
Figure 2.
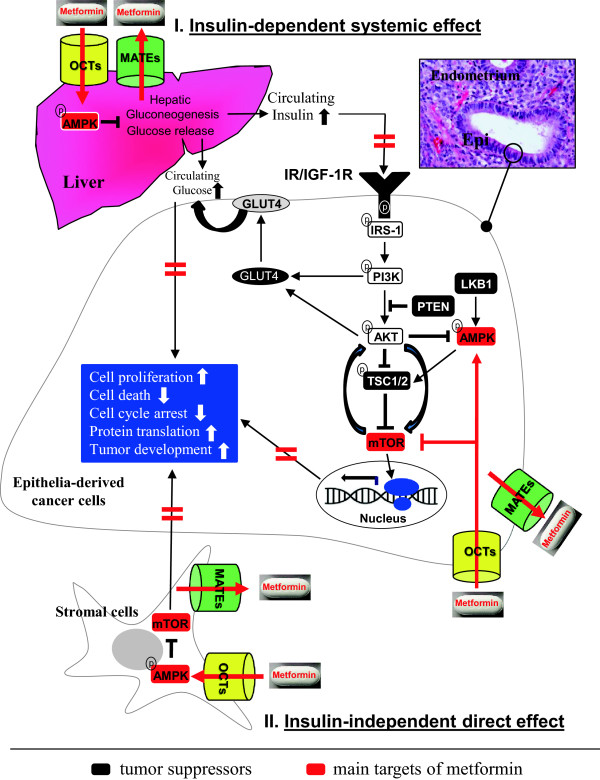
A schematic diagram representing the hypothetical mechanisms of the insulin-dependent systemic (I) and insulin-independent direct (II) effects of metformin in the endometrium. In the endometrium, binding of insulin and IGF-1 ligands to their receptors INSR and/or IGF-1R as homodimers or heterodimers leads to the activation of downstream signaling pathways, including the PI3K/AKT/mTOR pathway. A number of studies have demonstrated that in vitro enhancement of the PI3K/AKT/mTOR cascade in multiple cancer cells – including type I EC cell lines – ultimately results in specific cellular outcomes including cell proliferation, cell death, cell cycle arrest, and protein translation. Thus, activation of the PI3K/AKT/mTOR cascade might be the underlying mechanism behind the initiation and progression of EC in women with PCOS. Because AMPK, mTOR, and GLUT4 are considered to be central factors that are targeted by metformin, and because various OCTs and MATEs that mediate the metformin uptake and excretion are present in endometrial epithelial and stromal cells, we propose the following two mechanisms of metformin-induced inhibition of the PI3K/AKT/mTOR cascade in PCOS women with early stage EC. (1) Metformin activates the AMPK pathway in the liver and suppresses hepatic gluconeogenesis. This leads to reduced levels of circulating insulin and glucose, and this lack of substrates for IR/IGF-1R binding disrupts the activation of insulin/IGF-1 signaling pathways in the endometrial cancer cells. (2) In the endometrium, metformin either directly targets members of the AMPK, mTOR, and GLUT4 axis in endometrial cancer cells through the activity of epithelial OCTs and MATEs, or through stromal OCTs and MATEs in a paracrine manner to inhibit epithelia-derived cancer cell proliferation and growth. Thick horizontal red lines indicate inhibitory effects of metformin. For references, see the text.