

Abstract
Hepatic forkhead protein FoxO1 is a key component of systemic glucose homeostasis via its ability to regulate the transcription of rate-limiting enzymes in gluconeogenesis. Important in the regulation of FoxO1 transcriptional activity are the modifying/demodifying enzymes that lead to posttranslational modification. Here, we demonstrate the functional interaction and regulation of FoxO1 by herpesvirus-associated ubiquitin-specific protease 7 (USP7; also known as herpesvirus-associated ubiquitin-specific protease, HAUSP), a deubiquitinating enzyme. We show that USP7-mediated mono-deubiquitination of FoxO1 results in suppression of FoxO1 transcriptional activity through decreased FoxO1 occupancy on the promoters of gluconeogenic genes. Knockdown of USP7 in primary hepatocytes leads to increased expression of FoxO1-target gluconeogenic genes and elevated glucose production. Consistent with this, USP7 gain-of-function suppresses the fasting/cAMP-induced activation of gluconeogenic genes in hepatocyte cells and in mouse liver, resulting in decreased hepatic glucose production. Notably, we show that the effects of USP7 on hepatic glucose metabolism depend on FoxO1. Together, these results place FoxO1 under the intimate regulation of deubiquitination and glucose metabolic control with important implication in diseases such as diabetes.
Glucose homeostasis is maintained by action of the opposing pancreatic hormones glucagon and insulin. Glucagon serves in part to induce hepatic gluconeogenesis, the de novo synthesis of glucose from noncarbohydrate precursors, during states of nutrient deprivation. During feeding, insulin curtails the increase in blood glucose by increasing glucose uptake in peripheral tissues and suppressing gluconeogenesis in the liver. However, in diabetic individuals with insulin resistance, the liver functions as in the fasted state and inappropriately activates gluconeogenesis (1). Antidiabetic drugs, such as the widely prescribed metformin, are believed to lower blood glucose primarily through repression of hepatic gluconeogenesis (2).
Both insulin and glucagon lead to rapid changes in signaling pathways that converge on the transcriptional regulation of the rate-limiting enzymes in gluconeogenesis, glucose-6-phosphatase (G6Pc; G6Pase) and phosphoenolpyruvate carboxykinase (Pck1; PEPCK). Glucagon release in the fasted state initiates the gluconeogenic gene program via induction of the cAMP/protein kinase A (PKA) pathway and activation of transcription factors such as cAMP-responsive element binding protein (CREB), hepatic nuclear factor 4α, and forkhead box O1 (FoxO1) (3–6). As an additional level of regulation, coactivators peroxisome proliferator-activated receptor-γ coactivator 1 α (PGC-1α) and cAMP-regulated transcriptional coactivator 2 interact with and potentiate the transcriptional activity of these gluconeogenic transcription factors (3, 7, 8). In opposition, insulin suppresses hepatic gluconeogenesis primarily through its activation of phosphatidylinositol 3-kinase (PI3K) and Akt pathways, leading to negative regulation of PGC-1α and FoxO1 (9–11).
FoxO1, a member of the FoxO subfamily of forkhead winged/helix transcription factors, is critical for the insulin-mediated suppression of gluconeogenesis (12, 13). FoxO1 interacts with an insulin-responsive element (IRE) on the promoter region(s) of G6Pc and Pck1 (14–17). Insulin-signaling results in suppression of FoxO1 transcriptional activity through Akt-dependent phosphorylation of FoxO1 on specific conserved residues (T24, S256, and S319 in human FoxO1) (15, 18). Once phosphorylated, FoxO1 associates with 14-3-3 proteins, leading to cytoplasmic sequestration (19) followed by ubiquitination and degradation (20, 21). Consistent with the suppressive effect of insulin on FoxO1 activity, insulin resistance in diabetes leads to hyperactivation of FoxO1 with consequent elevation of FoxO1 target genes (5). Mice lacking hepatic FoxO1 display reduced gluconeogenic gene expression, increased glucose tolerance, and reduced hepatic glucose production (13). Moreover, inhibition of hepatic FoxO1 has been shown to ameliorate fasting hyperglycemia in several diabetic animal models (5, 12, 22). Thus, the rapid repression of FoxO1 and subsequent decrease in gluconeogenic gene expression is a key component of glucose homeostasis and may provide a strategy for intervention in the management of insulin-resistant diabetes.
FoxO family members govern a variety of cellular processes, some unique and some overlapping, and exhibit considerable levels of regulation. In general, FoxO activity is regulated by posttranslational modifications that affect subcellular localization, protein stability, and DNA binding. These include phosphorylation, acetylation, methylation, glycosylation, and ubiquitination (reviewed in [23]). Some of these modifications and the responsible kinases/enzymes are shared among family members, but the extent to which they have been characterized is by no means exhaustive. Thus, a better grasp on the effectors and their mode of regulation has the potential to reveal fresh alternatives for controlling FoxO1 activity.
Ubiquitination, the covalent addition of ubiquitin moieties to a target protein, can lead to decreased stability (through ubiquitin-targeted proteasomal degradation) or alteration of localization and/or activity of the modified protein. And the process of ubiquitin modification can be reversed by the action of deubiquitinating enzymes (24). Recently, the deubiquitinating enzyme ubiquitin-specific protease 7 (USP7; also known as herpesvirus-associated ubiquitin-specific protease, HAUSP) has been identified in the negative regulation of FoxO3/4 transcriptional activity. Monoubiquitination of FoxO3/4 results in relocalization to the nucleus, which is counteracted by USP7-induced deubiquitination (25).
USP7 belongs to the ubiquitin-specific proteases family of deubiquitinating enzymes and contains a characteristic cysteine motif in its catalytic domain (26). USP7 is most notable for its complex role in regulating stability of the p53 tumor suppressor, where it directly binds to and deubiquitinates both p53 and Mdm2, the E3 ubiquitin ligase responsible for p53 destabilization (27, 28). In fact, USP7 regulates several tumor suppressors, and a role for inhibition of USP7 in cancer is an active area of study (29). FoxO proteins are considered tumor suppressors (30), making the interaction of USP7 with FoxO3/4 highly attractive to cancer research. However, it is unknown whether USP7 controls the action of FoxO proteins in other biological areas, namely, glucose metabolism.
Although the effect of polyubiquitination and ubiquitin-targeted proteasomal degradation of FoxO1 is well appreciated, a role for monoubiquitination of FoxO1 per se has not been examined. We hypothesized that a similar interaction with USP7 might exist for FoxO1, which led us to interrogate a mechanistic link between USP7 and FoxO1 metabolic function. Here, we expand the list of USP7 targets to include FoxO1, showing that USP7 deubiquitinates monoubiquitinated FoxO1 to affect its association with the G6Pc and Pck1 promoters. We demonstrate that knockdown of USP7 elevates gluconeogenic genes whereas overexpression of USP7 leads to suppression of gluconeogenesis in cell culture and in the whole animal. Notably, we determine a requirement of FoxO1 activity for USP7's effects on gluconeogenic gene expression. Taken together, our findings reveal USP7 activation as a powerful inhibitor of FoxO1 activity and a potential therapeutic route in the amelioration of hyperglycemia associated with insulin-resistant malignancies.
Materials and Methods
DNA constructs and adenoviruses
The pcDNA3 Flag-FoxO1, 3X IRS luciferase, and myristoylated Akt reporter constructs have previously been described (31). Myc-tagged USP7 was a gift from Dr Pier Pandolfi, and pCl-neo Flag-tagged USP7 was provided by Dr Bert Vogelstein via Addgene plasmid 16655 (32). pRK5 HA-Ubiquitin (Ub) wild-type and lysine-less KO (Addgene plasmids 17608 and 17603, respectively) were provided by Dr Ted Dawson (33). A Flag-tagged noninsulin-sensitive FoxO1 mutant (pcDNA3 Flag-FoxO1 T24A/S256A/S319A; Addgene plasmid 13508) was provided by Dr Kunliang Guan (31). Plasmids carrying cDNAs encoding FoxO1-GFP fusion proteins were gifts from Dr Alexander Banks and have previously been described (34). Point mutant generation of USP7 C223S was conducted by PCR-based mutagenesis. Short-hairpin RNA (shRNA) constructs targeting USP7 (with 100% sequence complementarity to both human and mouse) were generated in a pLKO.1 backbone with the following target sequences: shUSP7 #1, 5′-TGTATCTATTGACTGCCCTTT-3′; and shUSP7 #2, 5′-GGCAACCTTTCAGTTCACTGT-3′.
Adenoviruses were generated with the pAd-Track/pAd-Easy system unless otherwise noted. Flag-USP7 adenoviruses were subcloned from pCl-neo Flag-USP7 constructs and express USP7 under a cytomegalovirus promoter. Adenoviruses expressing shRNAs were subcloned from pLKO.1 vectors and are driven by a U6 promoter. A nontargeting control shRNA adenovirus containing a scrambled sequence has previously been described (35). Adenoviruses encoding FoxO1 shRNA and corresponding control shRNA were gifts from Dr Alexander Banks and were generated as previously described (36). All adenoviruses were amplified in HEK293A cells, purified by CsCl gradient centrifugation, and dialyzed in buffer containing 10 mM Tris pH, 8.0; 2 mM MgCl2; and 4% glycerol. Adenoviral titer was determined by serial dilution/infection of HEK293A cells and quantifying the number of fluorescing cells.
Cell culture and treatment
HEK293A cells were maintained in DMEM containing 10% fetal bovine serum and penicillin/streptomycin. Transfections were performed with Lipofectamine 2000 transfection reagent (Invitrogen; Life Technologies) according to the manufacturer's recommendations. Where indicated, HEK293A cells were rinsed with PBS and switched to DMEM-lacking serum. Live-cell visualization of FoxO1-GFP localization was imaged by fluorescence microscopy on a Leica DMI6000B microscope (Leica Microsystems). Images were processed in Adobe Photoshop CS4 software to increase brightness for publication; consistent threshold settings were maintained across images.
Mouse primary hepatocytes were isolated from male C57BL/6 mice by perfusion with liver digest medium (Gibco, Life Technologies; pH, 7.4) followed by 70 μm filtration exclusion and Percoll (Sigma) gradient centrifugation. Cells were seeded in DMEM containing 10% fetal bovine serum, 2 mM sodium pyruvate, 1 μM dexamethasone, 0.1 μM insulin, and penicillin/streptomycin. After cell attachment, medium was replaced with DMEM supplemented with 0.2% BSA, 2 mM sodium pyruvate, 0.1 μM dexamethasone, 1 nM insulin, and penicillin/streptomycin (maintenance medium). Adenovirus infections were performed the following day for 4 hours with 3.5×106 infectious particles per 4×105 cells. Fresh maintenance medium was replenished daily. Cells were collected 48 hours postinfection. Where indicated, cells were serum-starved in DMEM containing 0.2% BSA, 2 mM sodium pyruvate, and penicillin/streptomycin prior to stimulation with insulin (Sigma) and/or forskolin (Fisher). For measurement of glucose production, cells were cultured for 3 hours in 0.2% BSA, phenol red-free, glucose-free DMEM. Amount of glucose in the medium was quantified using a colorimetric glucose assay (EnzyChrom, BioAssay Systems) and normalized to total protein amount in the whole-cell lysates. Assays were conducted with or without 20 mM sodium lactate and 2 mM sodium pyruvate, and glucose production in the absence of lactate/pyruvate subtracted from that produced in the presence of lactate/pyruvate.
Transcriptional reporter assays
HEK293A cells were transfected to a fixed amount of DNA as corrected with empty vector plasmid. Cells were collected 24 hours posttransfection with 1× Passive Lysis Buffer (Promega). Firefly luciferase reporter was determined by addition of Luciferase Assay Substrate (Dual-Luciferase Reporter Assay System, Promega) and quantification of luminescence on a FLUOstar Omega plate reader (BMG Labtech). Of note, cytomegalovirus-driven Renilla luciferase vector was cotransfected as an internal control; however, despite a range of experimental vector to control vector ratios tested, Renilla luciferase activity was sensitive to cotransfected plasmids (data not shown) and, thus, withheld from normalization. Data are presented as firefly luciferase reporter values alone and are representative of at least two independent experiments.
Chromatin immunoprecipitation
Primary hepatocytes stimulated for 1.5 hours with 10 μM forskolin or dimethylsulfoxide vehicle were fixed in 1% formaldehyde for 10 minutes at room temperature. Crosslinking was quenched by adding glycine to a final concentration of 125 mM and rinsing twice with cold PBS. Cells were collected in PBS containing protease inhibitors followed by mild lysis (10 mM HEPES, pH, 7.9; 10 mM KCl; 1.5 mM MgCl2; 0.5% Igepal; 1 mM phenylmethanesulfonyl fluoride (PMSF); and protease inhibitors) and centrifugation. The resulting nuclear pellets were resuspended in chromatin immunoprecipitation (ChIP) buffer (50 mM HEPES pH, 7.9; 140 mM NaCl; 1 mM EDTA; 1% Triton X-100; 0.1% NaDOC, 0.1% sodium dodecyl sulfate (SDS); 1 mM PMSF, and protease inhibitors) and chromatin sheared by sonication with a Diagenode Biorupter for three cycles of 5 minutes (30 seconds on, 30 seconds off). Samples were clarified and chromatin immunoprecipitated overnight at 4 °C with anti-FoxO1 (C29H4, Cell Signaling Technologies) or isotype rabbit immunoglobulin G (Abcam). Immunecomplexes were recovered with Protein A magnetic beads (Dynabeads; Novex, Life Technologies) preblocked with salmon sperm DNA (Invitrogen, Life Technologies). Following extensive washes, immunoprecipitated DNA was then isolated with a Chelex 100-based DNA purification method described in (37). Input DNA was prepared from 10% of respective chromatin prior to precipitation. Immunoprecipitated DNA and input DNA were analyzed by quantitative real-time PCR with primers specific for the G6Pc IRE (forward: 5′-TGGCTTCAAGGACCAGGAAG-3′ and reverse: 5′-TGCAAACATGTTCAGGGTGA-3′), Pck1 IRE (forward: 5′-TGGCTCAGAGCTGAATTTCC-3′ and reverse: 5′-CCTGTTGCTGATGCAAACTG-3′), and a control genomic region 17.9 kb upstream of Cycs (forward: 5′-GGCTCTCCTTGCAGTTTTTG-3′ and reverse: 5′-CCGACCTTTACATCGCCTAA-3′). Enrichment of specific promoter regions after immunoprecipitation was calculated as percent of input.
Animal experiments and procedures
Mouse experiments were performed with 10-week-old male C57BL/6 mice purchased from Taconic Farms and allowed at least 1 wk of acclimation to our facilities. All mice were maintained on normal chow and housed under a 12 hours light/12 hours dark cycle at 22 °C. Mice were handled for 3 days prior to adenovirus infection (1.5×109 infectious particles per mouse) by tail vein injection under isofluorane anesthesia. All analyses were performed 4 days after infection. For gene expression analysis, mice were killed after a 48-hours fast; the livers were snap-frozen in liquid nitrogen and stored at −80 °C until processing. Pyruvate tolerance tests were performed on animals fasted for 16 hours prior to ip injection of 2 g/kg sodium pyruvate dissolved in sterile PBS. Glycemia was measured by tail bleed at the indicated times using a glucometer (Precision Xtra, Abbott Diabetes Care). All studies were performed according to protocols approved by Dana-Farber Cancer Institute's Animal Care and Use Committee.
Quantitative real-time PCR analysis
Total RNA was extracted from cells or pulverized liver using TRIzol reagent (Ambion, Life Technologies), followed by cDNA preparation from 2 μg of total RNA with a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) on an MJ Mini thermal cycler (Bio-Rad). cDNA products were quantified by real-time PCR using Power SYBR Green PCR Master Mix (Applied Biosystems) on a CFX384 Real-Time PCR System (Bio-Rad). Gene expression was determined by generation of a standard curve and normalized for the expression of 36B4. All primer sequences are available upon request.
Western blotting
Whole-cell extracts were prepared in radioimmunoprecipitation assay buffer containing phosphatase inhibitors (1 mM glycerol-2-phosphate, 5 mM NaF, and 1 mM Na orthovanadate), 1 mM PMSF, and protease inhibitors (Roche). Cytoplasmic and nuclear fractionation from cells and pulverized liver were performed as previously described with minor modifications (38). Briefly, cytoplasmic fractions were obtained from a buffer A containing 10 mM HEPES, pH, 7.9; 10 mM KCl; 1.5 mM MgCl2, 0.5% Igepal; phosphatase inhibitors; 1 mM PMSF; and protease inhibitors. After washing with buffer A, nuclear pellets were resuspended in a buffer B containing 20 mM HEPES, pH, 7.9; 150 mM NaCl; 1.5 mM MgCl2; 0.2 mM EDTA; 15% glycerol; 0.5% Igepal; 0.3% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; phosphatase inhibitors; 1 mM PMSF; and protease inhibitors. Protein concentration was determined by detergent-compatible protein assay (Bio-Rad) and lysates separated by SDS-PAGE, transferred to Immobilon-P transfer membranes (Millipore), and blotted according to manufacturer's recommendations for the indicated antibodies. Antibodies for the detection of FoxO1, phospho-FoxO1 (Ser256), phospho-FoxO1 (Thr24), Akt, phospho-Akt (Thr308), phospho-Akt (Ser473), GSK3β and phospho-GSK3β (Ser9) were from Cell Signaling Technologies. Anti-USP7 antibody was purchased from Bethyl Laboratories, Inc. Anti-HA, anti-Myc, and anti-Flag peroxidase conjugates were from Sigma. Horse peroxidase-conjugated secondary antibodies were from Jackson ImmunoResearch. Anti-Lamin B1 (Abcam) and anti-β-Tubulin (Millipore) were used as loading controls.
Coimmunoprecipitations
To assess the endogenous interaction of USP7 with FoxO1 from primary hepatocytes, nuclear extracts were prepared in radioimmunoprecipitation assay buffer without SDS containing phosphatase inhibitors, 1 mM PMSF, and protease inhibitors. Clarified lysates were precleared with 20 μL of a protein A sepharose (GE Healthcare Bio-Sciences) slurry prior to incubation with anti-FoxO1 or isotype rabbit immunoglobulin G overnight at 4 °C. The immunecomplexes were then precipitated by addition of 20 μL protein A sepharose slurry and incubation for 2 hours at 4 °C. Coimmunoprecipitations (co-IPs) of epitope-tagged USP7 and FoxO1 were performed with clarified whole-cell extracts from HEK293A cells resuspended in co-IP buffer (20 mM HEPES, pH, 7.9; 125 mM NaCl; 1 mM EDTA; 0.1% Igepal; 0.3% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate; phosphatase inhibitors; 1 mM PMSF; and protease inhibitors), where complexes were captured overnight by immunoprecipitation with anti-Flag agarose (Sigma). Immunoprecipitated proteins were washed four times with respective lysis buffer, eluted with 2X SDS sample buffer and boiling, and detected by immunoblotting after separation by SDS-PAGE.
Deubiquitination assays
For detection of FoxO1 deubiquitination in vivo, HEK293A cells transfected with the indicated plasmids were lysed in 1% SDS buffer containing 50 mM Tris, pH, 7.5; 150 mM NaCl; 0.1% Triton X-100; phosphatase inhibitors; 1 mM PMSF; protease inhibitors; and 0.5 mM N-ethylmaleimide (Boston Biochem); boiled for 10 min, and sonicated. Lysates were immunoprecipitated with anti-Flag agarose after dilution to 0.1% SDS and clarification. Immunoprecipitates were washed with high-salt buffer (50 mM Tris, pH, 7.5; 250 mM NaCl; 1% Triton X-100; 1 mM EDTA; and inhibitors), eluted as above, and analyzed by SDS-PAGE. Following transfer, membranes were autoclaved prior to immunoblotting for ubiquitinated species. For in vitro deubiquitination assays, Flag-tagged FoxO1 and USP7 were anti-Flag affinity purified from HEK293A cells transfected with plasmids encoding the respective proteins. HA-tagged monoubiquitinated Flag-FoxO1 was immunoprecipitated as described above, and Flag-USP7 was immunoprecipitated from cells lysed in co-IP buffer. After extensive washing, immunoprecipitates were eluted in deubiquitination buffer (50 mM Tris, pH, 7.5; 150 mM NaCl; 0.001% Triton X-100; 1 mM EDTA; 10 mM dithiothreitol, 5% glycerol; and inhibitors) with 200 μg/mL 3X Flag-peptide (Sigma). Purified USP7, wild-type or C223S mutant, was added to aliquots of purified monoubiquitinated FoxO1 and reactions incubated at 30 °C for 1 hour. Reactions were terminated using 6X SDS sample buffer and analyzed by western blotting.
Statistical analysis
Data were analyzed using Prism software (GraphPad Software, Inc) and are expressed as mean ± SEM. Two-tailed Student t tests and one-way ANOVA with Newman-Keuls Multiple Comparison test were used to compare means between groups as indicated; P < .05 was considered significant.
Results
USP7 interacts with and deubiquitinates monoubiquitinated FoxO1
It has been reported that USP7 targets FoxO family members FoxO3 and FoxO4, where deubiquitination of monoubiquitinated FoxO3/4 leads to nuclear exclusion and decreased activity (25). However, FoxO1 as a USP7 substrate and the effect of monoubiquitination on FoxO1 activity have not been explored. To assess whether FoxO1 is also targeted by USP7, we first determined whether USP7 interacts with FoxO1. Immunoprecipitation of FoxO1 from the nuclear fraction of cultured mouse hepatocytes showed binding of endogenous USP7 to FoxO1 (Figure 1A). For further mechanistic analysis, we verified this interaction in an amenable transient expression system. HEK293A cells were cotransfected with Myc-tagged USP7 (Myc-USP7) and Flag-tagged FoxO1 (Flag-FoxO1) followed by coimmunoprecipitation. As shown in Figure 1B, immunoprecipitation of Flag-FoxO1 clearly revealed Myc-USP7 bound to FoxO1. Notably, USP7 activity is not necessary for this interaction, as both wild-type and a catalytically inactive mutant of USP7 (C223S; CS) efficiently interacted with FoxO1.
Figure 1.

FoxO1 is a substrate of USP7. A, Endogenous USP7 and FoxO1 interact. Western blot analysis of immunoprecipitated FoxO1 from nuclear extracts of primary hepatocytes that were serum-starved overnight. B, USP7 catalytic activity is not required for interaction with FoxO1. Western blot analysis of whole-cell extracts (input) or following coimmunoprecipitation with anti-Flag agarose (IP: Flag) of HEK293A cells transfected with Flag-FoxO1 and Myc-USP7 constructs. C, Wild-type USP7 deubiquitinates FoxO1. Western blot analysis of whole-cell extracts (input) or following immunoprecipitation with anti-Flag agarose (IP: Flag) of HEK293A cells transfected with indicated constructs. D, FoxO1 is monoubiquitinated. Western blot analysis of whole-cell extracts (input) or following immunoprecipitation with anti-Flag agarose (IP: Flag) of HEK293A cells transfected with Flag-FoxO1 and HA-tagged wild-type ubiquitin or a lysine-less ubiquitin mutant (Ub KO). E, USP7 deubiquitinates monoubiquitinated FoxO1 in vitro. Ubiquitinated FoxO1 (in the presence of HA-Ub KO) was affinity purified from HEK293A cells. Purified Flag-USP7 constructs were added and reactions incubated at 30 °C for 1 hour prior to termination and analysis by SDS-PAGE.
Having established an interaction between USP7 and FoxO1, we next sought to determine whether USP7 deubiquitinates FoxO1. In evaluating the effects of USP7 on FoxO1 ubiquitination, we utilized coexpression of HA-tagged ubiquitin for visualization of ubiquitinated FoxO1 species. Immunoprecipitation of Flag-FoxO1 under denaturing conditions revealed a pattern of ubiquitination that was decreased upon overexpression of USP7 (Figure 1C). In contrast to this, the catalytically inactive USP7 CS actually enhanced the level of FoxO1 ubiquitination, which is likely a result of this mutant acting as a dominant negative (27) (Figure 1C). Further, to assess whether the pattern of ubiquitinated bands in the immunnoprecipitate of Flag-FoxO1 reflects true monoubiquitination of FoxO1, we cotransfected FoxO1 with a ubiquitin construct in which all seven lysine residues have been mutated to arginines (Ub KO). Given that this mutant ubiquitin cannot form polyubiquitin chains, the resulting similarity in pattern of FoxO1 ubiquitination in the presence of wild-type Ub and Ub KO suggests that the observed ubiquitination of FoxO1 is indeed monoubiquitination (Figure 1D). To confirm the direct deubiquitination of monoubiquitinated FoxO1 by USP7, we incubated affinity-purified monoubiquitinated FoxO1 with purified USP7 in an in vitro deubiquitination assay. Shown in Figure 1E, purified wild-type USP7, but not the USP7 CS mutant, markedly reduced the levels of monoubiquitinated FoxO1 in this cell-free assay. Taken together, these results support the conclusion that USP7 directly deubiquitinates monoubiquitinated FoxO1.
FoxO1 transcriptional activity is suppressed by USP7
To investigate the functional consequence of FoxO1 as a USP7 target, we next analyzed the effect of USP7 on the transcriptional activity of FoxO1. Unlike polyubiquitination, which impedes FoxO activity by signaling FoxO proteins for proteasomal degradation, several studies have shown that monoubiquitination of FoxO4 increases its transcriptional activity (25, 39, 40). Thus, and similar to its reported function on FoxO3/4, we hypothesized that removal of monoubiquitin moieties from FoxO1 by USP7 would attenuate FoxO1 activity. Indeed, overexpression of wild-type USP7 suppressed FoxO1 activation on a FoxO1-responsive reporter construct, whereas USP7 CS led to an increase in activation (Figure 2A, top). Importantly, these effects on transcriptional activity were in absence of an effect on FoxO1 protein levels (Figure 2A, bottom).
Figure 2.

USP7 affects FoxO1 transcriptional activity. A, USP7 suppresses transcriptional activation of FoxO1 on the 3X IRS FoxO1 response element-luciferase reporter construct in HEK293A cells. Data presented as relative activity and shown as ± SEM; n = 3. ***, P < .001 by one-way ANOVA with Newman-Keuls Multiple Comparison test. Effect is noted in absence of changes on FoxO1 protein levels, as indicated by included Western blot analysis of whole-cell extracts. B, Constitutively active Akt (Myr Akt) suppresses FoxO1 transcriptional activity to a further degree than USP7 alone. Data presented as in (A). C, USP7 suppresses transcriptional activation of a noninsulin-sensitive FoxO1 mutant (FoxO1 mut.). Cells were deprived of serum for 6 h prior to harvest. Data presented as relative activity and shown as ± SEM; n = 3. *, P < .05; **, P < .01; and ***, P < .001 by one-way ANOVA with Newman-Keuls Multiple Comparison test. D, USP7 fails to affect FoxO1 nuclear/cytoplasmic localization. Fluorescence microscopy images showing typical nuclear and cytoplasmic localization of FoxO1-GFP. Cells were treated as in (C).
Given that insulin is known to inhibit FoxO1 activity through Akt-mediated phosphorylation and nuclear exclusion (14, 19, 41), we wanted to examine whether USP7 could be acting downstream of insulin. A constitutively active myristoylated Akt construct potently suppressed FoxO1 activity, which was to a greater degree than USP7 alone (Figure 2B). However, when we used a FoxO1 mutant that is resistant to Akt-mediated phosphorylation and suppression, we found that expression of wild-type USP7 was still capable of attenuating FoxO1 transcriptional activity (Figure 2C). Based on previous reports that USP7-mediated deubiquitination of FoxO3/4 results in its nuclear exclusion (25), we cotransfected USP7 with a GFP-fused FoxO1 to test the effect of USP7 on FoxO1 localization. Under conditions identical to that where USP7 overexpression decreased FoxO1 transcriptional activation, we failed to observe an appreciable change in FoxO1 localization. This was true of both wild-type FoxO1 and a noninsulin-sensitive mutant (Figure 2D and Supplemental Figure 1). These data suggest that USP7-mediated deubiquitination of FoxO1 leads to a suppression of its transcriptional activity.
USP7 suppresses gluconeogenesis in primary hepatocytes
Given the effect of USP7 on FoxO1 transcriptional activity, we hypothesized that USP7 might control FoxO1 activation of gluconeogenesis. To interrogate a link between USP7 and gluconeogenesis, we examined the effect of targeting USP7 with short hairpin RNA (shRNA) on gluconeogenic gene expression in primary culture of mouse hepatocytes. Primary hepatocytes were infected with adenoviruses expressing either a nontargeting scrambled control shRNA (shSCR) or one of two shRNA sequences against USP7 (designated shUSP7 #1 and shUSP7 #2) and treated with forskolin, a cAMP activator used to mimic the condition of fasting, alone or in combination with insulin (Figure 3A). These stimuli were chosen considering roles of FoxO1 in both the cAMP induction of gluconeogenic genes and the insulin-mediated inhibition of cAMP-induced gluconeogenesis (9, 10, 13, 42). Adenoviral-mediated expression of shUSP7 sequences resulted in dramatic reduction of USP7 protein levels without altering FoxO1 amount or localization (Figure 3B). Supporting a role for USP7 on expression of FoxO1-target genes, knockdown of USP7 potentiated the forskolin response of G6Pc and Pck1 (Figure 3C). These effects on gene expression corresponded with efficiency of knockdown, with increased knockdown of USP7 having the most pronounced increase on gene expression (Figure 3, B and C). Pretreatment with insulin prior to and during the forskolin treatment suppressed expression of G6pc and Pck1 to a similar extent in both control and shUSP7 cells (Figure 3C). Importantly, the knockdown of USP7 did not lead to a general activation of the cAMP/PKA pathway, as the cAMP-inducible gene Nurr77 exhibited a similar pattern of expression in shUSP7 and shSCR cells upon forskolin treatment (Supplemental Figure 2A). To evaluate the physiological outcome coinciding with these effects on gene expression, hepatic glucose production was measured from primary hepatocytes adenovirally infected with shUSP7 and incubated with medium including lactate and pyruvate as gluconeogenic substrates. Consistent with the observed increase in gluconeogenic genes, USP7 knockdown enhanced forskolin-induced glucose production (Figure 3D).
Figure 3.

USP7 knockdown increases gluconeogenic gene expression and glucose production in primary hepatocytes. A, Schematic of forskolin (FSK) and insulin (INS) treatment of primary hepatocytes. To mimic fasting conditions, cells were serum-starved in 0.2% BSA for 5 hours prior to a 1.5 hours incubation with 10 μM forskolin. To analyze the effect of insulin on suppression of gluconeogenic gene induction, cells were serum-starved for 4 hours prior to a 1 hour pretreatment with 100 nM insulin followed by a 1.5 hours incubation with both insulin and forskolin. B, Insulin signaling in primary hepatocytes with USP7 knockdown. Western blot analysis of nuclear and cytosolic fractions from hepatocytes as treated in (A). C, USP7 knockdown increases gluconeogenic gene expression upon forskolin treatment. Primary hepatocytes were infected with control (shSCR) or USP7 shRNA adenoviruses and subjected to the conditions presented in (A). mRNA levels are graphed relative to expression of unstimulated/serum-starved shSCR and presented as average ± SEM; n = 3. *, P < .05; and ***, P < .001 by one-way ANOVA with Newman-Keuls Multiple Comparison test. Data are representative of at least two independent experiments. D, USP7 knockdown increases glucose levels in medium after overnight forskolin treatment. Glucose in medium of primary hepatocytes treated overnight with 10 μM forskolin was measured 3 hours after culturing in glucose-free medium supplemented with pyruvate and lactate. Presented as average ± SEM; n = 3. **, P < .01 by one-way ANOVA with Newman-Keuls Multiple Comparison test.
To better assess the role of USP7 activity on gluconeogenesis, we performed complementary gain-of-function experiments in primary hepatocytes with adenoviruses expressing USP7. A modest increase of wild-type USP7 over endogenous levels caused a significant suppression of forskolin-induced increases in G6Pc and Pck1 expression (Figure 4, A and B). This was in contrast to overexpression of the catalytically inactive USP7 CS, which had a tendency to potentiate gluconeogenic gene expression (Figure 4B). Again, these changes in gene expression were in absence of a global alteration of cAMP responsiveness, as observed by similar forskolin-induced Nurr77 expression (Supplemental Figure 2B). USP7 overexpression did not affect insulin activation of the PI3K/Akt pathway, nor did it alter the ability of insulin to suppress gluconeogenic genes (Figure 4, A and B). Furthermore, USP7 had no effect on total protein levels of FoxO1 (Figure 4A). These effects on gluconeogenic genes corroborated the results noted for transient expression of USP7 on FoxO1 transcriptional activation.
Figure 4.
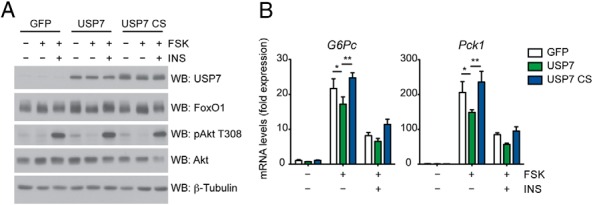
USP7 overexpression suppresses gluconeogenic gene expression in primary hepatocytes. A, USP7 does not alter insulin signaling in primary hepatocytes. Western blot analysis of whole-cell extracts from primary hepatocytes infected with control (GFP), USP7 wild-type, or USP7 catalytically inactive mutant (CS) adenoviruses and subjected to the conditions presented in Figure 3A. B, Wild-type USP7 suppresses gluconeogenic gene expression upon forskolin treatment. Hepatocytes were treated as in (A). mRNA levels are graphed relative to expression of unstimulated/serum-starved GFP and presented as average ± SEM; n = 3. *, P < .05; **, P < .01; and ***, P < .001 by one-way ANOVA with Newman-Keuls Multiple Comparison test. Data are representative of at least two independent experiments.
USP7 suppresses gluconeogenesis in mouse liver
We next sought to confirm whether these effects of USP7 could be recapitulated in vivo by altering hepatic USP7 levels. Tail vein delivery of adenoviral USP7 constructs resulted in significant increases in USP7 protein in the livers of C57BL/6 mice (Figure 5A). Given that liver-specific FoxO1 knockout mice exhibit hypoglycemia only after a prolonged fast (13), we decided to observe the effect of USP7 on mice that had been similarly fasted. Indeed, coincident with a role for USP7 in suppression of gluconeogenesis, mice receiving tail vein injection of wild-type USP7, and not the catalytically inactive USP7 CS, exhibited decreased expression of hepatic gluconeogenic genes after prolonged fasting (Figure 5B). Of note, nuclear FoxO1 levels were variable but not affected by treatment (Figure 5A). Also, suppression of gluconeogenic genes was not due to a disruption of insulin signaling in the livers of these animals, as the phosphorylation status of key targets in the insulin/PI3K/Akt pathway was unaffected by USP7 overexpression (Figure 5A).
Figure 5.

USP7 overexpression in C57BL/6 mouse liver suppresses gluconeogenesis. A, Insulin signaling remains relatively unchanged in livers of mice overexpressing USP7. Western blot analysis of nuclear and cytosolic fractions from livers of 48-h fasted mice. B, Hepatic overexpression of wild-type USP7 suppresses gluconeogenic genes. Liver mRNA levels from mice in (A) are graphed relative to expression of GFP and presented as average ± SEM; n = 6–7. *, P < .05; **, P < .01; and ***, P < .001 by one-way ANOVA with Newman-Keuls Multiple Comparison test. C, USP7 overexpression improves pyruvate tolerance. Intraperitoneal (IP) pyruvate tolerance test from mice infected with GFP, wild-type USP7, or USP7 CS adenoviruses. Mice were fasted overnight prior to injection of 2 g/kg sodium pyruvate. Data are presented as change in glycemia following pyruvate injection and are from two independent experiments. Presented as average ± SEM. n = 9–19. *, P < .05 GFP vs USP7; and #, P < .05 USP7 vs USP7 CS by one-way ANOVA with Newman-Keuls Multiple Comparison test per time point.
Next, to determine the physiological consequence of USP7 perturbation in mouse liver, we performed pyruvate tolerance tests on mice with hepatic overexpression of GFP, wild-type USP7, or USP7 CS. In accordance with the change in gluconeogenic genes, overexpression of wild-type USP7 led to a decreased conversion of pyruvate to glucose after pyruvate challenge, indicating suppressed hepatic gluconeogenesis in these animals (Figure 5C). Notably, overexpression of the USP7 CS mutant did not alter pyruvate tolerance, highlighting the importance of USP7 catalytic activity in regulation of glucose homeostasis. In aggregate, these data support a suppressive effect of hepatic USP7 on gluconeogenesis.
Effect of USP7 on gluconeogenesis is dependent on FoxO1 activity
To determine a requirement of endogenous FoxO1 for USP7's effect on gluconeogenesis, we performed double knockdown experiments with shUSP7 adenovirus and an shRNA adenovirus targeting FoxO1 (shFoxO1). As previously reported (13), shFoxO1 dramatically reduced FoxO1 levels, with a concordant reduction of gluconeogenic genes (Figure 6, A and B). Importantly, knockdown of FoxO1 abolished the increase in gluconeogenic gene expression observed by both shUSP7 adenoviruses (Figure 6B). These results suggest that USP7 depends on the presence of FoxO1 in order to affect gluconeogenic gene expression.
Figure 6.

USP7's effect on gluconeogenic gene expression is dependent on FoxO1. A, Efficient knockdown of FoxO1 protein upon double knockdown with USP7. Western blot analysis of whole-cell extracts from primary hepatocytes infected with the indicated adenoviruses (in the absence of shFoxO1, cells received coinfection with corresponding control shRNA adenovirus) and serum starved in 0.2% BSA for 3 hours prior to a 1.5 hours incubation with 10 μM forskolin. B, Knockdown of FoxO1 abolishes the effect of USP7 knockdown on gluconeogenic gene expression. mRNA levels are graphed relative to expression of unstimulated/serum-starved control shSCR and presented as average ± SEM; n = 3. *, P < .05; and ***, P < .001 by one-way ANOVA with Newman-Keuls Multiple Comparison test. Data are representative of at least two independent experiments.
This then begged the question: what controls USP7 activity on FoxO1? Neither stimulation of primary hepatocytes with forskolin nor insulin, conditions where FoxO1 is active and inactive, respectively, produced significant change in USP7 protein levels (Supplemental Figure 3A). Also, USP7 protein and mRNA levels remained constant in livers of fasted and refed mice (Supplemental Figure 3, B and C). In addition, assessment of USP7 activity in whole-cell, nuclear, and cytoplasmic lysates of primary hepatocytes failed to recognize an appreciable effect of hormonal stimulation on catalytic activity (Supplemental Figure 3, D and E). Thus, stimuli that dramatically affect FoxO1 transcriptional activation do not seem to alter USP7 levels or activity. In summation, these results suggest that USP7 modulation of FoxO1 transcriptional activity occurs subsequent to nuclear FoxO1 availability.
USP7 modulates FoxO1 occupancy on promoters of gluconeogenic genes
We had originally hypothesized that USP7-mediated deubiquitination of FoxO1 would lead to nuclear exclusion, as indicated for the mechanism in its interaction with FoxO3/4 (25). However, we have not seen such an effect with transient overexpression of USP7 and FoxO1 in HEK293A cells (Supplemental Figure 1), and have failed to detect a significant change in the level of endogenous nuclear FoxO1 upon USP7 knockdown (Figure 3B) or overexpression (Figure 5A). Because USP7 did not alter the nuclear/cytoplasmic localization of FoxO1, we examined the ability of USP7 to modulate the association of FoxO1 with gluconeogenic promoters. Using primers that specifically span the IRE(s) of G6Pc and Pck1 (Figure 7A), we performed ChIP of FoxO1 from primary hepatocytes infected with shSCR or shUSP7 and stimulated with forskolin. Compared with unstimulated cells, forskolin treatment resulted in increased ChIP of endogenous FoxO1 with G6Pc and Pck1 promoters (Figure 7B). Knockdown of USP7 further potentiated the forskolin-induced FoxO1 occupancy at these promoters (Figure 7B). Notably, this increase in FoxO1 promoter binding by USP7 knockdown occurred irrespective of unaltered nuclear FoxO1 levels (Supplemental Figure 4A). And consistent with elevated gluconeogenic gene expression, histone H3 acetylation at Lys9 (H3K9Ac), a marker of increased transcriptional activity recently shown to be elevated over G6Pc and Pck1 in livers of fasting and diabetic animals (43), was found to be significantly higher on the G6Pc promoter upon USP7 knockdown (Supplemental Figure 4B).
Figure 7.

USP7 alters FoxO1 occupancy at gluconeogenic gene promoters. A and B, ChIP-qPCR analysis reveals increased association of FoxO1 with G6Pc and Pck1 promoters at IRE regions upon USP7 knockdown. Schematic in (A) indicates primers used for amplification of FoxO1 binding sites (black boxes in promoters of G6Pc and Pck1) and a control region (Cycs). In (B), cells were infected with the indicated adenoviruses and treated with 10 μM forskolin for 1.5 hours prior to harvest and ChIP assay of FoxO1. Results graphed as average ± SEM; n = 3. *, P < .05 by one-way ANOVA with Newman-Keuls Multiple Comparison test. Data are representative of at least two independent experiments. C, Model illustrating novel role for USP7 in deubiquitination and suppression of FoxO1 activity. Transcriptionally active FoxO1 is presented in yellow and inactive forms of FoxO1 are presented in shades of green. Fasting-induced glucagon stimulus through cAMP/PKA activates FoxO1 transcription of gluconeogenic genes G6Pc and Pck1. Monoubiquitination of FoxO1, through an as-of-yet unidentified E3 ligase, promotes FoxO1 activity through enhanced occupancy over binding sites (IRE) on G6Pc and Pck1 promoters. During feeding, insulin suppresses hepatic gluconeogenesis through the PI3K/Akt-mediated phosphorylation and nuclear exclusion of FoxO1. Our results presented here suggest that USP7 acts as a break on nuclear FoxO1 through deubiquitination to suppress its association with promoters of gluconeogenic genes.
Discussion
Our studies expand the role of the USP7 deubiquitinating enzyme to FoxO1 modulation and regulation of glucose homeostasis. The findings presented here suggest that FoxO1 transcriptional activity is controlled by USP7-mediated deubiquitination. FoxO1-target genes were increased upon USP7 knockdown and conversely suppressed with overexpression of wild-type USP7. These changes in gene expression led to altered hepatic glucose output as assessed in cells and mouse liver. Importantly, the effect of USP7 on gluconeogenic genes required activity of FoxO1. In light of these data, we propose a model where deubiquitination of FoxO1 by USP7 leads to decreased occupancy of FoxO1 at promoters of gluconeogenic genes, thereby suppressing gluconeogenesis (Figure 7C).
Previous studies have investigated the effect of stress-induced monoubiquitination of FoxO proteins, having found that monoubiquitination promotes FoxO-dependent transcription. However, these studies focused on FoxO4 in the cell cycle (25, 39, 40). The present data support this model of monoubiquitination-induced increase in FoxO transactivation and broaden its biologic effects to the area of hepatic glucose metabolism. Our findings, in combination with others, suggest a common regulatory mechanism for USP7 and the family of FoxO proteins. However, contrary to its reported mechanism for FoxO3/4 (25), we found that USP7 suppresses FoxO1 activity in absence of an effect on its nuclear/cytoplasmic localization.
Altered FoxO1 transactivation despite unchanged nuclear accumulation is not an unusual concept, as FoxO proteins may exhibit reduced activity by mechanisms that do not depend on cellular redistribution. For example, insulin is still able to inhibit a mutant of FoxO1 that is rendered constitutively nuclear by mutation in the nuclear export signal (44). In addition, phosphorylation of FoxO1 residue S256 alters its in vitro binding activity by introducing a negative charge that interferes with basic residues in the DNA-binding domain of FoxO1 (45). This disruption of FoxO1 affinity for target chromatin has also been seen with acetylation, where acetylated FoxO1 has attenuated ability to bind DNA (46). Because we observed increased FoxO1 binding to gene promoters upon USP7 knockdown when monoubiquitination of FoxO1 is presumably elevated, the presence of ubiquitin moieties might facilitate FoxO1 association with chromatin. Related to this, a recent report has implicated a role for USP7 in the dissociation of minichomosome maintenance complex from chromatin at the end of S-phase (47), which the authors suggest might occur subsequent to monodeubiquitination of minichomosome maintenance. Thus, the ability of monoubiquitination to alter FoxO1 recruitment to DNA deserves further exploration.
An intriguing study by Nardini et al (48) has shown that monoubiquitination of the transcription factor NF-Y is necessary for active transcription of target genes by mimicking and, thus, facilitating monoubiquitination of histone H2B at Lys120, an epigenetic mark associated with transcriptional activation. This is important to our current findings given work originally performed in flies that revealed a role for USP7 in epigenetic silencing through deubiquitination of H2B (49). Although it is disputed whether USP7 deubiquitinates H2B in mammalian cells, USP7 has also been shown to deubiquitinate and stabilize Polycomb repressive complex 1, which confers gene repression by H2A monoubiquitination (50). Taken together, it is interesting to speculate a function for monoubiquitination of FoxO1 in the activation of permissible histone marks and, coordinately, monodeubiquitination of FoxO1 by USP7 in transcriptional silencing.
The binding of cofactors to transcription factors can also facilitate a transcriptionally favorable chromatin environment. Thus, a third possibility exists where USP7 might modulate FoxO1 activity by affecting the association of FoxO1 with coregulators, such as CREB binding protein (CBP) and its related protein, p300 (CBP/p300) or PGC-1α. The binding of histone acetyltransferases CBP/p300 to FoxO proteins is essential for the transactivation of target gene promoters (51–53). This is true also of PGC-1α, which coactivates FoxO1 on gluconeogenic promoters in an insulin-sensitive fashion (10). Whether USP7-mediated deubiquitination of FoxO1 inhibits these interactions, or whether monoubiquitinated FoxO1 enhances them, is unknown.
In some cases, the same lysine residues are alternately acetylated or ubiquitinated. Of note, the specific lysines that are acetylated in FoxO1 are not the ones that get polyubiquitinated (54). Interestingly, the effect of deubiquitination on FoxO1 as reported here is the opposite of that observed by deacetylation of FoxO1, where deacetylation increases FoxO1 transcriptional activity (55, 56). Does monoubiquitination antagonize acetylation or vice versa? Van der Horst and colleagues (25) reported a reduction of FoxO4 monoubiquitination with mutation of lysine residues Lys199 and Lys211, which are sites of acetylation that are conserved among FoxOs. These findings might suggest overlapping and alternately monoubiquitinated/acetylated FoxO residues. However, this mutant did not exhibit altered transcriptional activity, and the authors did not address its function in the presence of USP7, questioning whether these residues are true targets for USP7-mediated deubiquitination. Nevertheless, the extent that monoubiquitination affects other posttranslational modifications, and where it falls in the hierarchy for eliciting functional change, requires future attention.
Our findings of unaltered USP7 protein and activity upon fasting and feeding stimuli have led us to propose a model whereby USP7 suppresses FoxO1 transactivation mainly under fasting conditions, when FoxO1 is predominantly nuclear. Given that insulin/Akt overrode effects shown by USP7 loss- and gain-of-function experiments, a role for USP7 action on FoxO1 during feeding cannot be concluded. More specifically, our data suggest that nuclear FoxO1 is requisite for USP7 function. Although a physiological role for USP7-mediated suppression of gluconeogenesis during fasting might seem counterintuitive, it is not unprecedented. In fact, a negative feedback pathway has been suggested where FoxO amplification of insulin/growth factor signaling limits prolonged FoxO activation (reviewed in [57]). Our data suggest that USP7 is active regardless of hormonal stimulus, thus, acting as a constitutively accessible break on nuclear FoxO1. However, it is reasonable to surmise that the specific activity of USP7 on FoxO1 (as opposed to its enzymatic activity, as assessed by our assays) is subject to hormonal control. USP7 activity can be altered through interactions with different binding partners and through posttranslational modification (49, 58–60); future studies will be required to determine whether USP7-mediated deubiquitination of FoxO1 can be controlled at either of these levels. All together, the results presented here reveal USP7 as an additional node for fine-tuning FoxO1 activity, and suggest that activation of USP7 could be useful in alleviating states of excess hepatic glucose production.
Recently, Lee et al (61) reported that overexpression of USP7 in mouse liver decreased blood glucose by increasing hepatic levels of peroxisome proliferator-activated receptor γ (PPARγ). This conclusion is complicated by the fact that PPARγ levels are extremely low in nonobese liver (62), which questions the physiological relevance of a hepatic USP7-PPARγ interaction. In addition, although the authors suggested that elevated PPARγ led to the decreased glycemia upon USP7 overexpression, a causal role in glucose homeostasis was not fully explored. Given our current findings, it is likely that USP7-mediated inactivation of FoxO1 and suppression of hepatic gluconeogenesis contributed to their observed phenotype.
FoxO1 function is also of critical importance for other metabolic tissues, such as skeletal muscle and adipose tissue. Mice overexpressing FoxO1 in skeletal muscle exhibit muscle atrophy and impaired glycemic control (63), whereas inhibition of FoxO transcriptional activity prevents muscle loss (64). In adipose tissue, expression of a dominant negative FoxO1 leads to increased energy expenditure and improved glucose tolerance (65). Thus, under both scenarios, suppression of FoxO1 would be a favorable approach to improve metabolic imbalance. Whether USP7 can inhibit FoxO1 function in muscle and/or adipose tissue will be addressed in future studies. Of note, a functional interaction between USP7 and Tip60 has been reported to be required for 3T3-L1 adipocyte differentiation (66), further supporting a role for USP7 targets in the maintenance of energy/nutrient homeostasis.
The human genome encodes approximately 100 deubiquitinating enzymes, which counteract several hundred ubiquitin ligases, suggesting that ubiquitination and its reversal are subject to considerable specialization and specificity. Because of their high specificity, deubiquitinating enzymes are attractive targets for drug development (24). Our data suggest that USP7 may be important for the regulation of FoxO1 transcriptional activity during fasting/refeeding, making USP7 an excellent candidate for translational applications. The identification here of a deubiquitinase involved in glucose metabolism could provide a potent target for clinical intervention of not only hepatic glucose output in the context of diabetes, but also for broader therapeutic pathways in the treatment of obesity and other metabolic disorders.
Additional material
Supplementary data supplied by authors.
Acknowledgments
The authors thank members of the Puigserver laboratory for experimental assistance and mechanistic insight, with special thanks to Yoonjin Lee for invaluable aid in animal experiments. We are grateful to initial work performed by Drs J. Wade Harper and Sebastian Hayes, for whom without this project would not have been conceived. We also thank Drs Bruce Spiegelman and John Blenis for helpful discussions, and Drs Alexander Banks and Pier Pandolfi for donation of reagents that were of critical importance to this work.
This work was supported by an American Heart Association predoctoral fellowship (J.A.H.) and postdoctoral fellowship (M.T.). P.P. received support from American Diabetes Association and National Institute of Health (NIH)/National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01 069966).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CBP
- CREB-binding protein
- ChIP
- chromatin immunoprecipitation
- CREB
- cAMP-responsive element-binding protein
- co-IP
- coimmunoprecipitation
- CS
- C223S
- FoxO1
- forkhead box O1
- IRE
- insulin-responsive element
- PGC-1α
- proliferator-activated receptor-α coactivator 1 α
- PKA
- protein kinase A
- PI3K
- phosphatidylinositol 3-kinase
- PMSF
- phenylmethanesulfonyl fluoride
- PPARγ
- peroxisome proliferator-activated receptor γ
- SDS
- sodium dodecyl sulfate
- shRNA
- short-hairpin RNA
- Ub
- ubiquitin
- Ub KO
- ubiquitin in which all seven lysine residues are mutated to arginines
- USP7
- ubiquitin-specific protease 7.
References
- 1. Newgard CB. 2004. Regulation of glucose metabolism in the liver. In: DeFronzo RA, Ferrannini E, Keen H, Zimmet P, eds. International Textbook of diabetes, 3rd ed Chichester: John Wiley, Sons; 253–275. [Google Scholar]
- 2. Zhou G, Myers R, Li Y, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Herzig S, Long F, Jhala US, et al. CREB regulates hepatic gluconeogenesis through the coactivator PGC-1. Nature. 2001;413:179–183. [DOI] [PubMed] [Google Scholar]
- 4. Quinn PG, Granner DK. Cyclic AMP-dependent protein kinase regulates transcription of the phosphoenolpyruvate carboxykinase gene but not binding of nuclear factors to the cyclic AMP regulatory element. Mol Cell Biol. 1990;10:3357–3364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Altomonte J, Richter A, Harbaran S, Suriawinata J, Nakae J, Thung SN, Meseck M, Accili D, Dong H. Inhibition of Foxo1 function is associated with improved fasting glycemia in diabetic mice. Am J Physiol Endocrinol Metab. 2003;285:E718–728. [DOI] [PubMed] [Google Scholar]
- 6. Hall RK, Sladek FM, Granner DK. The orphan receptors COUP-TF and HNF-4 serve as accessory factors required for induction of phosphoenolpyruvate carboxykinase gene transcription by glucocorticoids. Proc Natl Acad Sci U S A. 1995;92:412–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Yoon JC, Puigserver P, Chen G, et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131–138. [DOI] [PubMed] [Google Scholar]
- 8. Koo SH, Flechner L, Qi L, et al. The CREB coactivator TORC2 is a key regulator of fasting glucose metabolism. Nature. 2005;437:1109–1111. [DOI] [PubMed] [Google Scholar]
- 9. Nakae J, Kitamura T, Silver DL, Accili D. The forkhead transcription factor Foxo1 (Fkhr) confers insulin sensitivity onto glucose-6-phosphatase expression. J Clin Invest. 2001;108:1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Puigserver P, Rhee J, Donovan J, et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550–555. [DOI] [PubMed] [Google Scholar]
- 11. Li X, Monks B, Ge Q, Birnbaum MJ. Akt/PKB regulates hepatic metabolism by directly inhibiting PGC-1alpha transcription coactivator. Nature. 2007;447:1012–1016. [DOI] [PubMed] [Google Scholar]
- 12. Dong XC, Copps KD, Guo S, et al. Inactivation of hepatic Foxo1 by insulin signaling is required for adaptive nutrient homeostasis and endocrine growth regulation. Cell Metab. 2008;8:65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Matsumoto M, Pocai A, Rossetti L, Depinho RA, Accili D. Impaired regulation of hepatic glucose production in mice lacking the forkhead transcription factor Foxo1 in liver. Cell Metab. 2007;6:208–216. [DOI] [PubMed] [Google Scholar]
- 14. Nakae J, Park BC, Accili D. Insulin stimulates phosphorylation of the forkhead transcription factor FKHR on serine 253 through a Wortmannin-sensitive pathway. J Biol Chem. 1999;274:15982–15985. [DOI] [PubMed] [Google Scholar]
- 15. Guo S, Rena G, Cichy S, He X, Cohen P, Unterman T. Phosphorylation of serine 256 by protein kinase B disrupts transactivation by FKHR and mediates effects of insulin on insulin-like growth factor-binding protein-1 promoter activity through a conserved insulin response sequence. J Biol Chem. 1999;274:17184–17192. [DOI] [PubMed] [Google Scholar]
- 16. Goswami R, Lacson R, Yang E, Sam R, Unterman T. Functional analysis of glucocorticoid and insulin response sequences in the rat insulin-like growth factor-binding protein-1 promoter. Endocrinology. 1994;134:736–743. [DOI] [PubMed] [Google Scholar]
- 17. Ayala JE, Streeper RS, Desgrosellier JS, et al. Conservation of an insulin response unit between mouse and human glucose-6-phosphatase catalytic subunit gene promoters: transcription factor FKHR binds the insulin response sequence. Diabetes. 1999;48:1885–1889. [DOI] [PubMed] [Google Scholar]
- 18. Rena G, Guo S, Cichy SC, Unterman TG, Cohen P. Phosphorylation of the transcription factor forkhead family member FKHR by protein kinase B. J Biol Chem. 1999;274:17179–17183. [DOI] [PubMed] [Google Scholar]
- 19. Brunet A, Bonni A, Zigmond MJ, et al. Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell. 1999;96:857–868. [DOI] [PubMed] [Google Scholar]
- 20. Huang H, Regan KM, Wang F, et al. Skp2 inhibits FOXO1 in tumor suppression through ubiquitin-mediated degradation. Proc Natl Acad Sci U S A. 2005;102:1649–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Insulin-induced phosphorylation of FKHR (Foxo1) targets to proteasomal degradation. Proc Natl Acad Sci U S A. 2003;100:11285–11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang K, Li L, Qi Y, et al. Hepatic suppression of Foxo1 and Foxo3 causes hypoglycemia and hyperlipidemia in mice. Endocrinology 2012;153:631–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhao Y, Wang Y, Zhu WG. Applications of post-translational modifications of FoxO family proteins in biological functions. J Mol Cell Biol. 2011;3:276–282. [DOI] [PubMed] [Google Scholar]
- 24. Reyes-Turcu FE, Ventii KH, Wilkinson KD. Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu Rev Biochem. 2009;78:363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. van der Horst A, de Vries-Smits AM, Brenkman AB, et al. FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol. 2006;8:1064–1073. [DOI] [PubMed] [Google Scholar]
- 26. Everett RD, Meredith M, Orr A, Cross A, Kathoria M, Parkinson J. A novel ubiquitin-specific protease is dynamically associated with the PML nuclear domain and binds to a herpesvirus regulatory protein. EMBO J. 1997;16:566–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J, Gu W. Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization. Nature. 2002;416:648–653. [DOI] [PubMed] [Google Scholar]
- 28. Li M, Brooks CL, Kon N, Gu W. A dynamic role of HAUSP in the p53-Mdm2 pathway. Mol Cell. 2004;13:879–886. [DOI] [PubMed] [Google Scholar]
- 29. Nicholson B, Suresh Kumar KG. The multifaceted roles of USP7: new therapeutic opportunities. Cell Biochem Biophys. 2011;60:61–68. [DOI] [PubMed] [Google Scholar]
- 30. Dansen TB, Burgering BM. Unravelling the tumor-suppressive functions of FOXO proteins. Trends Cell Biol. 2008;18:421–429. [DOI] [PubMed] [Google Scholar]
- 31. Tang ED, Nuñez G, Barr FG, Guan KL. Negative regulation of the forkhead transcription factor FKHR by Akt. J Biol Chem. 1999;274:16741–16746. [DOI] [PubMed] [Google Scholar]
- 32. Cummins JM, Vogelstein B. HAUSP is required for p53 destabilization. Cell Cycle. 2004;3:689–692. [PubMed] [Google Scholar]
- 33. Lim KL, Chew KC, Tan JM, et al. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J Neurosci. 2005;25:2002–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Qiang L, Banks AS, Accili D. Uncoupling of acetylation from phosphorylation regulates FoxO1 function independent of its subcellular localization. J Biol Chem. 2010;285:27396–27401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dominy JE, Jr., Lee Y, Jedrychowski MP, et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell. 2012;48:900–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Matsumoto M, Han S, Kitamura T, Accili D. Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism. J Clin Invest. 2006;116:2464–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–185. [DOI] [PubMed] [Google Scholar]
- 38. Rodgers JT, Puigserver P. Fasting-dependent glucose and lipid metabolic response through hepatic sirtuin 1. Proc Natl Acad Sci U S A. 2007;104:12861–12866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Brenkman AB, de Keizer PL, van den Broek NJ, Jochemsen AG, Burgering BM. Mdm2 induces mono-ubiquitination of FOXO4. PLoS One. 2008;3:e2819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Brenkman AB, de Keizer PL, van den Broek NJ, et al. The peptidyl-isomerase Pin1 regulates p27kip1 expression through inhibition of Forkhead box O tumor suppressors. Cancer Res. 2008;68:7597–7605. [DOI] [PubMed] [Google Scholar]
- 41. Biggs WH, 3rd, Meisenhelder J, Hunter T, Cavenee WK, Arden KC. Protein kinase B/Akt-mediated phosphorylation promotes nuclear exclusion of the winged helix transcription factor FKHR1. Proc Natl Acad Sci U S A. 1999;96:7421–7426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wondisford AR, Xiong L, Chang E, et al. Control of Foxo1 Gene Expression by Co-activator P300. J Biol Chem. 2014;289:4326–4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ravnskjaer K, Hogan MF, Lackey D, et al. Glucagon regulates gluconeogenesis through KAT2B- and WDR5-mediated epigenetic effects. J Clin Invest. 2013;123:4318–4328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tsai WC, Bhattacharyya N, Han LY, Hanover JA, Rechler MM. Insulin inhibition of transcription stimulated by the forkhead protein Foxo1 is not solely due to nuclear exclusion. Endocrinology. 2003;144:5615–5622. [DOI] [PubMed] [Google Scholar]
- 45. Zhang X, Gan L, Pan H, et al. Phosphorylation of serine 256 suppresses transactivation by FKHR (FOXO1) by multiple mechanisms. Direct and indirect effects on nuclear/cytoplasmic shuttling and DNA binding. J Biol Chem. 2002;277:45276–45284. [DOI] [PubMed] [Google Scholar]
- 46. Matsuzaki H, Daitoku H, Hatta M, Aoyama H, Yoshimochi K, Fukamizu A. Acetylation of Foxo1 alters its DNA-binding ability and sensitivity to phosphorylation. Proc Natl Acad Sci U S A. 2005;102:11278–11283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jagannathan M, Nguyen T, Gallo D, et al. A Role for USP7 in DNA Replication. Mol Cell Biol. 2014;34:132–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nardini M, Gnesutta N, Donati G, et al. Sequence-specific transcription factor NF-Y displays histone-like DNA binding and H2B-like ubiquitination. Cell. 2013;152:132–143. [DOI] [PubMed] [Google Scholar]
- 49. van der Knaap JA, Kumar BR, Moshkin YM, et al. GMP synthetase stimulates histone H2B deubiquitylation by the epigenetic silencer USP7. Mol Cell. 2005;17:695–707. [DOI] [PubMed] [Google Scholar]
- 50. Maertens GN, El Messaoudi-Aubert S, Elderkin S, Hiom K, Peters G. Ubiquitin-specific proteases 7 and 11 modulate Polycomb regulation of the INK4a tumour suppressor. EMBO J. 2010;29:2553–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Nasrin N, Ogg S, Cahill CM, et al. DAF-16 recruits the CREB-binding protein coactivator complex to the insulin-like growth factor binding protein 1 promoter in HepG2 cells. Proc Natl Acad Sci U S A. 2000;97:10412–10417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Perrot V, Rechler MM. The coactivator p300 directly acetylates the forkhead transcription factor Foxo1 and stimulates Foxo1-induced transcription. Mol Endocrinol. 2005;19:2283–2298. [DOI] [PubMed] [Google Scholar]
- 53. Yang Y, Zhao Y, Liao W, et al. Acetylation of FoxO1 activates Bim expression to induce apoptosis in response to histone deacetylase inhibitor depsipeptide treatment. Neoplasia. 2009;11:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kitamura YI, Kitamura T, Kruse JP, et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell Metab. 2005;2:153–163. [DOI] [PubMed] [Google Scholar]
- 55. Daitoku H, Hatta M, Matsuzaki H, et al. Silent information regulator 2 potentiates Foxo1-mediated transcription through its deacetylase activity. Proc Natl Acad Sci U S A. 2004;101:10042–10047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Frescas D, Valenti L, Accili D. Nuclear trapping of the forkhead transcription factor FoxO1 via Sirt-dependent deacetylation promotes expression of glucogenetic genes. J Biol Chem. 2005;280:20589–20595. [DOI] [PubMed] [Google Scholar]
- 57. Hay N. Interplay between FOXO, TOR, and Akt. Biochim Biophys Acta. 2011;1813:1965–1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fernández-Montalván A, Bouwmeester T, Joberty G, et al. Biochemical characterization of USP7 reveals post-translational modification sites and structural requirements for substrate processing and subcellular localization. FEBS J. 2007;274:4256–4270. [DOI] [PubMed] [Google Scholar]
- 59. Epping MT, Meijer LA, Krijgsman O, Bos JL, Pandolfi PP, Bernards R. TSPYL5 suppresses p53 levels and function by physical interaction with USP7. Nat Cell Biol. 2011;13:102–108. [DOI] [PubMed] [Google Scholar]
- 60. Khoronenkova SV, Dianova II, Ternette N, Kessler BM, Parsons JL, Dianov GL. ATM-dependent downregulation of USP7/HAUSP by PPM1G activates p53 response to DNA damage. Mol Cell. 2012;45:801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lee KW, Cho JG, Kim CM, et al. Herpesvirus-associated ubiquitin-specific protease (HAUSP) modulates peroxisome proliferator-activated receptor γ (PPARγ) stability through its deubiquitinating activity. J Biol Chem. 2013;288:32886–32896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tontonoz P, Hu E, Graves RA, Budavari AI, Spiegelman BM. mPPAR gamma 2: tissue-specific regulator of an adipocyte enhancer. Genes Dev. 1994;8:1224–1234. [DOI] [PubMed] [Google Scholar]
- 63. Kamei Y, Miura S, Suzuki M, et al. Skeletal muscle FOXO1 (FKHR) transgenic mice have less skeletal muscle mass, down-regulated Type I (slow twitch/red muscle) fiber genes, and impaired glycemic control. J Biol Chem. 2004;279:41114–41123. [DOI] [PubMed] [Google Scholar]
- 64. Reed SA, Senf SM, Cornwell EW, Kandarian SC, Judge AR. Inhibition of IkappaB kinase alpha (IKKα) or IKKbeta (IKKβ) plus forkhead box O (Foxo) abolishes skeletal muscle atrophy. Biochem Biophys Res Commun. 2011;405:491–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Nakae J, Cao Y, Oki M, et al. Forkhead transcription factor FoxO1 in adipose tissue regulates energy storage and expenditure. Diabetes. 2008;57:563–576. [DOI] [PubMed] [Google Scholar]
- 66. Gao Y, Koppen A, Rakhshandehroo M, et al. Early adipogenesis is regulated through USP7-mediated deubiquitination of the histone acetyltransferase TIP60. Nat Commun. 2013;4:2656. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.