogen), 1% glutamine, 2% pyruvate, and an antibiotics mixture (Bio-Lab, Jerusalem, Israel). Cytotoxicity assays were performed as described before (22). Briefly, cells were incubated with diluted anti-K562 antiserum (rabbit or mouse) for 30 min at 4 °C and then with complement (NHS or HIS, final: 50%) for 60 min at 37 °C. Cell lysis was determined after propidium iodide staining and analysis by flow cytometry (FACSort; BD Biosciences). Specific percentage cell lysis was calculated by subtraction of cell death observed after treatment with HIS from that of NHS-treated cells (NHS-HIS/100-HIS). In cell imaging experiments, cells were treated first with a sublytic dose of antibody (yielding 10–20% cell lysis) for 30 min at 4 °C and then with C9D-NHS supplemented with C9-AF555 or C9-AF488 (to 70 μg/ml) for 10 min at 37 °C. Then, the cells were washed and observed under a confocal fluorescence microscope (see below).
Mortalin Plasmids and Recombinant Proteins
Human mortalin and mortalin lacking the 51 N-terminal amino acids corresponding to the mitochondrial targeting sequence (mortalinΔ51) were cloned into a pEGFP-N1 vector (Clontech) with a C-terminal EGFP. The vectors were introduced into the cells by electroporation. Full-length mortalin, mortalinΔ51, mortalin SBD, and mortalin ATPase domain were also cloned into pETDuet prokaryotic vector (Novagen, Merck) with an N-terminal His8 tag. Escherichia coli bacteria transformed with the latter plasmids were induced overnight with 1 mm isopropyl β-d-thiogalactopyranoside at 16 °C. Recombinant His-tagged mortalinΔ51, mortalin SBD, and mortalin ATPase domain were purified by anion exchange chromatography and over nickel-agarose columns (23). Purified recombinant mortalin V482F that has a mutation in its peptide-binding region and lost its p53 binding was prepared by Iosefson and Azem (23).
RNA Interference
K562 cells were transiently transfected with specific siRNA directed to mortalin (AUUGUAUUCUCCGAGUCAGUU) or with nonspecific control siRNA (ACUCUAUCUGCACGCUGACUU) (Dharmacon, Lafayette, CO) using Oligofectamine (Invitrogen). In brief, the cells were washed with serum-free medium and plated in a 24-well plate (50 × 103 cells/well). siRNA (300 nm) mixed with Oligofectamine (according to the manufacturer's instructions) was added to the cells. Cells treated without siRNA (NT) were also used as control. Cells were then incubated in culture medium for 48 h before being tested.
Western Blotting
Cell lysates were subjected to SDS-PAGE under reducing conditions (150 mm dithiothreitol (DTT)) in a 10% acrylamide gel and then transferred onto a nitrocellulose membrane (Schleicher & Schuell). The membrane was blocked with 5% skim milk (Tnuva, Rehovot, Israel) in Tris-buffered saline containing 0.05% Tween 20 (TBST) for 1 h at room temperature. The membrane was then treated with mouse anti-mortalin antibodies, mouse anti-actin antibodies, or mouse anti-EGFP antibodies followed by peroxidase-conjugated goat anti-mouse IgG. Bands were developed with an enhanced chemiluminescence reagent (Pierce) and exposed to a SuperRX film (Fuji, Tokyo).
Mortalin and C9 Imaging in Cells by Confocal Microscopy
Complement C9 was imaged in cells as described before (9). To image mortalin, cells were transfected with pEGFP-mortalin by electroporation. Then, transfected cells were incubated with anti-K562 antibodies and C9-depleted human serum supplemented with C9-AF555 (human C9 labeled with Alexa Fluor 555 (Molecular Probes)) for 10 min at 37 °C. Next, the cell were washed with HBSS and placed on a 22-mm coverslip (Assistant, Sondheim, Germany). Alternatively, nontransfected cells were treated with antibody and C9-depleted serum supplemented with C9-AF488 (human C9 labeled with Alexa Fluor 488) for 10 min at 37 °C. Next, the cells were fixed with 1% paraformaldehyde and permeabilized with saponin. The permeabilized cells were immune-treated with anti-mortalin antibody followed by a second Cy3-labeled antibody (Jackson ImmunoResearch). Labeled cells were analyzed under a Zeiss Laser Confocal Fluorescence Microscope C-LSM 510 (Oberkochen, Germany). Images and merged images were obtained with the LSM software (Carl Zeiss, GmbH, Germany). Images were processed further for display by using ImageJ (National Institutes of Health).
C9 Polymerization Assay
Purified human C9 (2 μg) was incubated with 42 or 100 μm ZnCl2 in 20 mm Tris (pH 7.2) for 2 h at 37 °C. C9 is known to undergo, under these conditions, accelerated and spontaneous polymerization (24). To test the effect of mortalin and its purified domains on C9 polymerization, C9 was pretreated with the recombinant proteins or BSA as control (2 μg) for 15 min at 37 °C and then with ZnCl2 for 2 h at 37 °C. The proteins were subjected to SDS-PAGE on a 3–10% acrylamide gradient gel under reducing conditions, and the gel was stained with Coomassie Blue.
Sucrose Gradient Sedimentation
To test the binding of mortalin and its purified domains to complement C9, purified human C9 (1 μg) was incubated with recombinant mortalin, SBD, or ATPase domain (2 μg) for 1 h at 37 °C. The samples were layered on top of a 13-ml 10–30% sucrose density gradient in buffer and were subjected to high speed centrifugation for 18 h at 40,000 rpm at 4 °C. Fractions (300 μl) were collected from the gradient top with an Auto Densi-Flow density gradient fractionator (Labconco, Kansas City, MO) and a Pharmacia Biotech RediFrac fraction collector. Samples (100 μl) from each fraction were blotted to a nitrocellulose membrane in a Bio-Dot Microfiltration Apparatus (Bio-Rad) and developed with mouse anti-His antibodies and peroxidase-conjugated goat anti-mouse IgG.
Analysis of Protein Interactions
Interaction between two proteins was measured by using an enzyme-linked immunosorbent assay (ELISA). Purified human C7, C8, or C9 (0.5 μg) was attached overnight at 4 °C to wells of a 96-well plate (Nunc, Rochester, NY). After the washes, His-tagged recombinant mortalin, SBD, or ATPase domain (1 μg) was added to the wells for 1 h at 37 °C. The wells were then washed, treated with mouse anti-His antibody (diluted 1:1000) for 1 h at 37 °C and peroxidase-labeled goat anti-mouse IgG (diluted 1:10,000) for 1 h at room temperature, developed with O-phenylenediamine (OPD) (Sigma), and analyzed in an ELISA reader at 492 nm. In competition assays, mortalin and its domains were preincubated with C8 or C9 before addition to C9-coated wells.
Statistical Analysis
Statistical significance was analyzed by using a two-sided unpaired Student's t test. Results are expressed as arithmetic mean ± S.D. Statistical significance was assumed when p < 0.05.
RESULTS
Mortalin Protects K562 cells from Complement-dependent Cytotoxicity
To examine the significance of mortalin to the protection of K562 cells from complement-dependent cytotoxicity, cells were transfected with mortalin-EGFP or with EGFP as control. After a 48-h incubation, analysis under a confocal microscope confirmed that mortalin-EGFP was located in the mitochondria (Fig. 1A). In contrast, EGFP was distributed evenly throughout the cells. Transfected cells were treated with anti-K562 antibodies for 30 min at 4 °C and then with NHS or HIS for 60 min at 37 °C. Notably, mortalin overexpression reduced cell death from 79 to 11% (Fig. 1B). We next examined whether C5b-9 deposition on the cell surface was affected by overexpression of mortalin. K562 cells transfected for 48 h with mortalin-EGFP were treated with anti-K562 antibodies and then with C9D-NHS supplemented with C9-AF555 for 10 min at 37 °C, as described under “Experimental Procedures.” Next, the cells were observed under a confocal microscope (Fig. 2A). The levels of bound C5b-9 and of mortalin-EGFP were quantified. Regression analysis of the relationship between the two variables showed that cells expressing higher mortalin-EGFP levels had lower C5b-9 deposition relative to cells expressing low levels of mortalin-EGFP (Fig. 2B). The strength of the correlation was intermediary (R2 = 0.72); however, this was probably due to a stepwise rather than gradual effect of mortalin on C9 deposition. By dividing the cell population into cells expressing low or high mortalin levels, the difference in C9 deposition between the two subpopulations became highly significant (p < 0.005) (Fig. 2C).
FIGURE 1.
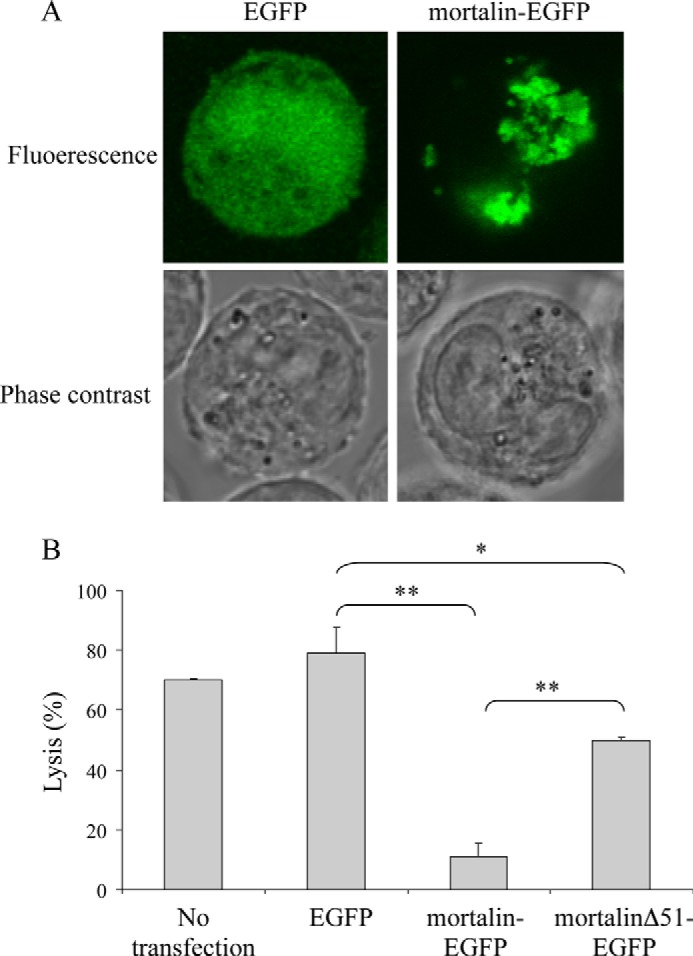
Overexpression of mortalin protects K562 cells from complement-dependent cytotoxicity. A, K562 cells transfected for 48 h with a mortalin-EGFP or EGFP plasmid were analyzed under a confocal microscope. A representative cell of each is shown in fluorescence and bright field. Transfected or nontransfected cells were treated with anti-K562 antibodies and NHS for 1 h at 37 °C. B, next, the cells were washed, mixed with propidium iodide, and analyzed by flow cytometry. Percentage of cell lysis was determined based on the percentage of cells labeled with propidium iodide. For correction of specific cell lysis, percentage lysis after treatment with HIS was subtracted from that of cells treated with NHS. *, p < 0.01; **, p < 0.001. Error bars, S.D.
FIGURE 2.

Overexpression of mortalin reduces the amount of bound C5b-9 on the cell surface. K562 cells, 48 h after transfection with a mortalin-EGFP plasmid, were treated with anti-K562 antibodies and then with C9D-NHS supplemented with C9-AF555 for 10 min at 37 °C. Next, the cells were washed and observed under a confocal microscope, and 30 randomly selected cell images were taken. A, a representative image is shown. Levels of C9-AF555 bound to the cell surface (representing C5b-9) and of mortalin-EGFP in the cell images were quantified by densitometry and analyzed with ImageJ. B, regression analysis for estimating the relationship between C9 deposition and the level of mortalin is shown. A.U., arbitrary units. C, levels of C9 deposition in cells having low (0–5 A.U.) or high (>5 A.U.) mortalin-EGFP level are shown. *, p < 0.005. Error bars, S.D.
The effect of down-regulation of mortalin level on complement-dependent cytotoxicity was next examined. K562 cells were transfected with specific siRNA directed to mortalin or with a control nonspecific siRNA (NS) or without siRNA (NT). After a 48-h incubation, cell lysates were analyzed by SDS-PAGE followed by Western blotting and detection with anti-mortalin antibodies. As shown in Fig. 3A, mortalin-specific siRNA significantly reduced mortalin level in the cells. Transfected cells were treated with anti-K562 antibodies for 30 min at 4 °C and then with C9D-NHS supplemented with C9-AF488 for 10 min at 37 °C. Then, the cells were washed, fixed, permeabilized, and labeled with anti-mortalin antibody and a second Cy3-labeled antibody. The cells were observed under a confocal microscope (Fig. 3B), and the amount of C9-AF488 and of mortalin-Cy3 in the cells was quantified. Cells with lower mortalin-Cy3 level had more C9-AF488 on them (Fig. 3C). Thus, mortalin silencing enabled elevated deposition of the complement MAC on the cells.
FIGURE 3.

Mortalin inhibition by siRNA increases the amount of cell bound C9. K562 cells were transfected with specific siRNA directed to mortalin. Cells treated with nonspecific control siRNA (NS) or without siRNA (NT) were used as control. A, 48 h after transfection, the cells were lysed with sample buffer and analyzed by SDS-PAGE and Western blotting with anti-mortalin or anti-actin antibodies. K562 cells transfected with mortalin-siRNA were treated, 48 h after transfection, with anti-K562 antibodies and then with C9D-NHS supplemented with C9-AF488 for 10 min at 37 °C. B, next, the cells were washed, fixed, permeabilized, and labeled with anti-mortalin antibody and a secondary Cy3-labeled antibody. The cells were observed under a confocal microscope. C, levels of C9-AF488 on the cell surface and of mortalin-Cy3 were quantified by densitometry and analyzed with ImageJ in 30 randomly selected cell images. The level of C9-AF488 in cells having low (0–5 arbitrary units (A.U.)) and high (>5 arbitrary units) mortalin-Cy3 is shown. *, p < 0.005. Error bars, S.D.
Mortalin Lacking the Mitochondrial Targeting Sequence Has a Lower Protective Activity
Mortalin has a 51-amino acid long mitochondrial-targeting signal peptide at its N terminus. This signal peptide is efficiently removed in the mitochondria; thus, all mortalin observed in K562 cells lacks it (Fig. 4A). Recombinant full-length mortalin and mortalinΔ51 (mortalin lacking its mitochondrial targeting sequence) expressed in E. coli were also analyzed by SDS-PAGE and Western blotting with anti-mortalin antibodies (Fig. 4A). The size of K562 mortalin was similar to that of recombinant mortalinΔ51, and the blot showed no unprocessed mortalin. The importance of mortalin trafficking through mitochondria to cell resistance was next examined. EGFP-tagged full-length mortalin and mortalin lacking its signal peptide (mortalinΔ51-EGFP) were cloned and expressed in K562 cells. Analysis by SDS-PAGE and Western blotting showed the expressed recombinant proteins in cell lysates (Fig. 4B). The location of these proteins in the cells was analyzed under a confocal microscope. As shown in Fig. 4C, absence of the mitochondria targeting sequence in mortalinΔ51-EGFP led to its expression in the cytoplasm rather than in mitochondria; this was in sharp contrast to the mitochondrial localization of mortalin-EGFP. To demonstrate that mortalin in located mainly in mitochondria, K562 cells transfected with mortalin-EGFP were labeled with MitoTracker. Co-localization of mortalin with mitochondria was observed (Fig. 4D). Next, the transfected cells were subjected to CDC. Overexpression of full-length mortalin reduced cell death from 79 to 11%, whereas overexpression of mortalinΔ51 reduced cell death only to 50% (Fig. 1B). Thus, cells overexpressing recombinant mortalin which lack mitochondrial targeting sequence were more sensitive to CDC relative to cells transfected with full-length mortalin, suggesting that trafficking of mortalin through the mitochondria is essential to its protective activity.
FIGURE 4.
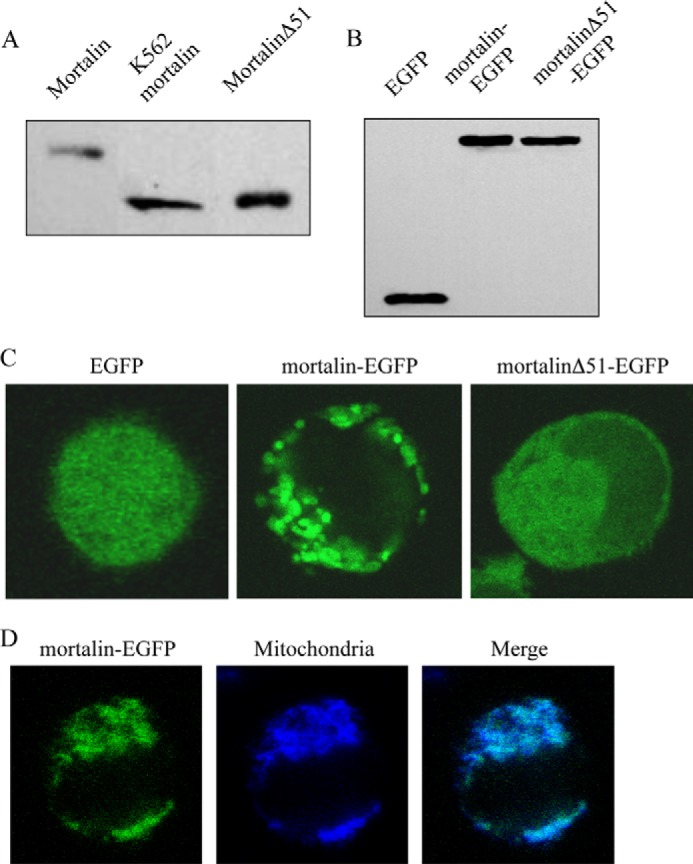
Expression of mortalinΔ51 in bacteria and in K562 cells. A, full-length mortalin or mortalinΔ51 in pETDuet in E. coli were induced by isopropyl β-d-thiogalactopyranoside, and the bacteria were lysed with sample buffer and analyzed by SDS-PAGE and Western blotting with anti-mortalin antibody. K562 cell lysate was analyzed together with them. B, K562 cells transfected for 48 h with pEGFP-N1 expressing mortalin-EGFP, mortalinΔ51-EGFP, or EGFP alone were lysed with sample buffer and analyzed by SDS-PAGE and Western blotting with anti-EGFP antibody. C, transfected K562 cells were analyzed under a confocal microscope, and a representative cell is shown. D, transfected K562 cells with mortalin-EGFP were treated with MitoTracker (25 nm) for 10 min at 37 °C to label their mitochondria. A confocal microscope image of a representative cell shows co-localization of mortalin with mitochondria.
Interaction of Mortalin ATPase Domain with C9
As we have reported previously, mortalin has the capacity to bind complement C8 and C9 and to inhibit Zn2+-induced C9 polymerization (20). To examine which domain of mortalin, the ATPase domain or SBD, expresses the inhibitory activity, we cloned and purified the mortalin domains as recombinant proteins. Binding of the mortalin domains to complement proteins was examined in an ELISA and by sucrose gradient sedimentation. As shown in Fig. 5A, the ATP-binding domain, but not the substrate-binding domain, bound to C9 and C8 but only weakly to C7. To exclude the possibility that mortalin binds to epitopes exposed only on plastic-attached C9, we tested the capacity of native C9 and C8 to compete with this binding. Both native C9 and C8 competed in a dose-dependent manner with the binding of mortalin and mortalin ATPase domain to plastic-attached C9 (Fig. 5B). In addition, binding in solution of mortalin to C9 and of mortalin ATPase domain to C9 was demonstrated in a pulldown experiment through sucrose gradient (Fig. 6). Human C9 protein was mixed with recombinant mortalin, SBD, or ATPase domain, layered on top of a sucrose density gradient, and spun down for 18 h at 40,000 rpm. Analysis by dot blotting of samples collected from the gradient showed that both mortalin and its ATPase domain, but not the SBD, were pulled down by C9 into denser sucrose concentrations.
FIGURE 5.

C9 binds to the ATPase domain of mortalin-ELISA. A, C9, C8, and C7 adsorbed onto microtiter plate wells were incubated with His-tagged recombinant full-length mortalin, SBD, or ATPase domain. The wells were washed, treated with anti-His antibody, and peroxidase-labeled second antibody, developed with OPD and analyzed in an ELISA reader. *, p < 0.005; **, p < 0.001 between ATPase domain and SBD. Error bars, S.D. B, microtiter plate wells were coated with C9 overnight at 4 °C. His-tagged mortalin or its domains were preincubated with C8 or C9 (0.25 μm, C81 and C91 or 0.5 μm, C82 and C92) for 1 h at 37 °C. The mixtures were then added to the C9-coated wells. Binding of His-tagged mortalin and its domains was quantified with anti-His antibody as in A. *, p < 0.05; **, p < 0.005; ***, p < 0.001 relative to control (− competitor).
FIGURE 6.

C9 binds to the ATPase domain of mortalin-sucrose density gradient. A, C9 (1 μg) was mixed in an Eppendorf tube with recombinant His-tagged mortalin, SBD, or ATPase domain (2 μg) for 1 h at 37 °C. The samples were layered on top of 13-ml 10–30% sucrose density gradients and were sedimented for 18 h at 40,000 rpm at 4 °C. Fractions (300 μl) were collected from the gradient top and analyzed by dot blotting with anti-His antibody. B, density of each scanned dot was quantified with ImageJ software. Plotted dot density of mortalin or mortalin domains in fractions 1–16 is shown. A.U., arbitrary units.
Next, a C9 polymerization assay was performed as described (24). Mortalin or its domain was mixed with C9 for 15 min at 37 °C; ZnCl2 was then added, and after an additional 2-h incubation at 37 °C the samples were analyzed by SDS-PAGE. As shown in Fig. 7, the ATPase domain of mortalin inhibited Zn2+-induced C9 polymerization whereas the SBD had no effect on C9 polymerization. The claim that mortalin interacts with C9 through its ATPase domain was further supported by showing that recombinant mortalin V482F, which has a nonfunctional substrate-binding domain, inhibited C9 polymerization (Fig. 7B) and could bind directly to C9 (Fig. 7C).
FIGURE 7.

Inhibition of C9 polymerization by the ATPase domain of mortalin. A, C9 was mixed with recombinant full-length mortalin, mortalin C-terminal SBD, mortalin N-terminal ATPase domain, or BSA as control for 15 min at 37 °C and then incubated with 42 or 100 μm ZnCl2 for 2 h at 37 °C. The samples were subjected to SDS-PAGE on a 3–10% acrylamide gradient gel and stained with Coomassie Blue. The bands of polyC9, monomeric C9, mortalin, SBD, and ATPase domain are indicated; the bands of C9 and BSA did not separate. B, C9 was mixed with recombinant full-length mortalin, mortalin-V482F (mV482F), or BSA as control for 15 min at 37 °C and then incubated with 42 or 100 mm ZnCl2 for 2 h at 37 °C. C9 polymerization was analyzed as described in A. C, mortalin or mV482F was attached to microtiter plate wells and then incubated with C9 for 1 h in 37 °C. The wells were then washed, treated with anti-C9 antibody and peroxidase-labeled second antibody, developed with OPD, and analyzed in an ELISA reader.
Factors Affecting the Interaction between Mortalin and C9
To characterize the binding between mortalin and C9, the effect of salt on the binding was first examined. Briefly, plastic-attached C9 was incubated with His-tagged mortalin in the presence of increasing NaCl concentrations for 1 h in 37 °C. Binding of mortalin was quantified by ELISA. As shown in Fig. 8A, binding of mortalin to C9 was inhibited by NaCl, indicating an ionic interaction between mortalin and C9. The possibility that the salt has removed some of the plastic-attached C9 was excluded in a direct C9 ELISA (Fig. 8B).
FIGURE 8.
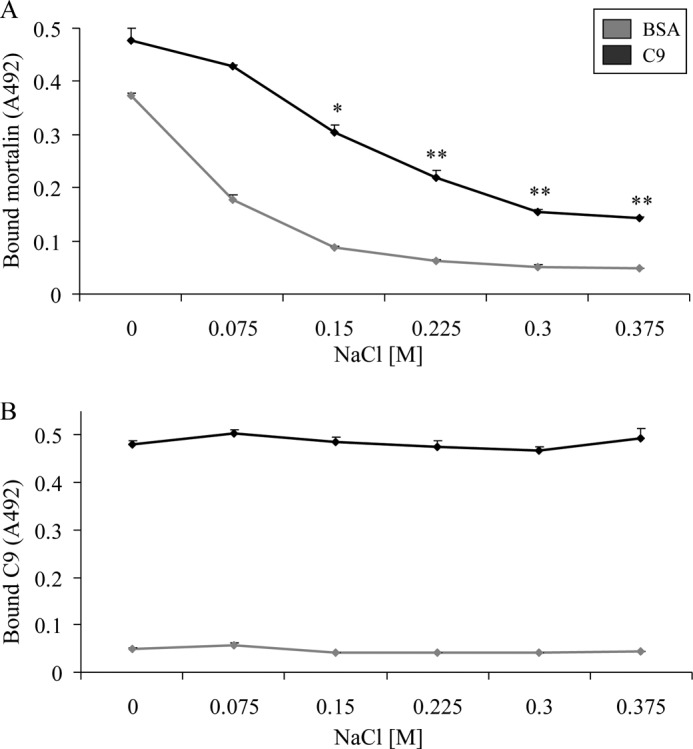
NaCl inhibits binding of mortalin to C9. A, human C9 (or BSA as control) (0.5 μg) was attached overnight to microtiter plate wells and then incubated with His-tagged mortalin in the presence of NaCl (75–375 mm) for 1 h in 37 °C. The wells were then washed, treated with anti-His antibody and peroxidase-labeled secondary antibody, developed with OPD, and analyzed in an ELISA reader. B, C9 or BSA (0.5 μg) attached to microtiter plate wells was incubated in the presence of NaCl (75–375 mm) without mortalin for 1 h in 37 °C. The wells were then washed, treated with anti-C9 antibody and peroxidase-labeled secondary antibody, developed with OPD, and analyzed in an ELISA reader. *, p < 0.05; **, p < 0.005 relative to control (C9 and mortalin without added NaCl). Error bars, S.D.
Because the ATPase domain of mortalin binds to C9, the possibility that nucleotides may affect this interaction was examined. Binding of His-tagged mortalin to plastic-attached C9 in the presence of various concentrations of nucleotides was tested (Fig. 9). As shown, ATP, ADP, and AMP-PNP inhibited binding of mortalin to C9. ID50 (50% inhibition) was achieved by approximately 200 μm ATP, 1100 μm ADP, and 600 μm AMP-PNP (calculated from regression line formulas). This strongly suggests involvement of the ATP-binding site in mortalin in its interaction with C9.
FIGURE 9.
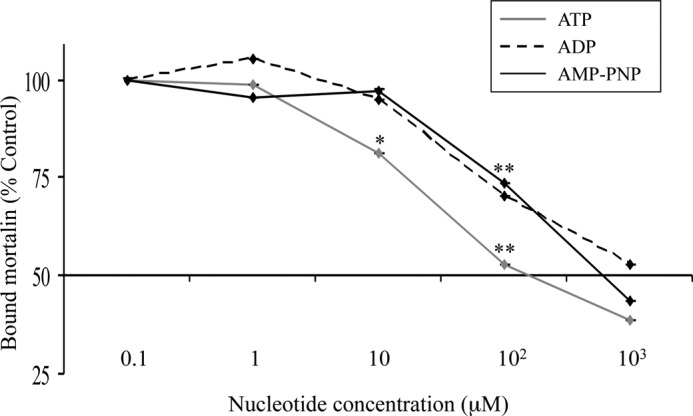
Nucleotides inhibit binding of mortalin to complement C9. Human C9 attached to microtiter plate wells was incubated with His-tagged mortalin in the presence of ATP, ADP, or AMP-PNP (0.1–103 μm) for 1 h in 37 °C. The wells were then washed, treated with anti-His antibody and peroxidase-labeled secondary antibody, developed with OPD, and analyzed in an ELISA reader. Results are presented as percentage control of binding at 0.1 μm nucleotide. *, p < 0.05; **, p < 0.005 relative to 0.1 μm same nucleotide.
DISCUSSION
The complement MACs exert dose-dependent effects on cells. At sufficiently large, lytic MAC doses, the outcome is an immediate programmed necrotic cell death (25, 26). In contrast, at lower, sublytic or nonlytic doses, the MAC can induce diverse nontoxic cellular responses (26–28), including induction of an elevated resistance to CDC (22). Mortalin is one of the cell protectors that restrain the lytic activity of the MAC. It appears to play a role in the process of MAC elimination by exovesiculation (19, 20), yet its mechanism of action is still unclear. Results presented here support the claim that mortalin restricts the MAC copy number deposited on the cell surface. Overexpression of mortalin is leading to fewer MACs deposited on the cells (Fig. 2), accompanied by a reduced sensitivity to CDC (Fig. 1). However, lowering the amount of mortalin in cells by RNAi enhances the quantity of cell-bound MACs (Fig. 3). The transient transfection of mortalin siRNA into K562 cells had no effect on the level of CD59 expressed on the cells (data not shown).
The plausible complement target proteins recognized by mortalin are the C8 and C9 components of the C5b-9 complex. Native mortalin in whole cell extracts binds to purified C9 and is shed from cells in vesicles together with C9 (19). As shown here, purified recombinant mortalin binds through ionic interactions directly to C8 and C9 (Figs. 5 and 8). By binding to C8 and C9, mortalin may possibly restrict the number of MAC on the cells in two ways: (a) facilitation of MAC elimination from the cell surface, and/or (b) disruption of MAC assembly. The potential capacity of mortalin to block MAC assembly was indicated by results showing that mortalin can inhibit zinc-induced polymerization of purified C9 and that exogenously added mortalin can block complement-mediated hemolysis of red blood cells (20). Whatever the exact mechanism(s) of protection by mortalin is, there is increasing evidence that it plays a role in cell resistance to CDC. In support, MKT-077, a mortalin inhibitor, was shown to inhibit binding of mortalin to C9 and to increase cell sensitivity to CDC in cells containing mortalin but not in mortalin-deficient cells (20).
Mortalin is located in K562 cells primarily in the mitochondria. Mortalin-EGFP labels mitochondria distributed throughout the cell. Interestingly, analysis by Western blotting shows that virtually all mortalin expressed in K562 cells lacks its mitochondrial signal peptide. This indicates that mortalin is translated in the vicinity of the mitochondria and its mitochondrial targeting sequence is rapidly and efficiently removed. Recombinant mortalin lacking the mitochondrial targeting sequence is not expressed in the mitochondria but is diffusely expressed throughout the cytoplasm (Fig. 4). Unlike cells overexpressing intact mortalin mRNA, cells overexpressing a mortalin which lacks its mitochondrial targeting sequence are less protected from CDC (Fig. 1). This suggests that the primary location of mortalin in mitochondria is essential to its protective activity against the MAC. It is possible that mortalin may have in the cytoplasm binding proteins or physiological conditions that reduce its complement protective activity. Investigations on the mode of action of mortalin, a mitochondrial matrix protein, on the MAC, possibly at the plasma membrane level, are ongoing. Mortalin may perhaps respond to MAC-induced stress by exiting the mitochondria, singly or as part of a protein complex, and moving to the plasma membrane where it interacts with intracellular domains of membrane-penetrating C9. Alternatively, mitochondrial vesicles containing mortalin may perhaps reach the plasma membrane and release mortalin to the extracellular milieu where it can inhibit MAC formation and facilitate its removal from the cells.
Like all Hsp70 family proteins, mortalin has two principal domains, an N-terminal ATPase and a C-terminal SBD, joined by a protease-sensitive site (21). Its chaperone activities require ATP hydrolysis. The ATPase domains of the Hsp70 proteins are conserved whereas the SBDs show a greater sequence variation leading to the diversification of the client proteins and substrate specificities. Based on the predicted structure of mortalin (21, 29), we constructed two potentially functional domains of mortalin, the ATPase domain (amino acids 48–436) and the SBD (amino acids 434–679). Binding of the two purified recombinant domains of mortalin to C9 and C8 was examined. Interestingly, intact mortalin and its purified ATPase domain, but not the SBD, bind to human C9 and C8 (Figs. 5 and 6). Accordingly, the ATPase domain, but not the SBD, inhibits zinc-induced C9 polymerization (Fig. 7). Furthermore, recombinant mutant mortalin with a nonfunctional substrate-binding domain, mortalin V482F, was shown to bind to C9 (Fig. 7) and to inhibit its polymerization (data not shown), whereas it failed to bind p53 (23). Through its ATPase domain, mortalin can recycle between ATP-bound and ADP-bound states. Binding of client proteins to mortalin through the SBD largely depends on this nucleotide binding activity. Interestingly, the interaction between mortalin and C9, through the ATPase domain, was found to be nucleotide-sensitive. This binding is inhibited by ATP, ADP, and AMP-PNP, in a dose-dependent manner (Fig. 9), suggesting that the association between mortalin and C9 is not related to the chaperone activity of mortalin. In contrast, it appears that C9 binds directly to the ATP-binding site of mortalin. This may neutralize the chaperone activity in C9-bound mortalin. Further investigation is required to determine whether and where do mortalin and C9 interact within complement-attacked cells.
Acknowledgment
We thank Dr. Abdussalam Azem, The George S. Wise Faculty of Life Sciences, Tel Aviv University, for support and advice.
This work was supported in part by grants from the Israel Science Foundation, the German-Israel Science Foundation, and the Israel Cancer Association.
- MAC
- complement membrane attack complex
- AF488
- Alexa Fluor 488
- AF555
- Alexa Fluor 555
- AMP-PNP
- 5′-adenylyl-β,γ-imidodiphosphate
- C5b-9
- complement terminal complex composed of C5b, C6, C7, C8 and C9
- C9D
- C9-depleted
- CDC
- complement-dependent cytotoxicity
- EGFP
- enhanced GFP
- HIS
- heat-inactivated serum
- mortalinΔ51
- mortalin lacking the mitochondrial targeting sequence
- NHS
- normal human serum
- OPD
- O-phenylenediamine
- SBD
- substrate-binding domain of mortalin.
REFERENCES
- 1. Müller-Eberhard H. J. (1986) The membrane attack complex of complement. Annu. Rev. Immunol. 4, 503–528 [DOI] [PubMed] [Google Scholar]
- 2. Morris J. C., Waldmann T. A. (2009) Antibody-based therapy of leukaemia. Expert Rev. Mol. Med. 11, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Walport M. J. (2001) Complement: first of two parts. N. Engl. J. Med. 344, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 4. Gancz D., Fishelson Z. (2009) Cancer resistance to complement-dependent cytotoxicity (CDC): problem-oriented research and development. Mol. Immunol. 46, 2794–2800 [DOI] [PubMed] [Google Scholar]
- 5. Carney D. F., Koski C. L., Shin M. L. (1985) Elimination of terminal complement intermediates from the plasma membrane of nucleated cells: the rate of disappearance differs for cells carrying C5b-7 or C5b-8 or a mixture of C5b-8 with a limited number of C5b-9. J. Immunol. 134, 1804–1809 [PubMed] [Google Scholar]
- 6. Scolding N. J., Morgan B. P., Houston W. A., Linington C., Campbell A. K., Compston D. A. (1989) Vesicular removal by oligodendrocytes of membrane attack complexes formed by activated complement. Nature 339, 620–622 [DOI] [PubMed] [Google Scholar]
- 7. Morgan B. P., Dankert J. R., Esser A. F. (1987) Recovery of human neutrophils from complement attack: removal of the membrane attack complex by endocytosis and exocytosis. J. Immunol. 138, 246–253 [PubMed] [Google Scholar]
- 8. Stein J. M., Luzio J. P. (1991) Ectocytosis caused by sublytic autologous complement attack on human neutrophils: the sorting of endogenous plasma-membrane proteins and lipids into shed vesicles. Biochem. J. 274, 381–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moskovich O., Fishelson Z. (2007) Live cell imaging of outward and inward vesiculation induced by the complement C5b-9 complex. J. Biol. Chem. 282, 29977–29986 [DOI] [PubMed] [Google Scholar]
- 10. Moskovich O., Herzog L. O., Ehrlich M., Fishelson Z. (2012) Caveolin-1 and dynamin-2 are essential for removal of the complement C5b-9 complex via endocytosis. J. Biol. Chem. 287, 19904–19915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhattacharyya T., Karnezis A. N., Murphy S. P., Hoang T., Freeman B. C., Phillips B., Morimoto R. I. (1995) Cloning and subcellular localization of human mitochondrial Hsp70. J. Biol. Chem. 270, 1705–1710 [DOI] [PubMed] [Google Scholar]
- 12. Koehler C. M. (2004) New developments in mitochondrial assembly. Annu. Rev. Cell Dev. Biol. 20, 309–335 [DOI] [PubMed] [Google Scholar]
- 13. Wiedemann N., Frazier A. E., Pfanner N. (2004) The protein import machinery of mitochondria. J. Biol. Chem. 279, 14473–14476 [DOI] [PubMed] [Google Scholar]
- 14. Liu Y., Liu W., Song X. D., Zuo J. (2005) Effect of GRP75/mtHsp70/PBP74/mortalin overexpression on intracellular ATP level, mitochondrial membrane potential and ROS accumulation following glucose deprivation in PC12 cells. Mol. Cell. Biochem. 268, 45–51 [DOI] [PubMed] [Google Scholar]
- 15. Taurin S., Seyrantepe V., Orlov S. N., Tremblay T. L., Thibault P., Bennett M. R., Hamet P., Pshezhetsky A. V. (2002) Proteome analysis and functional expression identify mortalin as an antiapoptotic gene induced by elevation of [Na+]i/[K+]i ratio in cultured vascular smooth muscle cells. Circ. Res. 91, 915–922 [DOI] [PubMed] [Google Scholar]
- 16. Wadhwa R., Takano S., Taira K., Kaul S. C. (2004) Reduction in mortalin level by its antisense expression causes senescence-like growth arrest in human immortalized cells. J. Gene Med. 6, 439–444 [DOI] [PubMed] [Google Scholar]
- 17. Dundas S. R, Lawrie L. C., Rooney P. H., Murray G. I. (2005) Mortalin is overexpressed by colorectal adenocarcinomas and correlates with poor survival. J. Pathol. 205, 74–81 [DOI] [PubMed] [Google Scholar]
- 18. Wadhwa R., Takano S., Kaur K., Deocaris C. C., Pereira-Smith O. M., Reddel R. R., Kaul S. C. (2006) Up-regulation of mortalin/mtHsp70/Grp75 contributes to human carcinogenesis. Int. J. Cancer 118, 2973–2980 [DOI] [PubMed] [Google Scholar]
- 19. Pilzer D., Fishelson Z. (2005) Mortalin/GRP75 promotes release of membrane vesicles from immune attacked cells and protection from complement-mediated lysis. Int. Immunol. 17, 1239–1248 [DOI] [PubMed] [Google Scholar]
- 20. Pilzer D., Saar M., Koya K., Fishelson Z. (2010) Mortalin inhibitors sensitize K562 leukemia cells to complement-dependent cytotoxicity. Int. J. Cancer 126, 1428–1435 [DOI] [PubMed] [Google Scholar]
- 21. Deocaris C. C., Kaul S. C., Wadhwa R. (2006) On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones 11, 116–128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Reiter Y., Ciobotariu A., Fishelson Z. (1992) Sublytic complement attack protects tumor cells from lytic doses of antibody and complement. Eur. J. Immunol. 22, 1207–1213 [DOI] [PubMed] [Google Scholar]
- 23. Iosefson O., Azem A. (2010) Reconstitution of the mitochondrial Hsp70 (mortalin)-p53 interaction using purified proteins: identification of additional interacting regions. FEBS Lett. 584, 1080–1084 [DOI] [PubMed] [Google Scholar]
- 24. Tschopp J. (1984) Circular polymerization of the membranolytic ninth component of complement: dependence on metal ions. J. Biol. Chem. 259, 10569–10573 [PubMed] [Google Scholar]
- 25. Ziporen L., Donin N., Shmushkovich T., Gross A., Fishelson Z. (2009) Programmed necrotic cell death induced by complement involves a Bid-dependent pathway. J. Immunol. 182, 515–521 [DOI] [PubMed] [Google Scholar]
- 26. Fishelson Z., Attali G., Mevorach D. (2001) Complement and apoptosis. Mol. Immunol. 38, 207–219 [DOI] [PubMed] [Google Scholar]
- 27. Morgan B. P. (1989) Complement membrane attack on nucleated cells: resistance, recovery and nonlethal effects. Biochem. J. 264, 1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tegla C. A., Cudrici C., Patel S., Trippe R., 3rd, Rus V., Niculescu F., Rus H. (2011) Membrane attack by complement: the assembly and biology of terminal complement complexes. Immunol. Res. 51, 45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sriram M., Osipiuk J., Freeman B., Morimoto R., Joachimiak A. (1997) Human Hsp70 molecular chaperone binds two calcium ions within the ATPase domain. Structure 5, 403–414 [DOI] [PubMed] [Google Scholar]