

Abstract
Cord blood (CB) cells that express CD34 have extensive hematopoietic capacity and rapidly divide ex vivo in the presence of cytokine combinations; however, many of these CB CD34+ cells lose their marrow-repopulating potential. To overcome this decline in function, we treated dividing CB CD34+ cells ex vivo with several histone deacetylase inhibitors (HDACIs). Treatment of CB CD34+ cells with the most active HDACI, valproic acid (VPA), following an initial 16-hour cytokine priming, increased the number of multipotent cells (CD34+CD90+) generated; however, the degree of expansion was substantially greater in the presence of both VPA and cytokines for a full 7 days. Treated CD34+ cells were characterized based on the upregulation of pluripotency genes, increased aldehyde dehydrogenase activity, and enhanced expression of CD90, c-Kit (CD117), integrin α6 (CD49f), and CXCR4 (CD184). Furthermore, siRNA-mediated inhibition of pluripotency gene expression reduced the generation of CD34+CD90+ cells by 89%. Compared with CB CD34+ cells, VPA-treated CD34+ cells produced a greater number of SCID-repopulating cells and established multilineage hematopoiesis in primary and secondary immune–deficient recipient mice. These data indicate that dividing CB CD34+ cells can be epigenetically reprogrammed by treatment with VPA so as to generate greater numbers of functional CB stem cells for use as transplantation grafts.
Introduction
Cord blood (CB) HSCs have numerous phenotypic and functional characteristics that distinguish them from their adult counterparts (1–5). CB CD34+ cells are thought to be more primitive due to their extensive proliferative capacity, their increased ability to generate hematopoietic colonies in vitro, their capacity to produce erythroid cells, which contain fetal hemoglobins, and the ability of smaller numbers of such cells to reconstitute a myeloablated allogeneic recipient (1). The use of CB cells as HSC grafts for allogeneic stem cell recipients suffering from hematological malignancies and genetic disorders has been limited to children or smaller adult recipients due to the limited number of stem cells present in a single CB collection (1, 4, 5). These limitations have resulted in an unacceptably high rate of graft failure and delayed engraftment kinetics in adult recipients (1–7). Attempts to overcome these barriers have included several different strategies such as the infusion of two different CB grafts or the ex vivo expansion of CB CD34+ cells using a variety of cytokine combinations that are able to promote HSC cycling and the subsequent division of these CD34+ cells (2, 6–9). These initial attempts at ex vivo stem cell expansion have resulted in the generation of larger numbers of hematopoietic progenitor and precursor cells but reduced numbers of marrow-repopulating cells. HSCs are largely quiescent cells that slowly cycle in vivo (10–13). The rapid ex vivo cycling and division of CB CD34+ cells that occurs in the presence of such cytokine combinations results in HSC commitment, with the residual marrow-repopulating potential being attributed to a small fraction of stem cells that had remained quiescent or had undergone a limited number of cell divisions (10–13). More recently, mesenchymal cell-feeder layers or a number of molecules such as immobilized notch ligand, a copper chelator, histone deacetylase inhibitors (HDACIs), all-trans retinoic acid, an aryl hydrocarbon receptor antagonist, prostaglandin E2 (PGE2), or a c-MPL agonist have been added to these cytokine combinations with the hope of expanding the number of transplantable CB HSCs (2, 7, 14–19). Several of these approaches have been evaluated in clinical trials but have resulted in the generation of larger numbers of short-term, but not long-term, marrow-repopulating cells (2, 20–22). Alternatively, strategies to facilitate the efficiency of homing and engraftment of CB CD34+ cells are also being pursued to increase the efficacy of allogeneic CB transplantation (23–25).
Our laboratory has proposed an alternative approach to expand the numbers of functional CB HSCs. This approach is based on the hypothesis that prior attempts to expand HSCs ex vivo using serum-containing (SC) media and cytokine combinations actually result in the silencing of HSC genetic programs (2, 7, 9, 17, 26–31). This alternative strategy is consistent with the growing evidence that epigenetic mechanisms play important roles in determining whether an HSC undergoes symmetrical divisions and generates additional stem cells, asymmetrical divisions that at best maintain HSC numbers while generating hematopoietic progenitor cells (HPCs), or symmetrical commitment divisions that deplete HSC numbers and generate greater numbers of HPCs (26, 27, 32–35).
In the present study, HDACI-treated CD34+ cells under serum-free (SF) culture conditions were shown to be able to generate additional CD34+ cells that possessed many features associated with primitive stem cells including increased aldehyde dehydrogenase (ALDH) activity, increased expression of CD90, c-Kit (CD117), integrin α6 (CD49f), and CXCR4 (CD184), but that lacked CD45RA expression (36). In addition, upregulation of a number of pluripotency genes including SOX2, OCT4 (also known as POU5F1), NANOG, and zinc finger protein of the cerebellum family member 3 (ZIC3, also known as HTX), but not hTERT (telomerase reverse transcriptase), was associated with valproic acid (VPA) treatment (28). The knock down of SOX2, OCT4, and NANOG in HDACI-treated CD34+ cells led to a dramatic reduction of CD34+ and CD34+CD90+ cell numbers. We found that treatment with HDACIs under SF culture conditions was capable of programming dividing CB CD34+ cells so as to generate greater numbers of primitive cells, which were capable of repopulating both irradiated and secondary immune–deficient recipient mice without the development of hematological malignancies or teratomas. Limiting dilution analysis demonstrated that the number of SCID-repopulating cells (SRCs) was 36-fold greater in VPA-treated cells as compared with that in primary CB CD34+ cells (PCs). These data indicate that epigenetic strategies that upregulate stem cell–specific transcription factors result in the preservation of the self-renewal and multilineage differentiative capacity of dividing CB HSCs.
Results
The effects of HDACIs and SF media on the ex vivo expansion of CB CD34+ and CD34+CD90+ cells.
Defining the culture conditions that permit the ex vivo expansion of HSCs has been the subject of numerous investigations (2, 7, 14–19). We previously demonstrated that limited expansion of CB HSC numbers can occur using SC culture conditions and sequential treatment with a DNA methyl transferase inhibitor (DNMTI) and an HDACI (17, 26, 27, 29). To further optimize culture conditions that would additionally favor CB HSC expansion, we first evaluated the generation of CD34+ and CD34+CD90+ cells in SF and SC cultures supplemented with cytokines, referred to here as the control conditions. A schematic representation of the ex vivo expansion strategies used to expand CB CD34+ cells and the terminology used to refer to the cell populations studied are provided in Figure 1A and Table 1. We added a variety of HDACIs (VPA, scriptaid [SCR], trichostatin A [TSA], suberoylanilide hydroxamic acid [SAHA], CAY10433 also known as BML-210 [C433], CAY10398, also known as MD85, and CAY10603 [molecular formula: C22H30N4O6]) at varying doses and periods of incubation (5–9 days) and evaluated their ability to increase the numbers of CD34+ cells generated in vitro under SC or SF culture conditions. Of the eight HDACIs studied, treatment with VPA, SCR, and C433 for 7 days was shown to be most effective for this purpose. Treatment with each of these agents led to the generation of a similar percentage of CD34+CD90+ cells (SCR: 73.4% ± 13.9%, C433: 70.1% ± 18.4%, and VPA: 75.2% ± 10.7%) as compared with control conditions (16.2% ± 9.2%) (ANOVA, P < 0.0001). However, the percentages progressively declined if the cells were maintained beyond 7 days. Similarly, we found that each of these HDACIs were effective in generating a greater absolute number of CD34+ and CD34+CD90+ cells per CB collection (ANOVA, P ≤ 0.0007) (Figure 1, B and C) as well as promoting CXCR4 expression (CD184) by CD34+CD90+ cells and generating a greater absolute number of CD34+CD90+CD184+ cells as compared with control conditions (ANOVA, P < 0.0001) (Figure 1D). The effects of VPA, SCR, and C433 were not additive when combined at optimal and half-optimal concentrations (data not shown).
Figure 1. Effect of HDACIs on the ex vivo expansion of CB CD34+, CD34+CD90+, and CD34+CD90+CD184+ cells.

(A) Schematic representation of the ex vivo expansion strategy of primary CB CD34+ cells (PCs). Freshly isolated PCs were primed for 16 hours with cytokines either in SF or SC media. Cells were then further treated for 7 days under the mentioned culture conditions with or without additional cytokines and in the presence or absence of HDACIs. The expanded and reisolated CD34+ cells were used for further analyses. Individual PCs were treated in the absence (control) or presence of VPA, SCR, or CAY10433 (C433) for 7 days in SF media with cytokines. VPA led to the generation of a significantly greater absolute number of CD34+ cells (*P < 0.05; **P < 0.005) (B), CD34+CD90+ cells (*P < 0.05; **P < 0.005) (C), and CD34+CD90+CD184+ cells (***P = 0.0005) (D) per CB collection (mean ± SEM; ANOVA, P ≤ 0.0007 [B and C] and ANOVA, P < 0.0001 [D]) than did other HDACIs (n = 6–7). C, control.
Table 1.
Terminology used to refer to the cell populations studied
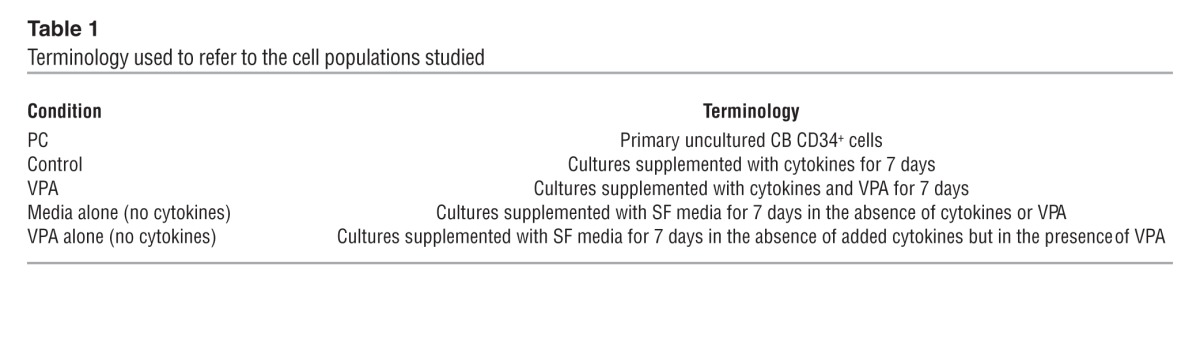
We more carefully examined the effect of serum on the ability of HDACIs to promote CD34+ cell expansion. The use of SF control culture conditions led to a greater expansion of CD34+ and CD34+CD90+ cell numbers than that achieved with SC control culture conditions (ANOVA, P < 0.0001, respectively) (Figure 2A). Under SF conditions, the addition of VPA led to a dramatic increase in the number of CD34+ (213-fold) and CD34+CD90+ (20,202-fold) cells as compared with SC conditions with VPA (a 78-fold expansion of the CD34+ cells and an 89-fold expansion of CD34+CD90+ cells; ANOVA, P ≤ 0.005) (Figure 2A).
Figure 2. Effect of VPA on the ex vivo expansion of CB CD34+ and CD34+CD90+ cells.

(A) The generation of CB CD34+ and CD34+CD90+ cells in the presence of cytokines occurred to a greater degree in SF than in SC cultures. A significant difference in the fold increase of CD34+ and CD34+CD90+ cells was observed in VPA-containing SF cultures as compared with SC cultures. *P < 0.05; **P < 0.005; ***P < 0.0005 (mean ± SEM; ANOVA, P < 0.0001; n = 6–7). (B) Phenotypic analysis of PC and CD34+ cells treated in SF media under control conditions or in the presence of VPA. Each cell population was analyzed for the expression of CD34, CD90, CXCR4 (CD184), CD49f, and CD45RA. The coexpression of CD184, CD49f, and CD45RA by CD34+CD90+ cells (red box) is depicted (n = 4).
We then pursued a more careful phenotypic analysis of the VPA-expanded CD34+ cells by analyzing the expression of an isoform of the leukocyte common antigen CD45RA and integrin α6 (CD49f) by CD34+CD90+ VPA-treated cells. Human HSCs have been shown to express CD49f but not CD45RA (36). In Figure 2B, we demonstrate that 47.0% ± 4.4% of the CD34+CD90+ cells from the cultures containing VPA expressed CD49f, while a minority of these cells expressed CD45RA (1.9% ± 2.1%).
Since the CXCR4/SDF1 axis is critical for HSC homing (31), we examined the migration of VPA-treated CD34+ cells in response to SDF1. As can be seen in Figure 3A, a significantly greater number of CD34+ cells treated with VPA migrated in response to SDF1 after 16 and 48 hours (P = 0.01 and P = 0.03). We next examined whether the upregulation of CXCR4 expression by CD34+ cells following VPA treatment was associated with increased homing of these cells to the marrow of NOD/SCID/γcnull (NSG) mice. As demonstrated in Figure 3B, VPA treatment led to increased homing of CD34+ cells as compared with CD34+ cells cultured under control conditions at both 16 hours (P < 0.0001) and 48 hours (P = 0.01) after infusion.
Figure 3. Effect of VPA on CD34+ cell migration and homing.

(A) SDF1 (100 ng/ml) induced migration of reisolated CD34+ cells generated in the absence (control) or presence of VPA (7 days). A significantly greater number of VPA-treated CD34+ cells migrated toward SDF1 after 16 and 48 hours (mean ± SEM, *P < 0.05, one-tailed t test; n = 4). (B) Homing in NSG mice of reisolated CD34+ cells generated in the absence (control) or presence of VPA (7 days) 16 and 48 hours after infusion (mean ± SEM; ****P < 0.0001; *P < 0.05). NSG recipient mice (n = 35).
We next examined the cell cycle status of the cultured CD34+ cells by labeling the cellular progeny within control cultures and cultures containing VPA with BrdU (2.5 hours) on day 7 of culture and compared the proportion of CD34+CD90+ cells that resided in different phases of the cell cycle (Figure 4A). We found that the cells treated with VPA contained a far greater proportion of CD34+CD90+ cells than did control cultures (75.2% ± 10.7% versus 16.2% ± 9.2%). In addition, a greater number of VPA-treated CD34+CD90+ cells resided within G2/M (18.0% ± 1.2%) than did the cells in control cultures (2.2% ± 1.0%, P < 0.0001), indicating that VPA-treated CD34+CD90+ cells continue to divide and retain their primitive phenotype, thereby accounting for the greater numbers of CD34+CD90+ cells observed on day 7. The CD34+CD90+ G0/G1 cell compartment was also increased in VPA-containing cultures (23.2% ± 13.8%) as compared with that seen in control cultures (5.0% ± 1.4%), suggesting that CD34+CD90+ cells exposed to VPA were capable of returning to G0/G1. Also, a smaller proportion of CD34+CD90+ cells from control cultures were in the S phase than those in the VPA-containing cultures (17.5% ± 1.8% versus 29.4% ± 7.9%) after 7 days of culture (Figure 4B). These data suggest that the control cells divided earlier during the culture period than did VPA-treated cells, which continued to divide and generate CD34+CD90+ cells on day 7.
Figure 4. Effect of VPA on the different cell cycle phases of CD34+CD90+ cells.

(A) Flow cytometric cell cycle analysis of CD34+CD90+ cells following SF culture under control (upper panel: 26.4%) conditions or cultures containing VPA (lower panel: 82.9%) for 7 days, with corresponding dot plots showing cells in different phases of the cell cycle by BrdU pulse labeling (2.5 hours) and staining with 7AAD (G0/G1, S, and G2/M). One of three representative experiments is shown. (B) Percentage of CD34+CD90+ cells that were in different phases of the cell cycle. A significant increase in the number of CD34+CD90+ cells was observed in G0/G1 (*P < 0.05), S (*P < 0.05), and G2/M (****P < 0.0001) phases in the VPA-containing cultures (mean ± SD; ANOVA, P < 0.002; n = 3).
In order to determine whether the effects of VPA were dependent on the continued exposure to cytokines, CB CD34+ cells underwent initial priming for 16 hours and were then cultured for 7 days in SF media alone or in SF media supplemented with VPA, but in the absence of additional cytokines (Figure 1A). These studies demonstrated that after an initial priming with cytokines, expansion of CD34+ and CD34+CD90+ cells occurred following incubation in SF media alone (no cytokines) (Figure 5A). An even greater absolute number of CD34+ (P < 0.0001) and CD34+CD90+ cells (P < 0.05), however, was generated when the cells were cultured in the presence of VPA alone (no cytokines), as compared with PCs (Figure 5A). This same degree of expansion of CD34+ cells, however, did not occur in cultures that were not initially primed and then incubated without additional cytokines, indicating the dependence of the expansion of CD34+ and CD34+CD90+ cell numbers on at least prior exposure to cytokines. On day 7, 21.2% ± 5.1% of cells cultured in media alone (no cytokines) were CD34+, and their phenotype was similar to that of PCs, while the cells that were exposed to VPA alone were characterized by a dramatic upregulation of CD90, CD184, and CD49f, but not CD45RA (Supplemental Figure 1; supplemental material available online with this article; doi:10.1172/JCI70313DS1), findings consistent with VPA leading to epigenetic reprogramming. The CD34+ and CD34+CD90+ cell numbers per CB collection underwent a 5.2-fold and 144-fold expansion when cultured in media alone (no cytokines) as compared with a 9.0-fold and 486-fold expansion, respectively, in cultures containing VPA alone (no cytokines (ANOVA, P < 0.0001) (Figure 5B). The degree of expansion of CD34+ (213-fold) and CD34+CD90+ (20,202-fold) cells was even more dramatically enhanced by the addition of cytokines to VPA-containing SF cultures during the 7-day incubation period (Figure 2A).
Figure 5. Effect of VPA on the ex vivo expansion of CB CD34+ and CD34+CD90+ cells in the absence of cytokines.

(A) PCs were primed with cytokines as indicated in Figure 1A and treated for 7 days under SF culture conditions in media alone or VPA alone without additional cytokines. Both cultures containing media alone (No cytokines) and VPA alone (No cytokines) led to a significantly greater number of CD34+ and CD34+CD90+ cells as compared with PCs. ****P < 0.0001; *P < 0.05 (mean ± SEM; ANOVA, P < 0.0001; n = 6). (B) A significant difference was observed in the fold increase of CD34+ and CD34+CD90+ cells in the SF cultures containing media alone (no cytokines) versus those with VPA alone (no cytokines). *P < 0.05; **P < 0.005 (mean ± SEM; ANOVA, P < 0.0001; n = 6).
Effect of HDACIs on HDAC levels.
HDACs exist in cells as subunits of multiprotein complexes and govern gene expression. The class I HDACs (HDAC1, -2, -3, and -8) possess sequence homology to the yeast transcriptional regulator RPD3; however, class II HDACs (HDAC4, -5, -6, -7, -9, and -10) share domains similar to those of deacetylase HDA1, which is found in yeast (30). Class I and II HDACs interact with the transcriptional corepressors mSIN3, NCoR, and SMRT, which recruit HDACs to transcription factors (30, 37). HDACIs have been previously shown by our laboratory to lead to increased H3 acetylation in CB CD34+ cells (29). HDAC activities, however, can be modulated not only by the binding of these inhibitors at the catalytic domain, but also by fine-tuned degradation of HDACs through the ubiquitin/proteasome pathway. Limiting the amounts of the E2 ubiquitin conjugase Ubc8 and the E3 ubiquitin ligase RLIM has been reported to maintain a balanced steady-state protein level of HDACs that is susceptible to modulation by VPA (37–39). In order to determine which HDACs were affected by the HDACIs, we evaluated the effects of SCR, C433, and VPA on both class I and II HDAC protein levels in CB mononuclear cells (CB-MNCs) and human embryonic kidney 293 (HEK293) cells after 2 and 24 hours of treatment. SCR, C433, and VPA did not inhibit HDAC expression after 2 hours of treatment of CB-MNCs (data not shown), but led to the inhibition of class I (HDAC1, -2, and -3), class IIa (HDAC4 and -5), and class IIb (HDAC6) HDACs to differing degrees after 24 hours of treatment. SCR and C433 were the most effective inhibitors of each of the HDACs (Figure 6 and Supplemental Table 1). Since individual HDACs play crucial roles in regulating intracellular processes and responding to the extracellular environment in cell-specific functions, the ability to predict the manner in which a particular cell type will respond to a given HDACI is limited (30, 38, 40, 41). We found that the pattern of HDAC inhibition following the treatment of HEK293 cells with these HDACIs was remarkably different than that of CB-MNCs and was more pronounced after 2 hours of treatment as compared with 24 hours (Supplemental Figure 2). Downregulation of HDAC2 and HDAC4 was common to the effects of each of the three HDACIs on HEK293 cells, while a reduction of HDAC1, -3, and -5 was common to CB-MNCs treated with the same agents. These findings indicate that SCR, C433, and VPA are each class I and II HDACIs, but that their effects on specific HDACs vary depending on the cell type being treated. It has been previously shown that downregulation of HDAC3 is essential for in vitro HSC expansion (42). Each of the HDACIs examined was capable of expanding CB CD34+ cell numbers, but VPA was the most effective compound in promoting CD34+ cell expansion, yet was not the most potent inhibitor of HDACs. The remaining experiments were carried out with VPA.
Figure 6. Effect of HDACIs on HDAC protein expression levels.

CB-MNCs were freshly isolated and treated in the absence and presence of SCR, C433, or VPA for 24 hours. Total cell lysates were prepared, and Western blotting was performed using HDAC mAbs specific to class I (HDAC1, -2, and -3), class IIa (HDAC4 and -5), and class IIb (HDAC6) HDACs as described in Methods. β-Actin was used as a loading control. One of four representative experiments is shown. Un-, untreated freshly isolated CB-MNCs.
VPA alters ALDH activity in cultured CB CD34+ cells.
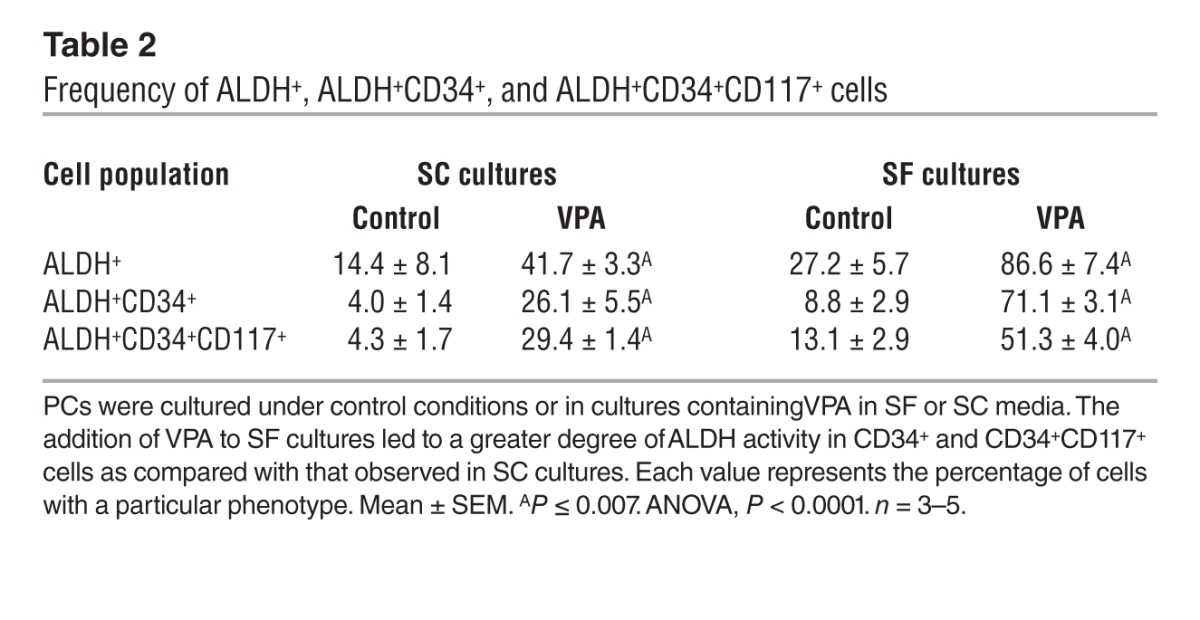
Since the phenotype of CB and marrow cells expanded in vitro with cytokines does not always correlate with function, we used ALDH activity as a functional marker of HSC (43–47). We observed a higher fraction of cells with ALDH activity in cells cultured in SF cultures than in those cultured in SC cultures in the presence of cytokines. Furthermore, the addition of VPA to cultures containing cytokines under SF culture conditions led to an even greater proportion of ALDH+ cells compared with that observed in SC cultures (Figure 7A and Table 2). The absolute number of ALDH+CD34+CD117+ cells generated in SF cultures plus VPA was greater than that achieved in SC cultures (P = 0.009) (Figure 7B).
Figure 7. ALDH functional activity in expanded CB CD34+ cells.

(A) PCs treated under control conditions or with VPA for 7 days with cytokines were assessed for ALDH activity. Contour plot analyses of various populations of cells including ALDH+CD34+ cells (left panel) and ALDH+ cells (middle panel). ALDH+ cells (blue box) were gated for coexpression of CD34 and c-Kit (CD117) (right panel). A greater degree of ALDH activity was observed in SF versus SC control cultures (P = 0.005) as well as in VPA-containing cultures (P = 0.001). Similarly, the percentage of ALDH+CD34+ and ALDH+CD34+CD117+ cells was significantly greater in SF than in SC cultures (P = 0.001 and P = 0.007, respectively). One of 3 to 5 representative experiments is shown. (B) A far greater number of ALDH+CD34+CD117+ cells was generated in the presence of VPA in SF cultures as compared with that in SC cultures (mean ± SEM; *P < 0.05; **P < 0.005; ANOVA, P = 0.009; n = 3–5).
Table 2.
Frequency of ALDH+, ALDH+CD34+, and ALDH+CD34+CD117+ cells
VPA influences the expression of genes associated with pluripotency.
The transcription factors SOX2, OCT4, and NANOG are the core regulatory players in determining both embryonic and induced pluripotent stem cell (iPS) fate decisions by co-occupying target genes including their promoters, thereby cooperating in both regulatory and autoregulatory feedback loops required to maintain self-renewal and pluripotency (48, 49). We explored the role of such master transcription factors in VPA-mediated HSC expansion by examining SOX2, OCT4, and NANOG expression in the CD34+ cells reisolated after 7 days of culture under control conditions or in those treated with VPA in SF and SC media. RT-PCR revealed the expression of SOX2, OCT4, and NANOG transcripts in CD34+ cells from SF cultures containing VPA, while OCT4 and SOX2 transcripts were barely detectable in the control cultures or SC cultures to which VPA was added (Figure 8A). Quantitative PCR (qPCR) demonstrated that expression of these pluripotency genes was upregulated in the presence of VPA in SF cultures (ANOVA, P = 0.0001) (Figure 8B) as compared with that observed in SC cultures. The possibility that OCT4 expression in adult stem cells is actually due to the expression of inactive pseudogenes rather than to a functional form of OCT4 has been reported by others (50, 51). Using RT-PCR, we found that one OCT4 pseudogene was not present in VPA-treated CD34+ cells (Figure 8A). qPCR analysis indicated that neither of the two pseudogene transcripts were present. Unlike OCT4, SOX2, and NANOG, telomerase reverse transcriptase (hTERT), another known marker of pluripotency in ES cells (52), was not upregulated in VPA-treated CD34+ cells (data not shown). We further analyzed downstream target genes of SOX2, OCT4, and NANOG, including SET, SMARCAD1, and MYST3, which play critical roles in chromatin remodeling, as well as an additional pluripotency gene, ZIC3 (48, 52, 53). SMARCAD1, MYST3, and ZIC3, but not SET, were also dramatically upregulated in VPA-treated CD34+ cells in SF cultures (ANOVA, P = 0.04). ZIC3 mRNA was not detected in control cultures supplemented with VPA in the presence of serum, but was exclusively upregulated in SF cultures supplemented with VPA (Figure 8C). The ZIC3 gene has been identified as a target of OCT4, SOX2, and NANOG in ES cells. ZIC3 overlaps with the OCT4, NANOG, and SOX2 transcriptional networks, is important in maintaining pluripotency, and can directly modulate the expression of NANOG (48, 53, 54).
Figure 8. Transcripts of pluripotency genes in VPA-expanded CD34+ cells.

(A) Expression of pluripotency genes in VPA-treated CD34+ cells. CD34+ cells were reisolated after treatment in the presence or absence of VPA in SF and SC cultures. cDNA was prepared from total RNA, and RT-PCR was performed. ES cells were a positive control for SOX2, OCT4, and NANOG transcripts and a negative control for CD34 expression. Expression of pseudo-OCT4 was not detected by RT-PCR in VPA-treated CD34+ cells under SF culture conditions. Lanes for control/VPA (SF) and VPA (SC)/ES cells were run on two different gels, and lanes for the pseudo- and genuine OCT4 gene were run on a single gel. GAPDH housekeeping gene, M-a 50-bp DNA ladder. One of four representative experiments is shown. (B) Quantitation of the effects of VPA on genes associated with pluripotency. CD34+ cells cultured under different conditions were isolated and processed as described in A. Relative transcript levels of SOX2, OCT4, and NANOG genes were calculated by SYBR Green qPCR. Fold change of mRNA expression by CD34+ cells reisolated from control cultures (left panel) and VPA-containing cultures (right panel) was calculated by normalizing to the level of corresponding transcripts present in PCs (mean ± SD; ANOVA, P = 0.0001; n = 4). (C) Quantitation of expression of genes associated with chromatin remodeling and pluripotency after VPA treatment under SF or SC culture conditions. Fold change of SET, MYST3, SMARCAD1, and ZIC3 mRNA expression levels was calculated as described above (mean ± SEM; ANOVA, P = 0.04; n = 3).
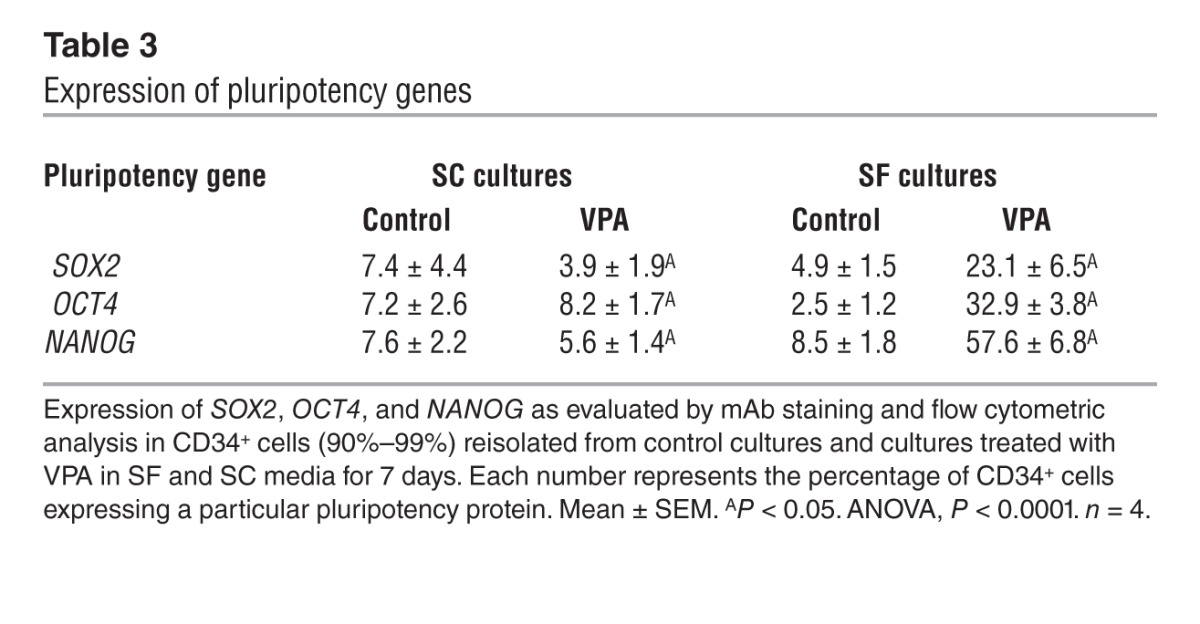
We then examined the expression of SOX2, OCT4, and NANOG proteins in CD34+ cells by flow cytometric analysis. SOX2, OCT4, and NANOG expression was greatest in the presence of VPA in SF cultures rather than in SC cultures (Figure 9A and Table 3). We further examined the expression of SOX2, OCT4, and NANOG proteins in CD34+ cells using mAb staining and confocal microscopy (Figure 9B and Supplemental Figure 3). The pluripotency genes were upregulated and localized to both the nucleus and cytoplasm in the VPA-treated CD34+ cells, while in ES cells, these proteins were predominantly localized to a subnuclear area. Although these proteins were not observed in CD34+ cells generated under control conditions or in PCs, a low level of ZIC3 protein was observed in PCs (data not shown). OCT4 and SOX2 are the major transcription factors that bind to the NANOG promoter and promote its transcription and that of related gene networks (48, 49). In the SF but not SC cultures, treatment with VPA led to the upregulation of NANOG, suggesting a possible functional interaction between SOX2 and OCT4 (Table 3). We then documented the physical interaction between NANOG and OCT4 by co-IP of proteins from VPA-treated cells (Figure 9C). Furthermore, Western blot analysis revealed that endogenous OCT4 and NANOG were expressed less abundantly in VPA-treated cells than in ES cells (Figure 9C).
Figure 9. Expression of pluripotency genes in VPA-expanded CD34+ cells.

(A) Representative flow cytometric analysis of SOX2, OCT4, and NANOG expression in reisolated CD34+ cells after 7 days of culture under control conditions or after exposure to VPA. Cells were fixed, permeabilized, and stained with isotype-matched IgG (red line) or SOX2, OCT4, and NANOG mAbs to assess the intracellular levels of protein in reisolated CD34+ cells from control (green line) and VPA (blue line) cultures. One of four representative experiments is shown. (B) Confocal microscopic analysis of pluripotency gene expression. CD34+ cells were reisolated after treatment with VPA in SF cultures and immunostained with isotype-matched IgG or OCT4, SOX2, NANOG, and ZIC3 antibodies (FITC, green) as described in Methods. Nuclei were stained with DAPI (blue). Shown is a single optical section of confocal z-stack series (scale bars: 10 μm) for OCT4, SOX2, and NANOG (original magnification, ×63) and IgG and ZIC3, as well as a higher magnification (×126) of OCT4. One of three representative experiments is shown. (C) Co-IP of pluripotency genes. ES (H9) cells and progeny of the VPA-treated cells (V) were lysed on day 7, and ES or V cell lysates were IP with NANOG pAb (or anti-IgG control) and fractionated by SDS-PAGE. Total protein lysates (input) from ES and VPA-treated cells were also included in the same gel but were noncontiguous. Western blot (WB) analysis was performed using an OCT4 pAb. Western blot analysis using NANOG mAb was also performed on ES and VPA-treated cell lysates. β-Actin was used as a loading control. One of three representative experiments is shown.
Table 3.
Expression of pluripotency genes
Pluripotency genes are essential for expansion of CD34+CD90+ cells.
To establish a functional link between the upregulation of pluripotency genes in VPA-treated cultures and the expansion of CD34+CD90+ cells, we transfected CD34+ cells either with individual siRNA or with a combined pool of siRNA directed against SOX2, OCT4, and NANOG (SON). We initially tested different concentrations of siRNA for the individual genes and their potential toxic effects on cells in control cultures or cultures treated with VPA (data not shown). The morphological appearance of cells treated with SON siRNA was not altered as compared with those treated with scrambled siRNA. We did not observe a significant reduction in the total number of cells generated in control cultures and VPA-containing cultures after transfection with scrambled, SON, and GAPDH siRNA (Supplemental Table 2). We monitored pluripotency gene expression after siRNA transfection in VPA-treated cultures using qPCR and RT-PCR (Figure 10, A and B). siRNA-mediated knock down led to markedly reduced expression of the transcripts (80%–84%) for SOX2, OCT4, NANOG, and ZIC3 (ANOVA, P < 0.0001). Confocal microscopy and mAb staining also revealed a marked reduction in SOX2, OCT4, and NANOG protein expression. Expression of ZIC3 protein, which is downstream of the OCT4, SOX2, and NANOG regulatory network, was reduced to a lesser extent (Figure 10C). We observed a significant reduction in the percentage of CD34+ (47.8% ± 4.4% versus 22.8% ± 8.6%) and CD34+CD90+ (20.5% ± 6.1% versus 11.7% ± 4.5%) (ANOVA, P = 0.0005) cells after treatment with SON siRNA as compared with individual siRNAs specific for each of the pluripotency genes or with scrambled siRNA (Figure 10D). Furthermore, after treating VPA-treated CD34+ cells with SON siRNA, we observed an 89.1% and 88.7% reduction in the absolute number of CD34+ and CD34+CD90+ cells per CB collection, respectively (ANOVA, P = 0.0008) (Figure 10E).
Figure 10. siRNA-mediated knock down of pluripotency genes.

(A) PCs were treated with VPA in SF cultures. After 3 days, cells were transfected with a pool of SOX2, OCT4, NANOG (SON), scrambled (negative control), and GAPDH siRNA (positive control) as described in Methods. SOX2, OCT4, NANOG, GAPDH, and ZIC3 transcripts were quantitated by SYBR Green qPCR and normalized to the level of CD34 transcripts *P < 0.05; **P ≤ 0.006; ***P = 0.0001 (mean ± SEM; ANOVA, P < 0.0001; n = 3). (B) Expression of SOX2, OCT4, NANOG, CD34, and GAPDH following siRNA-mediated knock down was analyzed by RT-PCR. M, DNA markers. ES cells were a positive control for SOX2, OCT4, NANOG, and ZIC3. Lanes were run on the same gel. (C) Pluripotency gene expression was analyzed by confocal microscopy. Upper panel: scrambled siRNA; lower panel: SON siRNA showing SOX2, OCT4, NANOG, and ZIC3 expression in VPA-treated cells. Images represent an optical section of confocal z-stack series. Scale bars: 10 μm (original magnification, ×40). Similar data were obtained in two additional experiments. (D) After 7 days of transfection, the percentage of cells that were CD34+ and CD34+CD90+ was analyzed using flow cytometry. Graph represents a comparative analysis of the percentage of CD34+ and CD34+CD90+ cells generated in VPA cultures after transfection with siRNA including scrambled and SON. *P ≤ 0.05 (mean ± SEM; ANOVA, P = 0.0005; n = 3). (E) Absolute numbers of CD34+ and CD34+CD90+ cells per CB collection generated in VPA-containing cultures following transfection with scrambled or SON siRNA were calculated. *P ≤ 0.05; ***P = 0.0001 (mean ± SEM; ANOVA, P = 0.0008; n = 3).
These data indicate that VPA treatment leads to an epigenetic reprogramming of cultured CD34+ cells that directs subsequent transcriptional activation of pluripotency genes, which is essential for the generation of CD34+ and CD34+CD90+ cells in SF cultures.
In vivo functional behavior of VPA-treated CD34+ cells in NSG mice.
We evaluated the marrow-repopulating potential of PC and CB CD34+ cells cultured under control conditions and with VPA and cytokines by assessing their engraftment within the marrow of NSG recipient mice. In all recipient mice, irrespective of the type of graft transplanted, human CD45+ and CD45+CD34+ cells were detected. Thirteen to 14 weeks after transplantation (Figure 11, A and B), 19.4% ± 4.9% of the marrow cells were donor-derived CD45+ cells in mice receiving PCs as compared with 13.2% ± 6.4% in mice receiving cells from control cultures. By contrast, transplantation of VPA-treated CB CD34+ cells resulted in a greater degree of human CD45+ cell chimerism (32.2% ± 11.3%) and CD45+CD34+ cells (13.0% ± 8.7%) compared with that achieved with control cells (P = 0.0008 and P = 0.004, respectively) (Figure 11, A and B). The degree of CD45+ cell chimerism with VPA grafts was also statistically greater than that achieved with PCs (P = 0.006).
Figure 11. Analysis of human cell chimerism in primary NSG mice.

NSG mice were transplanted with PCs, CD34+ cells reisolated from control cultures, and VPA-containing cultures. Mean ± SD percentage of chimerism with (A) human cells (CD45+), (B) CD45+CD34+ cells, (C) CD34+CD184+ cells, (D) CD33+ cells, (E) megakaryocytes (CD41+), erythroid cells (GPA+), granulocytes (CD14+), T cells (CD3+), and B cells (CD19+) was determined by flow cytometry. (F) Comparative analysis of the mean degree of human cell chimerism achieved with transplantation of PCs and CD34+ cells treated in the absence or presence of cytokines under SF culture conditions with or without VPA. (A) **P = 0.006; ***P = 0.0008 (ANOVA, P < 0.0001); (B) **P = 0.004 (ANOVA, P = 0.03); (C) *P = 0.01; **P = 0.0008 (ANOVA, P = 0.01); (D) **P = 0.003; ****P < 0.0001 (ANOVA, P < 0.0001); (E) median ± SD. *P < 0.05; **P < 0.005 (ANOVA, P < 0.0001); and (F) *P < 0.05, one-tailed t test. **P ≤ 0.002 (ANOVA, P < 0.0001). n = 27 NSG recipient mice.
A significantly greater number of donor-derived CD34+ cells within the marrow of mice receiving VPA-treated CD34+ cells coexpressed CD184 (9.9% ± 9.6%) as compared with the marrow of mice receiving PCs or grafts expanded under control conditions (ANOVA, P = 0.01) (Figure 11C). The pattern of VPA-treated CD34+ cell grafts differentiating into multiple hematopoietic lineages following transplantation was distinctly different from that in PCs or cells expanded under control conditions (ANOVA, P < 0.0001) (Figure 11, D and E), with higher proportions of CD41+, CD19+, and glycophorin A–positive (GPA+) cells appearing in mice receiving VPA-treated grafts.
We next evaluated the dependence of VPA-mediated SRC expansion on the continued exposure to cytokines during their generation. As can be seen in Figure 11F, the transplantation of CD34+ cells cultured in media alone (no cytokines) in the absence of cytokines and with VPA alone (no cytokines) achieved a similar degree of chimerism (8.2% ± 5.0% versus 12.5% ± 5.0%; P = 0.1, NS). The grafts generated in the absence of continued cytokine exposure retained the ability to generate cells belonging to multiple hematopoietic lineages (Supplemental Table 3). The degree of donor cell chimerism achieved with the transplantation of cells cultured under control conditions was similar to that achieved with cells that were not exposed to cytokines throughout the 7-day culture period in media alone (no cytokines) or VPA alone (no cytokines). A dramatic increase in the degree of human cell chimerism was, however, observed following the transplantation of grafts generated in the presence of cytokines plus VPA (32.2 ± 10.2%; ANOVA, P < 0.0001). These data suggest that SRCs persist if PCs are primed with cytokines for merely 16 hours and are then cultured in media alone or with VPA alone in the absence of cytokines. The presence of cytokines throughout the culture period, however, further enhanced the effectiveness of VPA, resulting in a higher degree of human cell chimerism in NSG recipients.
The self-renewal potential of the expanded grafts was evaluated by transplanting donor-derived cells present in primary recipients into secondary recipients. After 15 to 16 weeks, secondary recipients transplanted with marrow cells from primary recipients that had been transplanted with CB CD34+ cells treated with VPA achieved the greatest degree of human CD45+ cell chimerism (ANOVA, P < 0.0001) (Figure 12A). As shown in Figure 12B, the donor-derived cells in the secondary recipient mice belonged to multiple hematopoietic lineages, and this pattern was distinct from that observed in secondary recipients receiving marrow cells from primary recipients that received PC grafts or grafts expanded under control conditions (ANOVA, P < 0.0001). The degree of donor-derived erythroid cell engraftment was especially notable in the secondary recipients of marrow cells from mice receiving VPA-treated grafts (Figure 12B) (55). It is important to emphasize that none of the primary or secondary recipients of marrow cells receiving VPA-treated grafts developed evidence of blood cancer or developed teratomas. To further exclude the possibility of teratoma formation, CD34+ cells reisolated from control cultures, VPA-containing cultures, or ES cells were each subcutaneously injected into the hind limb of NSG mice and were evaluated after 8 weeks. Teratomas, composed of cells derived from three different germ layers, were observed exclusively in animals injected with the ES cells (Supplemental Figure 4).
Figure 12. Analysis of human cell chimerism in secondary NSG mice.

(A and B)Thirteen to 14 weeks after transplantation of PCs or grafts expanded for 7 days under the various conditions previously described, the primary recipient mice were sacrificed, and 2 × 106 BM cells were transplanted into secondary NSG mice. Each bar represents the median percentage of human donor cell engraftment that occurred in the marrow of the secondary NSG mice as determined by mAb staining and flow cytometric analysis and the multilineage hematopoietic cell engraftment that occurred in secondary NSG mice 15–16 weeks after transplantation of different types of grafts from primary recipients. Patterns of lineage development were statistically significantly different: *P < 0.05; **P < 0.005. Median ± SD (A and B). ANOVA, P < 0.0001. n = 18 NSG recipient mice.
In order to evaluate the persistence of the upregulation of pluripotency genes following VPA treatment, we evaluated the expression of these genes in donor cells in primary and secondary recipient NSG mice. Using qPCR, we did not detect transcripts for pluripotency genes including SOX2, OCT4, NANOG, and ZIC3 in the marrow cells of the primary and secondary recipients, indicating that the VPA-induced upregulation was transient (data not shown).
To assess the degree of HSC expansion achieved with VPA treatment, we used limiting dilution analysis to compare the frequency of SRCs in PCs, in cells derived from an equivalent number of PCs cultured under control conditions, or in cultures containing VPA. The transplantation of increasing numbers of PCs (50, 250, 500, 2,500, and 5,000) or the progeny of cells from an equivalent number of PCs after treatment under control conditions and with VPA resulted in increasing degrees of human cell chimerism following their transplantation (Figure 13A). Poisson distribution analysis revealed an SRC frequency of 1 in 1,115 (95% CI: 1/596 to 1/2,087) in PCs, 1 in 9,223 (95% CI: 1/3,419 to 1/24,879) in control cultures, and 1 in 31 (95% CI: 1/14 to 1/66) in cultures treated with VPA. The overall difference in stem cell frequencies between PC, control, and VPA-containing cultures was highly significant (P = 9.42 × 10–29), indicating the effective expansion of SRC numbers within VPA cultures (Figure 13B and Tables 4 and 5) as compared with PC or control cultures.
Figure 13. Comparison of the frequency of SRCs in PCs and the progeny of an equivalent number of CD34+ cells cultured under control conditions or treated with VPA.

(A) Increasing numbers of PCs (50, 250, 500, 2,500, and 5,000) and the progeny of cultures initiated with an equivalent number of cells cultured under control conditions or in the presence of VPA were individually transplanted into NSG mice. Percentage of human CD45+ cell engraftment in the BM of recipient mice after 12 to 13 weeks is shown. (B) Poisson statistical analysis was performed using the number of mice with or without evidence of human cell engraftment (Table 4). Graph represents the percentage of mice without human cell chimerism (negative) following the transplantation of PCs or the progeny of equivalent numbers of CD34+ cells from control cultures or cultures containing VPA. Dotted lines represent 95% CIs. (C) SRC numbers were calculated using Poisson statistical analysis and are represented as the number of SRCs per 1 × 106 CD34+ cells. **P ≤ 0.002; ANOVA, P = 0.003. n = 111 NSG recipient mice.
Table 4.
Limiting dilution analysis of human cell engraftment in NSG mice
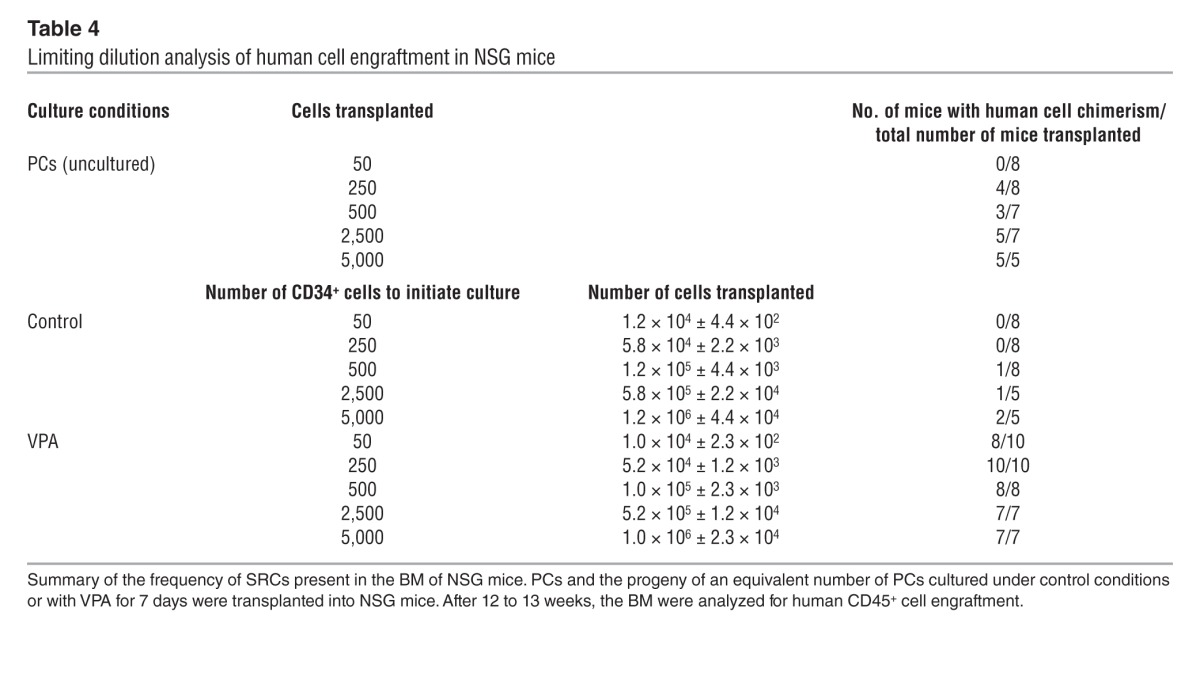
Table 5.
Frequency of SRC
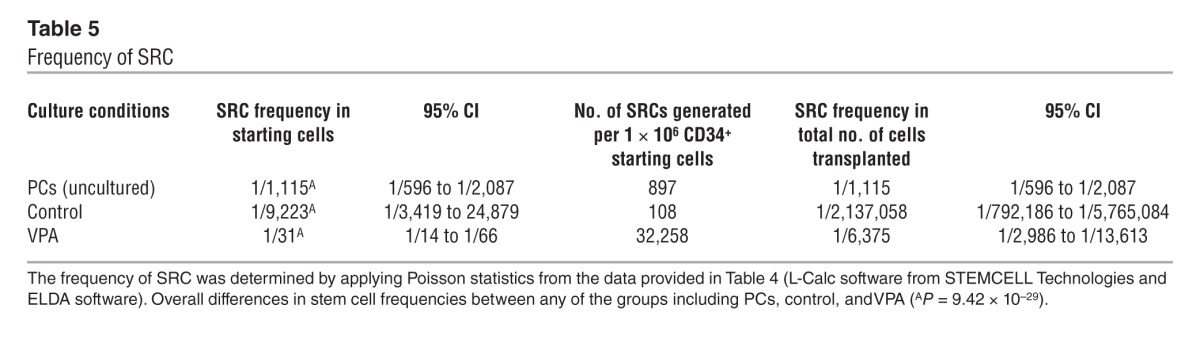
We calculated the presence of 897 SRCs and 108 SRCs in 1 × 106 PCs and cells cultured under control conditions, respectively. By contrast, we calculated the presence of 32,258 SRCs in 1 × 106 cells from VPA-containing cultures (Table 5). Therefore, incubation of PCs under control culture conditions led to a reduction in SRC numbers, while VPA treatment resulted in a 36-fold (P ≤ 0.002) increase in the number of SRCs compared with that in PCs and a 299-fold (P ≤ 0.002) increase in the number of SRCs compared with that in cells cultured under control conditions (Figure 13C).
Discussion
The multipotent nature of HSCs can be accounted for by the dynamic maintenance of HSC chromatin structure and epigenetic plasticity. The modification of chromatin structure is largely regulated by specific post-translational modifications of histones, which determine whether the resultant chromatin structure is permissive or repressive (38, 40, 41). The progressive loss of stem cell function by CB CD34+ cells following in vitro culture using SC culture conditions and cytokine combinations remains a barrier to the in vitro expansion of the numbers of transplantable HSCs (10–13). This decline of stem cell function is likely due to the removal of fully functional HSCs from a permissive environment that exists within the host and their placement into a hostile ex vivo environment, which leads to epigenetic modifications that alter the gene expression program that determines the critical functions of a stem cell, including self-renewal potential, marrow-repopulating capacity, and multilineage differentiative capacity. This loss of HSC function has been attributed to the rapid cell cycling and cell division that occur in response to the culture conditions used (54–56). Prior attempts at ex vivo stem cell expansion have met with limited success, due perhaps in part to their focus on creating a milieu that resembles the hematopoietic microenvironment and thereby favors retention of stem cell functional integrity (2, 3, 7). We have appreciated the difficulty of creating such a microenvironment ex vivo and have taken the alternative approach of attempting to directly maintain the epigenetic characteristics of HSCs using agents that affect chromatin structure. This approach was based on the understanding that dynamic changes in chromatin states are mediated by nucleosome remodeling, DNA methylation, and histone acetylation (38, 40–42). For this purpose, in this study we evaluated several HDACIs that are capable of inhibiting both class I and II HDACs, with the aim of achieving decondensation of the chromatin structure that harbors genes involved in retaining stem cell function following repeated cell division. HDAC1, -3, and -5 proteins were uniformly reduced by the three most active HDACIs, suggesting that a combination of these three HDACs plays a critical role in stem cell fate decisions that favor retention of stem cell function following division in vitro.
We have previously reported that HDACI treatment leads to increased H3 histone acetylation (29), while others have indicated that exposure to such agents results in the loss of repressive modifications either by binding to HDACs and/or by promoting DNA demethylation and sliding and/or displacement of nucleosomes, which may allow transcription factors to bind to related DNA, or by polyubiquitination, which leads to proteasomal degradation of particular HDACs (38, 40, 41). These interactions permit the recruitment of additional coactivators and/or histone-modifying enzymes that are required to form transcriptional machinery, leading to transcriptional activation (38, 40, 41). We report here that VPA was the most effective of all the HDACIs evaluated in generating cells capable of producing the greatest degree of human cell chimerism following their transplantation into primary and secondary NSG mice. VPA treatment led to a 36-fold increase in SRC frequency as compared with that of PCs, while retaining the high proliferative potential and multilineage differentiation capacity characteristic of HSCs. The use of SF rather than SC culture conditions was shown to be critical for our efforts to generate functional HSCs. VPA treatment under SF culture conditions resulted in the generation of 20,202-fold greater numbers of CD34+CD90+ cells than did VPA treatment under SC conditions. These findings indicate that serum contains factors that inhibit or repress the regulatory genes involved in the retention and/or expansion of functional HSCs and that the presence of serum favors the upregulation of genes involved in commitment and differentiation. These findings are remarkably similar to those reported by Hirai and coworkers, who showed that the efficiency of creating iPS cells can be drastically improved by changes in the composition of the culture media and the density at which transduced cells are seeded on feeder layers (57). We also showed that VPA treatment under SF culture conditions led to the persistent division of a higher fraction of dividing CD34+CD90+ cells after 7 days of incubation, thereby accounting for the expansion of CD34+CD90+ cell numbers. These findings indicate that VPA treatment results in the retention of the ability of CD34+CD90+ cells to continue dividing even over a 7-day culture period. This characteristic was, however, lost after more prolonged periods of incubation, which is likely indicative of the transient retention of a stem cell–defining gene expression program ex vivo under the conditions used. The primitive nature of the CB CD34+ cells generated using SF conditions and VPA treatment was further documented in our study by the greater degree of CD184 and integrin α6 (CD49f) expression, the lack of CD45RA expression, and the increased degree of ALDH activity. The increased expression of CXCR4 is of particular importance, since the interaction of CXCR4 with its ligand SDF1 plays a critical role in the homing of stem cell grafts to the marrow of transplanted recipients (58). The functionality of upregulated CD184 in VPA-treated CD34+ cells was documented in our study by the ability of these cells to migrate in vitro in response to SDF1 and to home to the marrow of NSG mice to a greater degree than CD34+ cells from control cultures. The enhanced marrow-repopulating potential of VPA-treated CB CD34+ cells can therefore be attributed to its effects not only on HSC generation, but also on promoting HSC homing to the marrow of recipient mice. We found that the VPA-treated CD34+ cells were also characterized by upregulation of the pluripotency-associated genes SOX2, OCT4, NANOG, and ZIC3, but not hTERT. These properties of VPA-treated CD34+ cells are characteristic of iPS cells and ES cells and have not been previously associated with normal HSCs. The knock down of these pluripotency genes (SOX2, OCT4, and NANOG) was demonstrated to impair the ex vivo generation of CD34+CD90+ cells by VPA. In addition, we observed the downregulation of ZIC3 following SON treatment, which is likely a reflection of its contribution to the maintenance of pluripotency by operating downstream of OCT4, SOX2, and NANOG (53, 54). The previous documentation that OCT4 is present in human tumors has been explored by others and has been attributed to OCT4 pseudogenes which lack OCT4 activity (50, 51). We were, however, unable to detect transcripts for these pseudogenes in VPA-treated cells. We further documented the physical interaction of NANOG with OCT4 by co-IP of these proteins from VPA-expanded cells, which was similar to that observed with ES cells. Interestingly, Yu and coworkers recently reported that OCT4 and SOX2 promote the expression of CD49f in human mesenchymal stem cells, which raises the possibility that many of the phenotypic markers that characterize VPA-treated CD34+ cells are related to the upregulation of these pluripotency genes (59). It is important to emphasize that the VPA-treated CB CD34+ cells were not immortalized and possessed distinctly different biological properties than did iPS and ES cells. Unlike iPS or ES cells, which can be maintained indefinitely in culture, VPA-treated CD34+ cell numbers declined after 8 days of culture. In addition, VPA-treated CD34+ cells did not form teratomas in NSG mice, a characteristic property of ES and iPS cells. The transient expression of these pluripotency genes by VPA-treated CD34+ cells was further documented by the absence of their transcripts in human cells that persisted in primary and secondary recipient NSG mice for a total of 30 weeks. These data indicate that the transient expression of such pluripotency genes induced in VPA-treated CD34+ cells likely influences the function of dividing CB HSCs without leading to their immortalization.
The ability of VPA to alter the phenotype of CB CD34+ cells was dramatically affected by the addition of a combination of cytokines that are known to influence the behavior of primitive cells along the hierarchy of hematopoietic cell differentiation. Unless PCs were at least primed with cytokines for a 16-hour period, HSC numbers declined. The expansion of CD34+ and CD34+CD90+ cell numbers was possible only if these cells were at least cytokine primed and then incubated for an additional 7 days in the presence of media alone (no cytokines) or VPA alone (no cytokines). Only those cells that were exposed to VPA without cytokines during the subsequent 7-day period, however, expressed an HSC phenotype similar to that observed in cultures containing VPA and cytokines (CD34+CD90+CD49f+CD184+CD117+CD45RA–). Importantly, the degree of expansion of CD34+ cells was further increased substantially when cytokines were added to the VPA for the entire 7-day incubation period. Remarkably, the CD34+ cells generated both in the presence and absence of cytokines retained the ability to establish multilineage hematopoiesis upon their transplantation into NSG mice, indicating that the brief cytokine exposure during the priming phase led to limited proliferation, with preservation of sufficient HSC function for hematological engraftment to occur. These data indicate that the ex vivo environment in which HSCs are placed in part determines their fate and that the retention of the HSC program responsible for its phenotype is a consequence of epigenetic reprogramming due to VPA in SF media, while the increase in cell numbers is a result of cellular proliferation promoted by cytokine exposure.
The studies reported here have important implications for the clinical pursuit of CB transplantation. The implementation of the approach outlined in this report has the potential to make available stem cell grafts that contain sufficient numbers of SRCs to allow adults to proceed with allogeneic CB stem cell transplantation with more favorable outcomes.
Methods
Isolation of CB CD34+ cells and their ex vivo culture.
CB collections were purchased from the Placental Blood Program at the New York Blood Center. CB-MNCs were isolated by Ficoll-Hypaque density centrifugation, and CD34+ cells were purified by immunomagnetic selection as previously described (29). Highly purified (90%–98%) PCs (4.0–5.0 × 104) were cultured in SF Stemline II (Sigma-Aldrich) culture medium or IMDM (Lonza) containing 30% FBS (HyClone Laboratories) supplemented with 150 ng/ml SCF, 100 ng/ml fms-like tyrosine kinase receptor 3 (FLT3 ligand), 100 ng/ml thrombopoietin (TPO), and 50 ng/ml interleukin 3 (IL-3) (R&D Systems) and incubated in a humidified incubator maintained at 37°C with 5% CO2. After 16 hours of incubation, the cells were exposed to varying concentrations of individual HDACIs including trichostatin A (TSA), suberoylanilide hydroxamic acid (SAHA), VPA (Sigma-Aldrich), SCR, and CAY10433 (C433, also termed BML-210), CAY10398 (also known as MD85), and CAY10603 (molecular formula: C22H30N4O6) (Cayman Chemical), either in the absence or continued presence of cytokines for an additional 7 days (Figure 1A). The following concentrations of the HDACIs reported in this study were utilized: VPA (1.0–1.25 mM), SCR (0.75–1.0 mM), and C433 (7.5–10 mM), unless otherwise specified. The cell populations studied and the various conditions under which they were cultured are referred to by specific terms listed in Table 1.
Viable PC and cultured cells were enumerated using the trypan blue exclusion method. The fold expansion of CD34+ cells or subpopulations was calculated based on the number of CD34+ cells determined to be present in an individual CB collection and the number of CD34+ cells that would have been generated if all of the CD34+ cells within that primary CB collection were cultured using the various culture conditions described.
Phenotypic analysis.
PC or cultured cells expanded in the presence or absence of HDACIs were stained with an anti-human CD34 mAb or an isotype-matched control mAb and analyzed using a FACSCanto II (BD). CD34+ cells were reisolated using a CD34+ cell isolation kit, as previously described, for further phenotypic and functional analyses. All mAbs were purchased from BD Biosciences and Cell Signaling Technology. Phenotypic analyses (CD34-APC, CD90-FITC, CD184-PE, CD117-PE, CD49f-PE, and CD45RA-PECy7) of the ex vivo–expanded cells after 7 days in control cultures or cytokines plus an HDACI were performed as previously described (29).
Migration assay.
The migratory behavior of CB CD34+ cells was evaluated as previously described using 6.5-mm-diameter, 5-μm-pore Transwell plates (Costar) (60). The lower compartments of the Transwells were filled with StemLine II SF medium supplemented with 100 ng/ml of stromal-derived factor 1 (SDF1) (R&D Systems). The Transwell filters were coated with Matrigel for 30 minutes at 37°C. Reisolated CD34+ cells (1 × 105) from the control and VPA-containing cultures were plated on the Matrigel-coated filters in 100 μl of Stemline II media. After 16 and 48 hours, cells that migrated to the lower compartment were enumerated, and the percentage of migration was calculated as follows: (number of cells migrated/total number of cells plated) × 100.
Homing assay.
The homing of reisolated CD34+ cells (5 × 105/mouse) after treatment under control conditions or with VPA was performed as previously described (60). The recipient NSG mice were purchased from the Jackson Laboratory and sublethally irradiated (300 cGy) 4 hours prior to infusion of the reisolated CD34+ cells via the tail vein. Sixteen and 48 hours after their infusion, BM cells were harvested from 2 femurs and 2 tibias from each recipient mouse and analyzed by flow cytometry for the presence of human CD34+ cells using a human CD34-APC mAb. The homing of these cell populations was determined by quantitating the number of CD34+ cells per 106 acquired events in the BM of the recipient mice. Four mice did not receive cells and were similarly analyzed in order to subtract the background from experimental samples (61). Eight NSG mice received CD34+ cells cultured under control conditions, and 9–10 mice received CD34+ cells from VPA-containing cultures.
Cell cycle analysis by BrdU labeling.
The cell cycle status of the cultured CD34+ CD90+ cells was assessed using the FITC-BrdU Kit (BD Pharmingen) according to the manufacturer’s instructions. CB CD34+ cells cultured for 7 days under control conditions in the presence of VPA were subsequently pulsed with BrdU for 2.5 hours. The cells were then washed with staining buffer (PBS plus 3% FBS) and stained with CD34-APC and CD90-PE mAbs, fixed and permeabilized with Cytofix/Cytoperm buffer and washed with Perm/Wash buffer (both from BD Pharmingen). After permeabilization, cells were treated with 30 μg of DNAse for 30 minutes at 37°C and then stained with an FITC-conjugated anti-BrdU antibody and 7AAD. The cell cycle status of CD34+CD90+ gated cells was then documented using a FACSCanto II Flow Cytometer with FACSDiva software (BD Biosciences).
Effects of HDACIs on HDACs.
Whole-cell extracts were prepared from freshly isolated CB-MNCs and human embryonic kidney 293 (HEK293) cells cultured in the presence of SCR (8 μM), C433 (80 μM), and VPA (1.25 mM). Cellular proteins were separated by SDS-PAGE using Novex (Invitrogen) and transferred by iBlot (Invitrogen). The membranes were probed with mAbs against individual histone deacetylases (HDAC1, -2, -3, -4, -5, and -6, and β-actin; Cell Signaling Technology) and developed using a chemiluminescence system with HRP-conjugated secondary antibodies (Amersham Biosciences) according to the manufacturer’s instructions. Densitometric analysis of Western blotting was performed with ImageJ software (NIH).
Isolation of primitive cells based on ALDH activity.
Increased ALDH activity is a characteristic of primitive hematopoietic cells and cancer stem cells (43, 44, 46, 62). To identify cell populations with high ALDH activity, an Aldefluor kit (StemCell Technologies Inc.) was used according to the manufacturer’s instructions. Cells (1 × 106/ml) were suspended in assay buffer, then half of the cells were added to the Aldefluor substrate (test sample) and the remaining half was added to DEAB inhibitor (control sample). The test and control samples were incubated for 40 minutes at 37°C. Cells were subsequently stained with CD34-APC and/or CD117-PE mAbs or an isotype-matched IgG for an additional 20 minutes. Cells were washed and analyzed by a BD FACSCanto II Flow Cytometer.
qPCR of pluripotency genes.
Total RNA was extracted from the human ES cell line H9 (WA09; WiCell Research Institute Inc., Madison, Wisconsin, USA), PCs, reisolated CD34+ cells from control cultures or cultures containing VPA in SF and SC media using TRIzol and an RNeasy kit from QIAGEN. Total RNA (0.5–1.0 μg) was reverse transcribed into cDNA using an RNA to cDNA EcoDry Premix kit (Clontech). The primer sequences are listed in Supplemental Table 4. qPCR was performed using SYBR Green (Thermo Fisher Scientific) and the Realplex thermocycler (Eppendorf). All experiments were performed in triplicate, and non-template controls (lacking cDNA template) were included in each assay. GAPDH served as an internal standard. The amplicons were run on a 2% agarose gel with a 50-bp-size (DNA ladder) marker.
Immunofluorescence staining.
PCs and reisolated CD34+ cells from control cultures and cultures containing VPA were fixed with methanol-free formaldehyde (2.8%) for 10 minutes at 37°C, briefly chilled on ice, and permeabilized with 100% ice-cold methanol. Cells were further incubated on ice for 20 minutes and blocked with incubation buffer (PBS containing 0.5% BSA) for 10 minutes and stained for SOX2 and OCT4 with an FITC-conjugated mAb or isotype controls for an hour at room temperature. Cells were also stained with a rabbit mAb for NANOG, followed by an FITC-conjugated anti-rabbit secondary antibody, and cells were washed and analyzed by flow cytometry.
PCs and reisolated CD34+ cells from control cultures and VPA-containing cultures were deposited onto glass slides, fixed with formaldehyde, and stained with antibodies according to the manufacturer’s instructions (Cell Signaling Technology) for SOX2, OCT4, NANOG, and ZIC3. ES cells, which express SOX2, OCT4, NANOG, and ZIC3, served as positive controls. Confocal microscopic analysis was performed using a Leica TCS SP5 (Wetzlar) and z-series. Images were acquired with LAS AF imaging software.
Co-IP of NANOG and OCT4.
ES cells and CD34+ cells treated with VPA for 7 days were lysed in RIPA buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton, 0.1% NP40, and 1.5 mM EDTA). Cell lysates from ES or VPA-treated cells (1.0 mg) were incubated with 6 μg of IgG (control), 20 μl (6 μg) of NANOG pAb (catalog AF1997; R&D Systems) and run overnight on a rolling platform at 4°C. Protein G beads (50 μl) (Cell Signaling Technology) were added the next day, and the samples continued rolling for an additional 4 hours. The beads were washed three times with lysis buffer, and the bound proteins were eluted by boiling the beads. Total cell lysates from ES (25 μg) and VPA-treated (125 μg) cells were fractionated by SDS-PAGE and analyzed by Western blotting using NANOG mAB (Cell Signaling Technology). Proteins from IP experiments were separated by SDS-PAGE, transferred using iBlot (Invitrogen), immunoblotted with goat pAb anti-OCT4 (Santa Cruz Biotechnology Inc.), washed, and developed using an ECL detection kit (SuperSignal West Pico; Thermo Scientific).
siRNA-mediated silencing of pluripotency genes.
CB CD34+ cells were treated with VPA under SF culture conditions. VPA-treated cells were transfected with individual SOX2, OCT4, NANOG, GAPDH, and scrambled siRNA or with a combination of SOX2, OCT4, and NANOG Silencer Select siRNAs (Invitrogen). A GFP plasmid was included to determine the transfection efficiency according to the manufacturer’s instructions for the Neon transfection system (Invitrogen).
After 72 hours of VPA treatment, cells (0.5 × 106 to 1 × 106) were washed in PBS and suspended in 8 μl of Neon resuspension buffer R (Invitrogen). Individual siRNAs (2 μl, 10–30 nM) or a combination of SOX2, OCT4, NANOG (SON), or pcDNA6.2/EmGFP plasmid (200 ng; Invitrogen) were mixed with the 8 μl of cells according to the manufacturer’s instructions. Cells were pulsed three times with a voltage of 1,400 and a width of 20 ms and immediately transferred to prewarmed media supplemented with cytokines plus VPA. Cell viability was assessed following 48 hours of transfection using the trypan blue exclusion method.
In addition, cells treated with scrambled or combined SON siRNA were stained using human CD34 and CD90 mAbs, and the percentages of CD34+ and CD34+CD90+ cells were determined by flow cytometric analysis. RNA was prepared after 7 days of transfection, and qPCR was performed using the SYBR Green method for SOX2, OCT4, NANOG, ZIC3, CD34, and GAPDH mRNA as described above. Relative expression levels were normalized to CD34 expression. SOX2, OCT4, NANOG, and ZIC3 protein expression levels following SON siRNA–mediated pluripotency gene knock down were assessed by confocal microscopy as described above.
Assay of in vivo marrow-repopulating potential of ex vivo–expanded CB CD34+ cells.
As described previously, NSG mice were sublethally irradiated with 300 cGy 4 hours prior to the infusion of PCs (2 × 105), and CD34+ cells reisolated after a week of culture under control conditions or cultures containing VPA in the presence or absence of cytokines were injected into NSG mice via the tail vein (17, 26, 27). Mice were sacrificed 13–14 weeks after transplantation. BM cells from each mouse were analyzed for the presence of cells expressing human CD45-PECy7 or APC, CD34-APC or FITC, CD36-APC, CD33-PECy7, CD14-FITC, CD19-PE, CD41-FITC, CD71-FITC, and glycophorin A–PE (GPA-PE). The presence of at least 0.1% human CD45+ cells in the marrow of each recipient mouse was considered indicative of donor human hematopoietic cell engraftment (17, 26, 27, 29). BM cells (2 × 106) from the primary recipient NSG mice were reinfused into sublethally irradiated secondary NSG recipient mice. Mice were sacrificed 15 –16 weeks after transplantation, and BM cells were stained with mAbs and analyzed by flow cytometry for evidence of human cell chimerism as described above.
Limiting dilution analysis.
The frequency of human SRCs in PCs and the progeny of an equivalent number of CD34+ cells that were expanded under control conditions or in the presence of VPA were analyzed by limiting dilution analysis as described previously (14). Increasing numbers of PCs (50, 250, 500, 2,500, 5,000) or the progeny of an equivalent number of PCs cultured with VPA for 7 days or under control conditions were infused into NSG mice (n = 111). The data from the limiting dilution experiments were pooled and analyzed by applying Poisson statistics to the single-hit model (n = 111). The frequency was calculated using L-Calc software (StemCell Technologies Inc.) and plotted using ELDA software (bioinf.wehi.edu.au/software/elda/), available at the Walter and Eliza Hall Bioinformatics Institute of Medical Research. The log fraction of nonresponding values was converted to the percentage of negative mice using the following formula: percentage of negative mice = e log fraction.
Assay for teratoma formation.
CD34+ cells (1 × 106) reisolated from control cultures or cultures containing VPA or a similar number of ES cells were suspended in 100 μl of PBS. These cells were mixed with an equal volume of ice-cold Matrigel and injected subcutaneously into the right hind limb of three NSG mice per group (reisolated CD34+ cells from control cultures, VPA-treated cultures, and ES cells). The mice were observed weekly for teratoma formation and sacrificed after 8 weeks. Teratomas were dissected, fixed, sectioned, and stained with H&E and examined morphologically (63).
Statistics.
Results are expressed as the mean ± SD or the mean ± SEM of varying numbers of individual experiments. Statistical differences were evaluated using the Student’s two-tailed t test unless otherwise specified. One-way ANOVA with pairwise comparison by Tukey’s test and/or Bartlett’s test for equal variance and the F test for variance comparison were also used. A P value less than or equal to 0.05 was considered significant.
Study approval.
All animal studies were approved by the Animal Care and Use Committee of the Icahn School of Medicine. Informed consent or subject approval was not required for this study, as low-volume unidentifiable CB units were purchased from the New York Blood Center.
Supplementary Material
Acknowledgments
This work was supported by a NYSTEM grant (C026431, to R. Hoffman). We thank James Godbold for his valuable advice on statistical analyses and Yan Li and Goar Mosoyan for providing technical assistance.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article:J Clin Invest. 2014;124(6):2378–2395. doi:10.1172/JCI70313.
See the related Commentary beginning on page 2365.
References
- 1.Cairo MS, Wagner JE. Placental and/or umbilical cord blood: an alternative source of hematopoietic stem cells for transplantation. Blood. 1997;90(12):4665–4678. [PubMed] [Google Scholar]
- 2.Dahlberg A, Delaney C, Bernstein ID. Ex vivo expansion of human hematopoietic stem and progenitor cells. Blood. 2011;117(23):6083–6090. doi: 10.1182/blood-2011-01-283606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Delaney C, Bollard CM, Shpall EJ. Cord blood graft engineering. Biol Blood Marrow Transplant. 2013;19(1 suppl):S74–S78. doi: 10.1016/j.bbmt.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Navarrete C, Contreras M. Cord blood banking: a historical perspective. Br J Haematol. 2009;147(2):236–245. doi: 10.1111/j.1365-2141.2009.07827.x. [DOI] [PubMed] [Google Scholar]
- 5.Stanevsky A, Goldstein G, Nagler A. Umbilical cord blood transplantation: pros, cons and beyond. Blood Rev. 2009;23(5):199–204. doi: 10.1016/j.blre.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 6.Barker JN, Scaradavou A, Stevens CE. Combined effect of total nucleated cell dose and HLA match on transplantation outcome in 1061 cord blood recipients with hematologic malignancies. Blood. 2010;115(9):1843–1849. doi: 10.1182/blood-2009-07-231068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Delaney C, Ratajczak MZ, Laughlin MJ. Strategies to enhance umbilical cord blood stem cell engraftment in adult patients. Expert Rev Hematol. 2010;3(3):273–283. doi: 10.1586/ehm.10.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ballen KK, et al. Double unrelated reduced-intensity umbilical cord blood transplantation in adults. Biol Blood Marrow Transplant. 2007;13(1):82–89. doi: 10.1016/j.bbmt.2006.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rao M, Ahrlund-Richter L, Kaufman DS. Concise review: Cord blood banking, transplantation and induced pluripotent stem cell: success and opportunities. Stem Cells. 2012;30(1):55–60. doi: 10.1002/stem.770. [DOI] [PubMed] [Google Scholar]
- 10.Giebel B, et al. Primitive human hematopoietic cells give rise to differentially specified daughter cells upon their initial cell division. Blood. 2006;107(5):2146–2152. doi: 10.1182/blood-2005-08-3139. [DOI] [PubMed] [Google Scholar]
- 11.Ho AD, Wagner W. The beauty of asymmetry: asymmetric divisions and self-renewal in the haematopoietic system. Curr Opin Hematol. 2007;14(4):330–336. doi: 10.1097/MOH.0b013e3281900f12. [DOI] [PubMed] [Google Scholar]
- 12.Huang S, Law P, Francis K, Palsson BO, Ho AD. Symmetry of initial cell divisions among primitive hematopoietic progenitors is independent of ontogenic age and regulatory molecules. Blood. 1999;94(8):2595–2604. [PubMed] [Google Scholar]
- 13.Srour EF, et al. Modulation of in vitro proliferation kinetics and primitive hematopoietic potential of individual human CD34+CD38–/lo cells in G0. Blood. 2005;105(8):3109–3116. doi: 10.1182/blood-2004-05-1773. [DOI] [PubMed] [Google Scholar]
- 14.Boitano AE, et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science. 2010;329(5997):1345–1348. doi: 10.1126/science.1191536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Felice L, et al. Histone deacetylase inhibitor valproic acid enhances the cytokine-induced expansion of human hematopoietic stem cells. Cancer Res. 2005;65(4):1505–1513. doi: 10.1158/0008-5472.CAN-04-3063. [DOI] [PubMed] [Google Scholar]
- 16.Himburg HA, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16(4):475–482. doi: 10.1038/nm.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Milhem M, et al. Modification of hematopoietic stem cell fate by 5aza 2′deoxycytidine and trichostatin A. Blood. 2004;103(11):4102–4110. doi: 10.1182/blood-2003-07-2431. [DOI] [PubMed] [Google Scholar]
- 18.Nishino T, et al. Ex vivo expansion of human hematopoietic stem cells by a small-molecule agonist of c-MPL. Exp Hematol. 2009;37(11):1364–1377. doi: 10.1016/j.exphem.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 19.North TE, et al. Prostaglandin E2 regulates vertebrate haematopoietic stem cell homeostasis. Nature. 2007;447(7147):1007–1011. doi: 10.1038/nature05883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.de Lima M, et al. Transplantation of ex vivo expanded cord blood cells using the copper chelator tetraethylenepentamine: a phase I/II clinical trial. Bone Marrow Transplant. 2008;41(9):771–778. doi: 10.1038/sj.bmt.1705979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Lima M, et al. Cord-blood engraftment with ex vivo mesenchymal-cell coculture. N Engl J Med. 2012;367(24):2305–2315. doi: 10.1056/NEJMoa1207285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Delaney C, Heimfeld S, Brashem-Stein C, Voorhies H, Manger RL, Bernstein ID. Notch-mediated expansion of human cord blood progenitor cells capable of rapid myeloid reconstitution. Nat Med. 2010;16(2):232–236. doi: 10.1038/nm.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goessling W, et al. Prostaglandin E2 enhances human cord blood stem cell xenotransplants and shows long-term safety in preclinical nonhuman primate transplant models. Cell Stem Cell. 2011;8(4):445–458. doi: 10.1016/j.stem.2011.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hoggatt J, et al. Differential stem- and progenitor-cell trafficking by prostaglandin E2. Nature. 2013;495(7441):365–369. doi: 10.1038/nature11929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.O’Leary H, Ou X, Broxmeyer HE. The role of dipeptidyl peptidase 4 in hematopoiesis and transplantation. Curr Opin Hematol. 2013;20(4):314–319. doi: 10.1097/MOH.0b013e32836125ac. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Araki H, et al. Expansion of human umbilical cord blood SCID-repopulating cells using chromatin-modifying agents. Exp Hematol. 2006;34(2):140–149. doi: 10.1016/j.exphem.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 27.Araki H, Yoshinaga K, Boccuni P, Zhao Y, Hoffman R, Mahmud N. Chromatin-modifying agents permit human hematopoietic stem cells to undergo multiple cell divisions while retaining their repopulating potential. Blood. 2007;109(8):3570–3578. doi: 10.1182/blood-2006-07-035287. [DOI] [PubMed] [Google Scholar]
- 28.Azuara V, et al. Chromatin signatures of pluripotent cell lines. Nat Cell Biol. 2006;8(5):532–538. doi: 10.1038/ncb1403. [DOI] [PubMed] [Google Scholar]
- 29.Chaurasia P, Berenzon D, Hoffman R. Chromatin-modifying agents promote the ex vivo production of functional human erythroid progenitor cells. Blood. 2011;117(17):4632–4641. doi: 10.1182/blood-2010-10-314567. [DOI] [PubMed] [Google Scholar]
- 30.Delcuve GP, Khan DH, Davie JR. Roles of histone deacetylases in epigenetic regulation: emerging paradigms from studies with inhibitors. Clin Epigenetics. 2012;4(1):5. doi: 10.1186/1868-7083-4-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gul H, Marquez-Curtis LA, Jahroudi N, Lo J, Turner AR, Janowska-Wieczorek A. Valproic acid increases CXCR4 expression in hematopoietic stem/progenitor cells by chromatin remodeling. Stem Cells Dev. 2009;18(6):831–838. doi: 10.1089/scd.2008.0235. [DOI] [PubMed] [Google Scholar]
- 32.Asai T, et al. Necdin, a p53 target gene, regulates the quiescence and response to genotoxic stress of hematopoietic stem/progenitor cells. Blood. 2012;120(8):1601–1612. doi: 10.1182/blood-2011-11-393983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J. Quiescence regulators for hematopoietic stem cell. Exp Hematol. 2011;39(5):511–520. doi: 10.1016/j.exphem.2011.01.008. [DOI] [PubMed] [Google Scholar]
- 34.Will B, et al. Satb1 regulates the self-renewal of hematopoietic stem cells by promoting quiescence and repressing differentiation commitment. Nat Immunol. 2013;14(5):437–445. doi: 10.1038/ni.2572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wilson A, et al. Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell. 2008;135(6):1118–1129. doi: 10.1016/j.cell.2008.10.048. [DOI] [PubMed] [Google Scholar]
- 36.Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333(6039):218–221. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- 37.Kramer OH, et al. The histone deacetylase inhibitor valproic acid selectively induces proteasomal degradation of HDAC2. EMBO J. 2003;22(13):3411–3420. doi: 10.1093/emboj/cdg315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cedar H, Bergman Y. Epigenetics of haematopoietic cell development. Nat Rev Immunol. 2011;11(7):478–488. doi: 10.1038/nri2991. [DOI] [PubMed] [Google Scholar]
- 39.Cunliffe VT. Eloquent silence: developmental functions of Class I histone deacetylases. Curr Opin Genet Dev. 2008;18(5):404–410. doi: 10.1016/j.gde.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kouzarides T. Chromatin modifications and their function. Cell. 2007;128(4):693–705. doi: 10.1016/j.cell.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Oh IH, Humphries RK. Concise review: multidimensional regulation of the hematopoietic stem cell state. Stem Cells. 2012;30(1):82–88. doi: 10.1002/stem.776. [DOI] [PubMed] [Google Scholar]
- 42.Elizalde C, et al. Histone deacetylase 3 modulates the expansion of human hematopoietic stem cells. Stem Cells Dev. 2010;21(14):2581–2591. doi: 10.1089/scd.2011.0698. [DOI] [PubMed] [Google Scholar]
- 43.Hess DA, et al. Functional characterization of highly purified human hematopoietic repopulating cells isolated according to aldehyde dehydrogenase activity. Blood. 2004;104(6):1648–1655. doi: 10.1182/blood-2004-02-0448. [DOI] [PubMed] [Google Scholar]
- 44.Lioznov MV, Freiberger P, Kroger N, Zander AR, Fehse B. Aldehyde dehydrogenase activity as a marker for the quality of hematopoietic stem cell transplants. Bone Marrow Transplant. 2005;35(9):909–914. doi: 10.1038/sj.bmt.1704928. [DOI] [PubMed] [Google Scholar]
- 45.Spangrude GJ, Brooks DM, Tumas DB. Long-term repopulation of irradiated mice with limiting numbers of purified hematopoietic stem cells: in vivo expansion of stem cell phenotype but not function. Blood. 1995;85(4):1006–1016. [PubMed] [Google Scholar]
- 46.Storms RW, et al. Distinct hematopoietic progenitor compartments are delineated by the expression of aldehyde dehydrogenase and CD34. Blood. 2005;106(1):95–102. doi: 10.1182/blood-2004-09-3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veeraputhiran M, Katragadda L, Balamurugan A, Cottler-Fox M. Aldehyde dehydrogenase as an alternative to enumeration of total and viable CD34(+) cells in autologous hematopoietic progenitor cell transplantation. Cytotherapy. 2011;13(10):1256–1258. doi: 10.3109/14653249.2011.602340. [DOI] [PubMed] [Google Scholar]
- 48.Boyer LA, et al. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell. 2005;122(6):947–956. doi: 10.1016/j.cell.2005.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loh YH, et al. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38(4):431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 50.Redshaw Z, Strain AJ. Human haematopoietic stem cells express Oct4 pseudogenes and lack the ability to initiate Oct4 promoter-driven gene expression. J Negat Results Biomed. 2010;9(1):2–8. doi: 10.1186/1477-5751-9-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zangrossi S, et al. Oct-4 expression in adult human differentiated cells challenges its role as a pure stem cell marker. Stem Cells. 2007;25(7):1675–1680. doi: 10.1634/stemcells.2006-0611. [DOI] [PubMed] [Google Scholar]
- 52.Takahashi K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131(5):861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- 53.Lim LS, et al. Zic3 is required for maintenance of pluripotency in embryonic stem cells. Mol Biol Cell. 2007;18(4):1348–1358. doi: 10.1091/mbc.E06-07-0624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Declercq J, Sheshadri P, Verfaillie CM, Kumar A. Zic3 enhances the generation of mouse induced pluripotent stem cells. Stem Cells Dev. 2013;22(14):2017–2025. doi: 10.1089/scd.2012.0651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sauvageau G, Iscove NN, Humphries RK. In vitro and in vivo expansion of hematopoietic stem cells. Oncogene. 2004;23(43):7223–7232. doi: 10.1038/sj.onc.1207942. [DOI] [PubMed] [Google Scholar]
- 56.Walasek MA, van Os R, de Haan G. Hematopoietic stem cell expansion: challenges and opportunities. Ann N Y Acad Sci. 2012;1266:138–150. doi: 10.1111/j.1749-6632.2012.06549.x. [DOI] [PubMed] [Google Scholar]
- 57.Hirai H, Katoku-Kikyo N, Karian P, Firpo M, Kikyo N. Efficient iPS cell production with the MyoD transactivation domain in serum-free culture. PLoS One. 2012;7(3):e34149. doi: 10.1371/journal.pone.0034149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Motabi IH, DiPersio JF. Advances in stem cell mobilization. Blood Rev. 2012;26(6):267–278. doi: 10.1016/j.blre.2012.09.003. [DOI] [PubMed] [Google Scholar]
- 59.Yu KR, et al. CD49f enhances multipotency and maintains stemness through the direct regulation of OCT4 and SOX2. Stem Cells. 2012;30(5):876–887. doi: 10.1002/stem.1052. [DOI] [PubMed] [Google Scholar]
- 60.Shivtiel S, et al. CD45 regulates homing and engraftment of immature normal and leukemic human cells in transplanted immunodeficient mice. Exp Hematol. 2011;39(12):1161–1170. doi: 10.1016/j.exphem.2011.08.012. [DOI] [PubMed] [Google Scholar]
- 61.Colvin GA, Lambert JF, Dooner MS, Cerny J, Quesenberry PJ. Murine allogeneic in vivo stem cell homing(,). J Cell Physiol. 2007;211(2):386–391. doi: 10.1002/jcp.20945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Aguila JR, et al. SALL4 is a robust stimulator for the expansion of hematopoietic stem cells. Blood. 2011;118(3):576–585. doi: 10.1182/blood-2011-01-333641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.O’Connor MD, Kardel MD, Eaves CJ. Functional assays for human embryonic stem cell pluripotency. Methods Mol Biol. 2011;690:67–80. doi: 10.1007/978-1-60761-962-8_4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.