

Abstract
Cobalt nitrile hydratases (Co-NHase) contain a catalytic cobalt (III) ion coordinated in an N2S3 first coordination sphere comprised of two amidate nitrogens and three cysteine-derived sulfur donors: a thiolate (-SR), a sulfenate (-S(R)O−), and a sulfinate (-S(R)O2−). The sequence of biosynthetic reactions that leads to the post-translational oxidations of the metal and the sulfur ligands is unknown, but the process is believed to be initiated directly by oxygen. Herein we utilize cobalt bound in an N2S2 first coordination sphere by a seven amino acid peptide known as SODA (ACDLPCG) to model this oxidation process. Upon exposure to oxygen, Co-SODA is oxidized in two steps. In the first fast step (seconds), magnetic susceptibility measurements demonstrated that the metallocenter remains paramagnetic, i.e. Co2+, and sulfur K-edge X-ray absorption spectroscopy (XAS) is used to show that one of the thiolates is oxidized to sulfinate. In a second process on a longer timescale (hours), magnetic susceptibility measurements and Co K-edge XAS show that the metal is oxidized to Co3+. Unlike other model complexes, additional slow oxidation of the second thiolate in Co-SODA is not observed, and a catalytically active complex is never formed. The likely reason is the absence of the axial thiolate ligand. In essence, the reactivity of Co-SODA can be described as intermediate between previously described models which either quickly convert to final product or are stable in air, and it offers a first glimpse into a possible oxidation pathway for nitrile hydratase biosynthesis.
Introduction
Post-translational modification of cysteinyl thiolates (RS−) to such products as disulfide bonds (RS-SR′), sulfenates (RSO−) or sulfinates (RSO2−) is relatively common in biology and has been found to play roles in signal transduction, protein structure, and enzyme functionality.1–3 Cysteine residues are also commonly used in metalloenzymes to ligate metals or metalloclusters. Nonetheless, the hydrolytic enzymes nitrile hydratase (NHase) and thiocyanate hydrolase (SCNase) are the only known examples of metalloenzymes requiring sulfenate and sulfinate ligands for activity.4–12 The active site of NHase consists of a low spin Fe(III) or Co(III) ion in an N2S3 coordination environment provided by two amide nitrogens from the protein backbone and three cysteines found in a highly conserved CXXC(SO2H)SC(SOH) motif (Figure 1). It is interesting to note that each of the three coordinating cysteines is present in a different oxidation state: one thiolate, one sulfenate and one sulfinate. The oxidized cysteines are found in equatorial coordination sites along with the amides, and the thiolate ligand occupies an axial position trans- to the presumed substrate-binding site. A number of roles have been proposed for the oxidized cysteines including modulating the Lewis acidity of the metal site, serving as a base during the catalytic cycle, and modifying substrate/product binding affinity.13
Figure 1.

Heterodimeric cobalt nitrile hydratase and its metal active site (PDB code: 1UGP, figure prepared using PyMOL software [The PyMOL Molecular Graphics System, Version 0.99rc6, Schrödinger, LLC]). The green color corresponds to the α chain and the blue color to the β chain.
Conversion of nitriles to amides is an industrially important reaction.14, 15 To help elucidate the mechanistic details of the enzymatic catalysis of these transformations, a number of small-molecule mimics of NHase have been synthesized.16–22 However, few of these have even modest nitrile hydrolysis activity, and most of those that utilize oxidized sulfur contain bis-sulfinate ligands since controlling the extent of oxidation of the metal-bound thiolates has proven difficult.23 This has been especially true for reactions of thiolates with oxygen, as opposed to O-atom transfer reagents, which tend to yield sulfinato complexes.18, 23–34 Nonetheless, oxygen is believed to be the oxidant in vivo for modification of the sulfur atoms of NHase.5, 12, 35–37 This stands in marked contrast to a wealth of existing synthetic inorganic literature describing synthesis of metal bound sulfinato species in which reactions with molecular oxygen are seldom reported. Generally, Co(III) and other metal sulfinato complexes have been prepared by one of three routes: direct reaction of a sulfinate salt and Co(III), SO2 insertion into a Co(III)-C bond, or oxidation of a Co(III) thiolate via hydrogen peroxide, a much stronger oxidant than molecular oxygen.38–44 Biosynthetic studies have shown that apo-NHase, i.e., the metal-free protein with cysteine thiolates, can be activated by incubation with Co2+ and appropriate activation mediating proteins via a novel mechanism that has been referred to as “self-subunit swapping”.35–37 First, the cobalt binding α-subunit is post-translationally modified and loaded with cobalt while the protein is part of a trimeric αe2 complex with NhlE, a maturation mediator. Then, the holo-α-subunit is exchanged with an apo-α-subunit from the tetrameric apo-α2β2 NHase to form the functional enzyme. Surprisingly, cobalt cannot be directly incorporated into apo-α2β2 NHase, and this swapping of apo- and holo- proteins is an obligatory part of the maturation. However, although this symphony of protein subunits is now well documented, the chemical steps allowing for specific cysteine modification, cobalt incorporation and oxidation, and exchange of the two forms of the α-subunit are unknown.
Recently, Shearer and coworkers demonstrated the first functional metallopeptide mimic of Co-NHase.22 The peptide coordinates Co in an N2S3 coordination sphere, and, as for the enzyme, two of the cysteines must be oxidized to observe modest activity with acrylonitrile. In this work, we utilize a related seven amino acid peptide known as SODA (ACDLPCG) to coordinate Co(II) in a less sulfur-rich N2S2 environment. We show that, upon exposure of the Co(II)-metallopeptide to air, the first oxidation occurs at the ligands without a change in the oxidation state of the metal (Scheme 1). This ligand-oxidized, sulfur bound Co(II) system is then further oxidized to Co(III) to produce a fully oxidized species (Scheme 1). We propose that these differentially oxidized states are relevant to understanding the biosynthesis of Co-NHase, and possible mechanisms for metal incorporation into the enzyme and the roles of the distinctively oxidized thiolates present in the first coordination sphere during this process are discussed (Scheme S1).
Scheme 1.
Hypothesized sequence for oxidation of Co(II)-SODA in air.
Experimental Section
All inorganic syntheses were performed under a nitrogen atmosphere using a double-manifold Schlenk vacuum line. Unless otherwise specified, all the chemicals and solvents, of highest available grades, were obtained from Sigma-Aldrich (Milwaukee, WI) and were used as received. Aqueous solutions were prepared using purified water (resistivity = 18.2 MΩ·cm).
Physical Measurements
Optical, FTIR, and CD spectroscopy
UV-visible measurements were executed on a Hewlett-Packard 8453 spectrophotometer using quartz cuvettes with a 1 cm pathlength. NMR spectra were recorded at 400 MHz (1H) using Varian Liquid-State NMR instruments in D2O (99.9%, Cambridge Isotopes Laboratories Inc.). FTIR (Fourier-transform infrared) spectra of the samples were recorded on a Thermo Nicolet Avatar-360 spectrometer in KBr pellets. A KBr pellet without sample was used as a reference for the background. For the samples, each spectrum is an average of 512 scans at 1 cm−1 resolution. The KBr pellets for the reduced samples were prepared in an anaerobic glove box. Circular dichroism (CD) spectroscopy was performed on a Jasco-815 spectropolarimeter using a rectangular quartz cell with a pathlength of 0.1 cm.
EPR spectroscopy
Electron paramagnetic resonance (EPR) spectra were recorded between 5K and 7K using a Bruker ELEXSYS E580 CW X-band spectrometer (Bruker, Silberstreifen, Germany) equipped with an Oxford model 900 EPL liquid helium cryostat (Oxford instruments, Oxfordshire, UK). The magnetic field modulation frequency was 100 kHz; the amplitude was 1 mT; the microwave power was between 1–4 mW and the microwave frequency was 9.42 GHz; the sweep time was 168 s. Samples for the EPR experiments were prepared in a 1:1 mixture of aqueous 50 mM N-ethyl morpholine (NEM) buffer (pH 7.5) and glycerol (BDH, West Chester, PA) before transfer to a quartz EPR tube. EPR spectra were simulated using EasySpin (v 3.1.1), a computational package developed by Stoll and Schweiger45 and based on Matlab (The MathWorks, Natick, MA, USA). The model used for the EPR simulations is based on a single high-spin Co2+ ion (S = 3/2). The variable parameters were: the g values (gx, gy and gz), the line widths (ΔBx, ΔBy and ΔBz), and the zero-field splitting parameters D and E. The fitting procedure was similar to the one previously described by Flores et al.46
Magnetic susceptibility
The molecular magnetic susceptibility (χM), effective magnetic moment (μeff) for the partially oxidized Co-SODA, and the time course measurement for the oxidation of reduced Co-SODA after exposure to air were measured via the Evans NMR method by monitoring shifts in the water solvent peaks at either 400 or 500 MHz (1H) using a Varian Liquid State NMR instrument.47, 48
Mass spectrometry
MALDI-MS (matrix assisted laser desorption/ionization mass spectrometry) characterization of apo-SODA was performed using α-cyano-4-hydroxycinnamic acid as matrix on a Voyager DE STR. ESI-MS (electrospray ionization mass spectrometry) was accomplished using a Thermo Quantum Discovery Max triple-quadrupole mass spectrometer. Measurements were conducted in positive (+) and negative (−) ionization modes, using a methanol/water (50:50 by volume) mobile phase at a flow rate of 10 μL min−1 and the following ionization conditions: spray voltage, 4000 (+, −); capillary temperature, 270°C; sheath gas pressures, 25 (+) and 15 (−); auxiliary gas pressure, 2 (+, −). Isotope Pattern Calculator v4.0, developed by Junhua Yan, was used for the calculation of the molecular mass with isotopic abundances.
Co K-edge and S K-edge X-ray Absorption Data Collection and Analysis
Solutions of Co-SODA were prepared as 1:1 CoSODA (50 mM NEM buffer, pH 7.5):glycerol samples, injected in aluminum sample holders between windows made from Kapton tape (3M; Minneapolis, MN, catalog no. 1205) and quickly frozen in liquid nitrogen. Co K-edge data were collected on beamline X3b at the National Synchrotron Light Source (Brookhaven National Laboratories; Upton, NY). The samples were maintained at 20 K throughout data collection in an evacuated He Displex cryostat and collected in fluorescence mode on a Canberra 31 element solid-state Ge detector. Total count rates for all channels were maintained under 30 kHz, and a deadtime correction was not applied. The data were collected in 10 eV steps below the edge (7609 – 7689 eV), 0.5 eV steps in the edge region (7689 – 7759 eV), 2 eV steps in the near edge region (7759 – 8009 eV) and 5 eV steps in the far edge region (8009 eV – 14 keV). The spectra represent the averaged sum of three independent data scans. The sulfur K-edge data were collected as solution samples at room temperature on beamline X19a at the National Synchrotron Light Source (Brookhaven National Laboratories; Upton, NY). Solutions of CoSODA (50 mM NEM buffer, pH 7.5) were injected into Lucite sample holders between two polyester film windows (2.5 μm thickness; SPI Supplies; West Chester, PA, catalogue no. 01865-AB) adhered to the sample holders using an epoxy resin. Data were collected in a sample chamber that was continuously purged with He(g) and data were recorded in fluorescence mode on a Canberra passivated implanted planar silicon (PIPS) detector. Data were collected from 200 eV below to 300 eV above the edge to obtain a good baseline for data analysis. In the pre-edge region, data were collected in 5 eV steps (2270 – 2465 eV); in the edge region, data were collected in 0.1 eV steps (2465 – 2475 eV); in the far edge region, data were collected in 2 eV steps (2475 – 2765 eV). All data were calibrated to the white line of Na2SO4. The spectra presented represent the summed average of 5 scans.
All Co and S K-edge spectra were processed and analyzed as previously described.22, 49 Briefly, S K-edge spectra were obtained by averaging the data sets, applying a baseline, and fitting the pre-edge region to a polynomial function. This baseline was then subtracted from the whole spectrum. The region above the edge jump was fit to a two knot cubic spline, and the data normalized to the edge height. The pre-edge and rising-edge features were modeled as pseudo-Voigt line shapes (a 1:1 sum of Gaussian and Lorentzian line shapes). Co K-edge spectra were processed and analyzed using the XAS data analysis suit EXAFS12350 which operates in Igor Pro51 and requires phase and amplitude functions generated within FEFF 8.20.52 All refinements are based on Fourier-filtered k3(χ) data over the energy range of k = 2.0 – 12.0 Å−1 and back-transformed over the range of r = 1.0 – 3.0 Å.
Peptide synthesis and purification
The seven amino acid peptide referred to as SODA (ACDLPCG) was synthesized, purified and quantitated as described previously.53 The reaction yield was 60% and the measured peptide mass (678 m/z, MALDI-MS (positive ion) [M]+) agreed well with the calculated peptide mass, 677.8 amu.
Synthesis and purification of NHasem1 (CCDLPCGVYDPA) and the subsequent preparation of Co-NHasem1 were performed as described by Shearer et al.22
Co-SODA preparation in reduced and oxidized forms
Metallopeptide samples were prepared analogously to the procedure described by Neupane et al. for the synthesis of the corresponding Ni-metallopeptide.54 One equivalent of an aqueous solution of Co(II) (ten times concentrated compared to SODA solution) was added dropwise to one equivalent of SODA in 50 mM NEM buffer (pH= 7.5) solution. The concentration of the SODA solution was determined using Ellman's test.55 Both solutions were degassed and saturated with nitrogen prior to the reaction. The reaction mixture immediately turned green upon addition of the cobalt solution. If exposed to oxygen, the reduced Co-SODA solution rapidly changes to brown.
Reduced Co-SODA
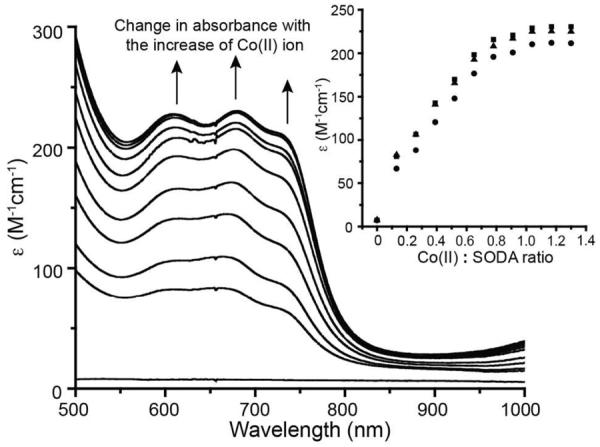
The electronic absorption spectrum (in buffered aqueous solution, pH 7.5) had the following features: λmax nm (ε M−1cm−1): 725 (210), 681 (230), 609 (225), 350 (shoulder); 303 (2250). The ESI MS (negative ion mode) yielded a [M-H]− peak with an m/z of 731.01 that corresponded well with the calculated mass of 731.1 amu for this species.
Partially oxidized Co-SODA
The features of the electronic absorption spectrum (in buffered aqueous solution, pH 7.5) are as follows: λmax nm (ε M−1cm−1): 600 (shoulder), 460 (shoulder), 355 (1100). ESI MS (positive ion mode) yielded an [M+O+3H]+ molecular ion peak at 752.04 m/z; the calculated mass was 752.17 amu. The measured effective magnetic moment was μeff = 4.38 B.M.
Fully oxidized Co-SODA
The electronic absorption spectrum features (in buffered aqueous solution, pH 7.5) included: λmax nm (ε M−1cm−1): 570 (shoulder), 450 (shoulder), 340 (1500). The ESI-MS (positive ion mode) yielded an [M+O+3H]+ molecular ion peak at 752.06 m/z, and the calculated mass was 752.17 amu.
Results and Discussion
Coordination of Co(II) by SODA
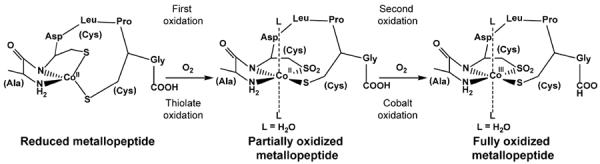
The seven amino acid peptide SODA (ACDLPCG) is derived from the nickel enzyme superoxide dismutase and is known to coordinate a Ni(II) ion via the two cysteines together with the amino terminus and a backbone amide nitrogen.56–58 Thus it seemed likely that a similar coordination set could accommodate Co(II), and the interactions between Co(II) and SODA were investigated via optical spectroscopy under both anaerobic and aerobic conditions. First, an anaerobic aqueous Co(II) solution was added dropwise to a buffered SODA solution (pH 7.5) resulting in a green solution. As shown in Figure 2, the UV-vis absorbance spectrum of this reduced, cobalt bound metallopeptide includes three distinct ligand field (LF) bands in the visible region at 725 nm (ε = 210 M−1cm−1), 681 nm (ε = 230 M−1cm−1) and 609 nm (ε = 225 M−1 cm−1). These bands have the appropriate energies and extinction coefficients for the 4T1(P) ← 4A2(F) transitions expected to arise from a tetrahedral Co(II) (d7) center (Table 1, Figure 2), and are split, as expected, due to spin-orbit coupling.59–61 The stoichiometry of the interaction between Co(II) and SODA was determined by monitoring the absorbances at these three wavelengths as a function of added Co(II) as shown in the inset of Figure 2. Titrations at all three wavelengths are consistent with a 1:1 interaction between Co(II) and SODA. Further evidence for 1:1 binding was provided by mass spectrometry. A [Co-SODA]− molecular ion peak with the expected isotopic pattern was observed in a negative-mode ESI-MS experiment (Figure S1). In addition to the LF bands, two stronger bands at 350 nm (ε = 1650 M−1cm−1) and 303 nm (ε = 2250 M−1cm−1) were observed in the far UV region of the optical spectrum for reduced Co-SODA (Figure 3). Based on comparison to other Co-substituted proteins such as Co-rubredoxin, Co-metallothionein, and Co liver alcohol dehydrogenase, these higher energy bands likely arise from cysteinyl sulfur to Co(II) charge transfer (CT).62–64 Comparison of these proteins has demonstrated that each Co-thiolate bond in a metalloprotein or peptide typically contributes ε = 900–1200 M−1cm−1 in such a ligand to metal charge transfer (LMCT) transition. Since ε303 (Co-SODA)=2250 M−1 cm−1, it is likely that Co-SODA includes two Co-thiolate bonds.65
Figure 2.
UV-vis spectrum of Co-SODA (solid line), partially oxidized Co-SODA (dotted line, 30 minutes of air oxidation), completely oxidized Co-SODA (dashed line, 24 hours of air exposure) All solutions are in aqueous 50 mM NEM buffer at pH 7.5. The inset shows an enlargement of the region between 500 and 1000 nm featuring bands associated with ligand field (LF) transitions for reduced Co-SODA.
Table 1.
Optical and CD data for various Co-SODA species
| Sample | Electronic absorption | CD absorption |
|---|---|---|
| λ (nm) (ε (M−1 cm−1)) | λ (nm) (Δε (M−1 cm−1)) | |
| Reduced Co-SODA | 725 (210) | 404 (0.20) |
| 681 (230) | 544 (−0.15) | |
| 609 (225) | 312 (0.54) | |
| 350 (shoulder, 1700) | 280 (−2.58) | |
| 303 (2250) | ||
| Partially oxidized Co-SODA | 600 (shoulder, 140) | 536 (−0.8) |
| 460 (shoulder, 430) | 420 (0.37) | |
| 355 (1100) | 306 (2.18) | |
| 267 (−3.92) | ||
| Fully oxidized Co-SODA | 570 (shoulder, 195) | 536 (0.7) |
| 450 (shoulder, 480) | 470 (0.96) | |
| 340 (1500) | 414 (shoulder, 0.36) | |
| 328 (−5.88) | ||
| 286 (−3.87) |
Figure 3.
Spectral changes in the UV-vis spectrum of SODA upon stepwise addition under anaerobic conditions of an aqueous solution (50 mM NEM buffer, pH 7.7) of Co(II). The inset shows the binding curves extracted at three different wavelengths from the spectra (squares 681 nm, circles 709 nm, and triangles 725 nm). The ratios of Co(II):SODA for which spectra are shown are 0:1, 0.13:1, 0.26:1, 0.39:1, 0.52:1, 0.65:1, 0.78:1, 0.91:1, 1.04:1, 1.17:1, and 1.3:1.
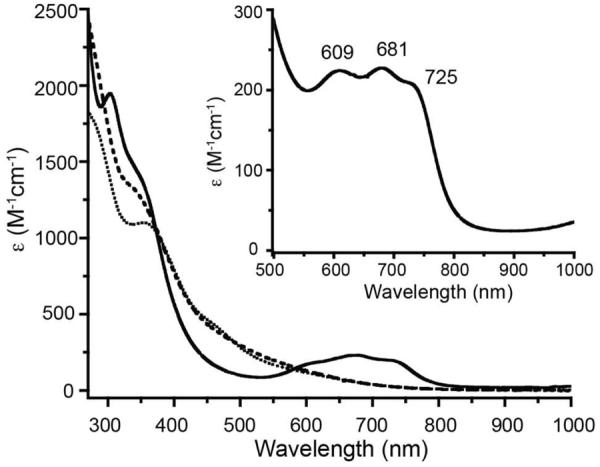
The CD spectrum of reduced Co-SODA consists of four transitions: a negative band at 280 nm, a positive band at 312 nm, a positive band at 404 nm, and a negative band at 544 nm (Figure 4, Table 1). The two highest energy bands are likely related to the S→Co LMCT transitions, which are CD active because the first coordination sphere of the cobalt is non-centrosymmetric. The neighboring chiral amino acid based transitions also induce chirality into the metal centered chromophores resulting in the two lower energy features and providing additional evidence that the peptide is involved in coordination of the metal.54, 66
Figure 4.
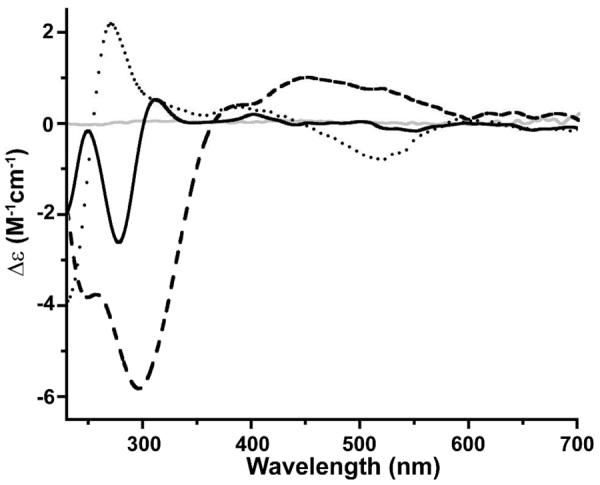
CD spectra for reduced Co-SODA (solid black line), partially oxidized Co-SODA (dotted line), completely oxidized Co-SODA (dashed line) and the SODA apo-peptide (solid grey line). All metallopeptide samples were in aqueous 50 mM NEM buffer at pH 7.5.
To evaluate the possibility that the other two ligands to the tetrahedral Co(II) in Co-SODA were nitrogenous, FTIR spectra in the region 400–1000 cm−1 were collected. Three characteristic (Co-N) stretching frequencies were observed at 452, 475 and 602 cm−1 for Co-SODA which are absent in the spectrum of the SODA apo-peptide (Figure 5). The two lower energy bands are consistent with symmetric and asymmetric Co-N stretching vibrations for two cis-positioned M-N bonds, for M=metal. The highest energy band likely arises from out of plane bending of C=O for a metal-bound amide, which is affected by the M-N bond strength since the two bonds are shared in a ring structure (Scheme 1).67, 68 Thus, it is very likely that the remaining two ligands of the primary coordination sphere are the amino terminus and a backbone amide, as seen already in the Ni-SODA peptide.
Figure 5.
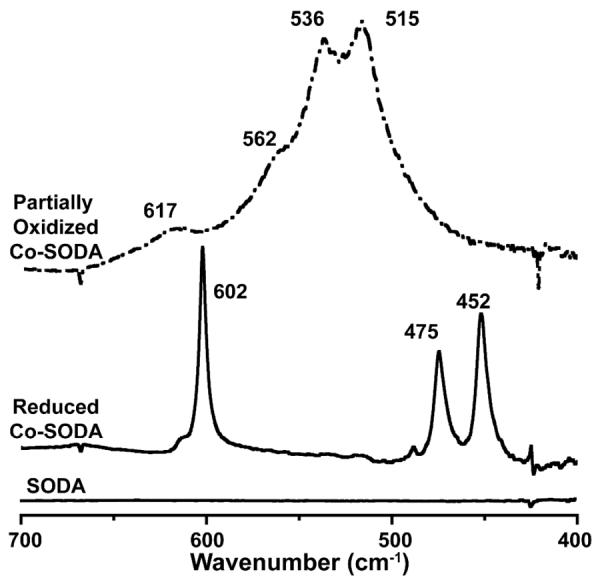
FTIR absorption spectra from the SODA apo-peptide (solid line at the bottom), reduced Co-SODA (solid line in the middle), and the partially oxidized Co-SODA (dash dotted line) in the 400–700 cm−1 region.
Aerobic oxidation of Co-SODA
The effects of molecular oxygen on the reduced Co-SODA sample were investigated by exposing it to air. The green, reduced Co-SODA was found to be oxygen sensitive. This was indicated by a color change to reddish-brown within a few minutes when exposed to air. The UV-vis spectrum recorded after 30 minutes of exposure to air consists of a nearly featureless, increase in absorption in the visible region with broad shoulders at 600 and 460 nm (Figure 3, Table 1). In the far UV region, a sharp band is seen at 355 nm. The prominent LF bands observed for the reduced Co-SODA bleached immediately upon air oxidation, suggesting changes in the tetrahedral coordination geometry around the cobalt center. Additional subtle changes in the optical spectra were observed on longer time scales (24–72 hours). The bands of the initially formed oxidized species were subtly shifted to 570 nm, 450 nm, and 340 nm, respectively (Figure 3, Table 1). After this second oxidized species was formed, no additional spectral changes were observed. These modifications of the optical spectra upon exposure to air suggest a stepwise oxidation of the reduced Co-SODA by oxygen; a partially oxidized intermediate forms first followed later by the fully oxidized product.
CD spectra were also recorded after exposure of the reduced Co-SODA to oxygen. After exposure of the sample to air for 30 minutes, a new spectrum with bands at 267 nm (negative), 306 nm (positive), 420 nm (positive) and 536 nm (negative) arose, suggesting changes in the interactions between the peptide and the cobalt ion. After 24 hours of exposure to air, the CD spectrum was further changed. In particular, both band positions and elliptical polarization (positive or negative) were changed, and a new positive feature appeared at 470 nm (Figure 4). This suggests further rearrangements in the metal-ligand electronic environment after extended interactions with oxygen. Thus CD spectroscopy provides evidence complementary to UV-vis absorption spectroscopy that two distinct species are sequentially formed by air oxidation of Co-SODA.
Magnetic susceptibility changes upon exposure of Co-SODA to oxygen
There are two distinct sites in Co-SODA at which oxidation could easily occur: the cysteinyl thiolates and the metal. Oxidation of the metal from Co(II) to Co(III) would result in a change from a paramagnetic (d7, S=3/2) starting material to a diamagnetic (d6, S=0) product. Thus we used the Evans NMR method to determine the effect of air oxidation on the magnetic susceptibility of the sample. In this experiment, the shift of a solvent NMR resonance in a sample containing the complex of interest is monitored relative to a pure solvent sample. The frequency difference between the solvent signal in the presence and the absence of the metal complex can be directly correlated to the magnetic susceptibility of the sample. As shown in Figure 6, the reduced sample is paramagnetic (0 minute data), resulting in two distinct 1H NMR resonances for the pure H2O and the H2O in the presence of Co-SODA (For a solution of ~ 1.9 mM metallopeptide in the 500 MHz NMR instrument, a shift of 30 Hz was observed.). Figure 6 shows analogous H2O 1H NMR spectra collected after the Co-SODA sample was exposed to air for varying lengths of time. The paramagnetism of the Co-SODA sample continued to be observed for several hours after exposure to air. The molecular magnetic susceptibility (χM) of the partially oxidized sample was evaluated using the Evans NMR method.47, 48 The calculated effective magnetic moment (μeff) was 4.38 Bohr magnetons. Based on the relation: μeff = √[n(n+2)], in which n= number of unpaired electrons, it is likely that the partially oxidized sample contains at least three unpaired electrons (i.e., an S=3/2 system), corresponding to Co(II). Finally, after 24 hours, the solvent signal collapsed into a single peak indicating that the system became diamagnetic. The persistence of paramagnetism for several hours suggests that the initial oxidation does not change the oxidation state of the cobalt. Instead, only on longer time scales is a diamagnetic, Co(III) species formed.
Figure 6.
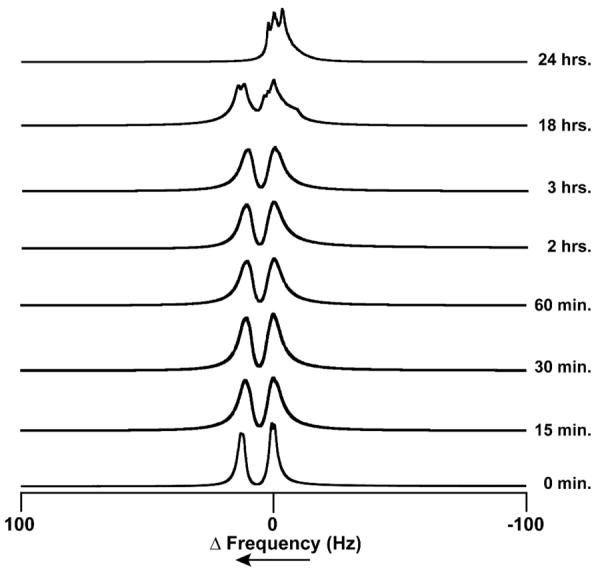
1H NMR resonances as a function of time exposed to air for H2O in an aqueous sample of reduced Co-SODA [in an NMR tube (O.D. ~ 3mm)]. The sample was placed within another NMR tube (O.D. ~ 5 mm) containing pure H2O, and the resonances are referenced such that the frequency of the signal of the blank H2O (measured independently) is zero.
To explore the hypothesis that the initial site of oxidation of the metallopeptide is the cysteine ligands, attempts were made to detect sulfinate or sulfenate groups via FTIR (see Figure S5). Although upon oxidation of the peptide extensive changes in the 1200–900 cm‒1 region of the spectrum were observed, the bands could not be unambiguously assigned, leaving interpretation relatively meaningless.
EPR spectroscopy
NMR experiments demonstrated paramagnetism in the reduced and partially oxidized Co-SODA samples. EPR spectra were recorded for these paramagnetic samples to gain insight into their electron spin distributions and corresponding changes upon oxidation. Figure 7A shows the EPR spectrum of the reduced Co-SODA sample. The spectrum was simulated using a spin Hamiltonian (see materials and methods) for a high spin Co(II) (d7 system, S=3/2) system.69, 70 The good agreement between the experimental and simulated data (σ = 0.94%) suggests, as expected, that the spectrum arises from a high spin Co(II) center. The parameters obtained from simulation, g values and zero field splitting (ZFS) constraints for the reduced Co-SODA species, are shown in Table 2. The small E/D value indicates the presence of an effectively axial field around the paramagnetic Co(II) center. The EPR spectrum of the partially oxidized sample (Figure 7B, Table 2) was also simulated with an S=3/2 spin Hamiltonian (σ = 1.20%). Although the spectra are quite similar, the g-values for the two species were not identical. This is most clearly seen from their rhombicity parameters [(gx−gy/gz) × 100%], which have values of 18.102 and 12.170 for the reduced and partially oxidized samples, respectively. The decrease in rhombicity after the partial oxidation of the complex is consistent with a change in the coordination geometry of the cobalt with no change in the oxidation state of the metal.
Figure 7.

Continuous wave (CW) EPR spectra of (A) reduced Co-SODA (in aqueous 50 mM NEM buffer at pH 7.5) at 5 K and (B) partially oxidized Co-SODA sample (in aqueous 50 mM NEM buffer at pH 7.5) at 7 K. The experimental data and the simulated data are shown as solid black line and dotted lines, respectively.
Table 2.
Simulation parameters for the EPR spectra of reduced and partially oxidized Co-SODA
| Parametera | Reduced Co-SODA | Partially oxidized Co-SODA |
|---|---|---|
| gx (±0.001) | 2.527 | 2.476 |
| gy | 2.111 | 2.199 |
| gz | 2.298 | 2.276 |
| ΔBx (MHz) | 3480 | 3695 |
| Δ By | 5544 | 6096 |
| Δ Bz | 2824 | 2491 |
| D (MHz) | ≥ 5 × 108 | ≥ 5 × 108 |
| E/D | 0.02 | 0.02 |
| [(gx−gy/gz) × 100%] | 18.102 | 12.170 |
The simulation parameters were the following: the principal g values (gx, gy and gz), the line widths (ΔBx, ΔBy and ΔBz) and the zero-field splitting parameters, D and E.
X-ray absorbance spectroscopy
Cobalt X-ray absorbance spectra (XAS) were obtained to confirm the oxidation state and characterize the first coordination sphere of the cobalt in the partially oxidized and completely oxidized samples. In the XAS spectrum of the fully oxidized sample (Figure 8A), a weak (0.06(1) eV refined area relative to edge height) pre-edge feature at ~7719 eV was observed that arises from the dipole forbidden Co(1s→3d) transition. This transition is ordinarily observed only for cobalt centers in a non-centrosymmetric environment. The edge energy of ~7723 eV corresponds to the presence of a Co(III) center in the fully oxidized metallopeptide sample, consistent with the diamagnetism observed via NMR. The extended X-ray absorbance fine structure (EXAFS) region of the XAS data is best modeled as a pseudo-octahedrally coordinated cobalt center (Figure 8D) in an S2(N/O)4 coordination environment (Figure 8B,C). The two sulfur scatterers are 2.24 Å from the Co(III) ion. The N/O ligands could be resolved into three shells: two N/O ligands at 2.13 Å, one N/O ligand at 1.97 Å, and one nitrogen ligand at 1.84 Å from the cobalt center. The last one is likely to be a deprotonated amide nitrogen ligand from the peptide backbone. The scatterer at 1.97 Å could be the amino terminus of the peptide, and the ligands at longer distances are likely solvent molecules or other loosely bound amides.
Figure 8.
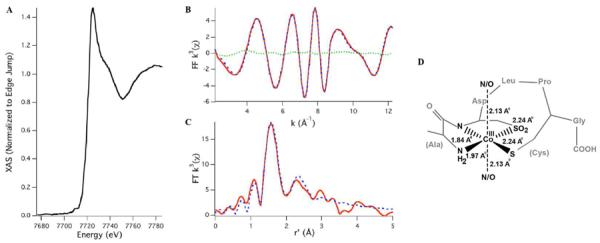
(A) XANES region of the Co K-edge X-ray absorption spectrum of fully oxidized Co-SODA. (B) FF k3 data and (C) FT k3 data (experimental data, simulated data, and the difference spectrum are represented as the red solid line, blue dashed line, and green dotted line, respectively). (D) Primary coordination geometry around cobalt in oxidized Co-SODA and metal-ligand bond distances from EXAFS. Refinement parameters: Co–S, n = 2 (restrained), r = 2.241(13) Å, σ2 = 0.0009(2) Å2; Co–N, n = 2 (restrained), r = 2.130(9) Å, σ2 = 0.0012(4) Å2; Co–N, n = 1 (restrained), r = 1.968(7) Å, σ2 = 0.0011(8) Å2; Co–N, n = 1 (restrained), r = 1.844(6) Å, σ2 = 0.0011(4) Å2; Co–C, n = 5 (restrained), r = 2.50(2) Å, σ2 = 0.0016(2) Å2; Eo = 7725.1 eV.
As shown in Figure 9 changes in the oxidation state of the Co and its first coordination sphere upon oxidation of Co-SODA were investigated by comparison of the XANES regions of the spectra for the fully oxidized, partially oxidized, and reduced (literally unoxidized) metallopeptides. The edge energy of the partially oxidized sample is 2.9(1) eV lower in energy than that of the fully oxidized sample. These edge energies are consistent with a change from Co(II) in the partially oxidized sample to Co(III) in the fully oxidized sample. The area under the pre-edge feature for the partially oxidized sample is 0.04(1) eV refined area/edge height, which is also consistent with a six coordinate geometry in the partially oxidized sample as observed in the fully oxidized sample. An EXAFS spectrum from a partially oxidized sample obtained via an experiment analogous to that shown in Figure 8 is shown in Figure S4. However, since the EXAFS data for the partially oxidized sample were of relatively poor quality at high k, the spectrum was only refined over the range 2–11 Å−1 and BT. Like the completely oxidized sample, the partially oxidized sample is best fit to a model including two sulfur ligands and four N/O ligands in the first coordination sphere (Figure S4). The Co K-edge energy for the reduced Co-SODA (7720.1 eV) is very similar to that of the partially oxidized sample, suggesting that both are in the Co(II) oxidation state (Figure 9). However, the pre-edge feature is most prominent in the spectrum from the reduced species (0.20(1) eV refined area/edge height (Figure 9) suggesting that the reduced sample features cobalt in the least centrosymmetric environment. Similarly, analysis of the EXAFS region of the spectrum from the reduced sample indicated that it consisted of Co in a four coordinate N2S2 site (Figures S6 and S7). Although the EXAFS data is relatively noisy so that it should be interpreted with caution, this observation is consistent with the assignment of the reduced species as a tetrahedral Co-N2S2 species predicted from the UV-vis and FTIR data above.
Figure 9.
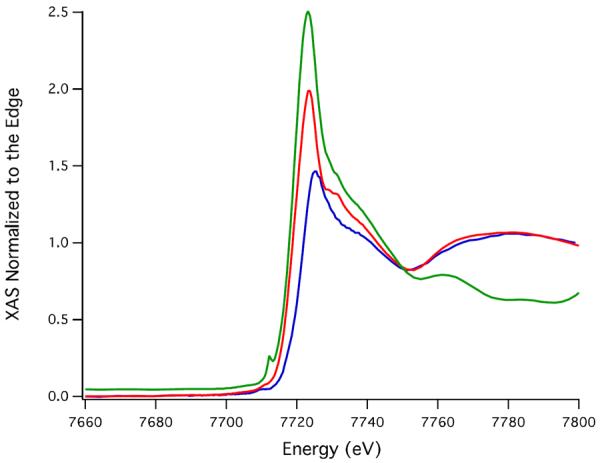
Comparison of the XANES region of the X-ray absorption spectrum of reduced (green), partially oxidized (red), and fully oxidized Co-SODA (blue).
Although FTIR could, in principle, be used to detect the SO stretches associated with oxidation of thiolates, as shown in Figure S5 in this case this approach was inconclusive in determining the initial site of oxidation in the peptide because a number of other vibrational modes of similar energies (1200–900 cm−1) were also impacted by oxidation of the peptide. Definitively assigning all of the observed changes simply proved untenable. On the other hand, S K-edge X-ray absorbance spectroscopy (XAS) is very sensitive to sulfur oxidation state, and a number of natural proteins and model complexes related to nitrile hydratase have been previously studied using this technique.71, 72 Sulfur K-edge X-ray absorbance spectra of the reduced, partially oxidized and fully oxidized metallopeptides described herein were obtained to unambiguously determine the oxidation states of the cysteinate ligands in each sample. As shown in Figure 10, the reduced sample contained, as expected, only unoxygenated cysteinate sulfurs; a pre-edge peak at 2470.9(1) eV was observed corresponding to the S(1s) → Co(3d) transition. At higher energy (2472.6(1) eV), the S(1s) → (C-S(σ*)) transition is observed. In contrast, the S K-edge results suggest the completely oxidized Co(III) complex contains one unmodified cysteinate and one that has been oxidized to a sulfinate (SO2−) ligand. The S K-edge spectrum for this sample includes a relatively small pre-edge feature at 2477.8(3) eV together with a strong peak at 2480.8(3) eV. These correspond to SO2(1s) → Co(3d) and SO2(1s) → C–S(σ*) transitions, respectively. In addition the S(1s) → Co(3d) transition is observed at 2470.1(1) eV and the S(1s) → (C-S(σ*)) transition at 2473.3(2) eV indicating the presence of a reduced thiolate sulfur ligand. The spectrum of the partially oxidized sample is remarkably similar to that of the completely oxidized metallopeptide and is also consistent with the sulfur being present as thiolate and sulfinate species. These data provide clear spectroscopic evidence that the first step in the oxidation process is modification of one of the cysteines to form sulfinate.
Figure 10.
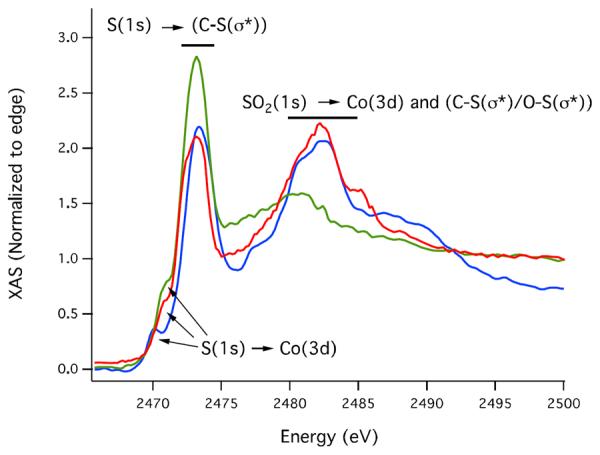
S K-edge X-ray absorption spectra of fully reduced (green), partially oxidized (red) and fully oxidized (blue) CoSODA.
Concluding Remarks
As depicted in Scheme 1, the data presented herein suggest the tetrahedral high spin d7 Co(II)-SODA is air oxidized in a two-step process. The immediate changes in the UV-vis and CD spectra upon exposure of Co-SODA to air suggest oxidation occurs rapidly and results in formation of a pseudo-octahedral species. However, perhaps surprisingly, NMR and EPR spectra indicate that the first oxidation product formed is still paramagnetic with three unpaired electrons. This indicates Co cannot be the first species oxidized. Instead, the S K-edge XAS spectrum of this intermediate shows that the first oxidation occurs at a single cysteine ligand to form a sulfinate. Then, it is only on longer time scales that Co oxidizes to the 3+ oxidation state becoming a diamagnetic low spin d6 system. EXAFS data suggest this fully oxidized metallopeptide contains a pseudo-octahedrally coordinate Co(III) ion ligated by an unmodified cysteinate as well as an oxidized sulfinate ligand. The remainder of the coordination sphere is four N/O donors present in two pairs at different distances. Two of those are likely to be the peptide ligands from the reduced peptide and the additional ones are expected to be solvent molecules. Thus air oxidation of Co-SODA causes two sequential types of changes. First oxidation of one of the cysteines occurs; then oxidation of the metal follows.
Oxidation of cysteinyl thiolates can result in a number of products including disulfide bonds, sulfinate and sulfenate. Thus the question of how each cysteine in the active site of nitrile hydratase achieves a unique oxidation state comes to the fore, and Co-SODA, in which one cysteinate is modified and the other remains reduced, is an interesting model. For these metalloproteins/peptides, formation of disulfide bonds is likely of little concern due to the relative inertness of Co(II) to substitution reactions. On the other hand, in the case of Co-SODA, a peptide originally designed to bind not Co but Ni, oxidation of one of the cysteines likely provides more stable bonding. This lowers the energies of the occupied Co d orbitals and increases the driving force for oxidation of the metal to form a low spin d6 system. Furthermore, binding of S-O ligands likely strains the tetrahedral geometry of Co(II)-SODA towards a more favorable octahedral environment, again providing ideal conditions for oxidation of the metal. We can speculate that once the metal is oxidized in Co-SODA there is relatively little energetic gain from oxidation of the second thiolate so that it remains unmodified.
Comparison of Co SODA to other model complexes for nitrile hydratase suggests reasons for the unique reactivity of this metallopeptide. Angelosante and coworkers reported the synthesis of an unsymmetrical, square planar S=1 Co(II) complex in which the metal is coordinated in an N2S2 environment consisting of two thiolates and two amidates.73 Interestingly, this complex mimics the asymmetric equatorial coordination environment of nitrile hydratase that consists of two five-membered rings and a single six-membered ring. However, in contrast to Co-SODA, the reported complex does not undergo air oxidation of either the thiolates or the metal.73 In essence, the square planar complex appears to be too stable to promote the oxidation. On the other hand, Shearer and coworkers reported the reactivity of an N2S3 coordinated Co-metallopeptide known as Co-NHasem1.74 Like Co-SODA, reduced Co-NHasem1 binds Co(II) in a tetrahedral environment consisting of two thiolates and two amidates. Upon exposure to air, both the metal and two of the three thiolates are oxidized. As shown in Fig. S3, we undertook NMR experiments with Co-NHasem1 to compare its oxidation pathway to that of Co-SODA. Unlike Co-SODA, upon oxidation Co-NHasem1 rapidly forms a diamagnetic Co(III) state. Furthermore, both equatorial thiolates are likely oxidized. The increased rate of metal oxidation in Co-NHasem1 is likely a result, in large part, of the additional axial thiolate ligand and the replacement of an amine with an amide ligand. As shown in Scheme S2, the strongly σ-donating axial thiolate likely interacts with the metal dz2 orbital, changing its character from non-bonding to antibonding – significantly raising its energy. Similarly, the amide of Co-NHasem1 provides stronger π-bonding than the amine of Co-SODA further increasing the ligand field stabilization energy of the N2S3 peptide and stabilizing the low spin d6 oxidized state. Thus oxidation of Co-NHasem1 is considerably more favorable energetically than oxidation of Co-SODA. Essentially, Co-SODA is intermediate between the relatively inert complex of Angelosante and coworkers and the reactive metallopeptide Co-NHasem1. Furthermore, it is likely the tetrahedral geometry of the reduced Co-SODA species and the lack of an axial thiolate that allow it to occupy this unique niche of reactivity. Nonetheless, more detailed computational investigation will be necessary to confirm this hypothesis.
Finally, we end by speculating as to the applicability of Co-SODA to understanding the details of metal incorporation and protein modification in the natural nitrile hydratase enzyme. Cobalt is generally transported into microbes as Co(II) ion, and it is likely that Co is incorporated into the apo-nitrile hydratase α subunit in this reduced oxidation state.75, 76 Biochemical evidence has arisen that, at least in vitro, post-translational modification of the nitrile hydratase protein and oxidation of the metal are autocatalytic.5 The reactivity of Co-SODA in air lends further credence to such a model. By analogy to Co-SODA, we hypothesize that interaction of the reduced NHase protein with air first results in oxidation of one or both of the equatorial sulfur ligands. Initial binding of the metal in a geometry that is more tetrahedral than square planar geometry would likely promote the specific protein modifications. The modified bonding with the oxidized ligands could then provide the driving force for a subtle change in geometry and oxidation of the metal to the active form of the enzyme. At the moment, these ideas are hypothetical, but further investigation of both the enzyme and relevant metallopeptide models is expected to clarify the remaining unknowns of this unique biosynthetic process. Moreover, we anticipate that designed metalloproteins will prove to be an exciting tool for exploring metallocenter biosynthesis in a biologically relevant, in vitro, chemical context.
Supplementary Material
Synopsis.
Air oxidation of the cobalt-bound seven amino acid peptide ACDLPCG (Co-SODA) is characterized as a model for maturation of nitrile hydratase. Co-SODA is oxidized in two steps. In the first step, the metallocenter remains unchanged while one of the thiolates is oxidized to sulfinate. In the second step, the cobalt center is oxidized to Co3+.
Table 3.
Summary of K-edge energies from Co and S XAS.
| Sample | Co K-edge energy (eV) | S K-edge energy (eV) | ||||
|---|---|---|---|---|---|---|
| Co (1s→3d) | Co edge | S(1s)→Co(3d) | S(ls)→C-S (σ*) | SO2(1s)→Co(3d) | SO2(1s)→C–S(σ*) | |
| Reduced Co-SODA | 7719.3(2) | 7720.1(3) | 2470.9(2) | 2472.6(2) | N/A | N/A |
| Partially oxidized Co-SODA | 7719.2(1) | 7720.1(4) | 2470.4(2) | 2473.2(2) | 2477.6(7) | 2480(1) |
| Fully oxidized Co-SODA | 7719.4(1) | 7723.2(3) | 2470.1(2) | 2473.2(2) | 2477.8(6) | 2480(1) |
ACKNOWLEDGMENT
This research was supported through the Center for Bio-Inspired Solar Fuel Production an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award Number DE-SC0001016. J.M.S. acknowledges the NSF for financial support (CHE-0844234). H.E.H. acknowledges NSF for financial support (EAR-0846188). XAS data collection at the National Synchrotron Light Source (NSLS) at Brookhaven National Laboratory was supported by the U.S. Department of Energy, Division of Materials Sciences and Division of Chemical Sciences. Beamline X3B at NSLS is supported by the NIH. This publication was made possible in part by the Center for Synchrotron Biosciences grant, P30-EB-009998, from the National Institute of Biomedical Imaging and Bioengineering (NIBIB). A.D. also thanks the Science Foundation of Arizona for a graduate fellowship. We are grateful to Dr. Ulrich Haussermann for technical assistance preparing anaerobic KBr pellets.
Footnotes
Supporting Information Additional mass spectral, NMR, and EXAFS data. Partial molecular orbital diagrams and hypothetical oxidation scheme. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.Paulsen CE, Carroll KS. ACS Chem. Biol. 2010;5:47–62. doi: 10.1021/cb900258z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claiborne A, Yeh JI, Mallett TC, Luba J, Crane EJ, Charrier V, Parsonage D. Biochemistry. 1999;38:15407–15416. doi: 10.1021/bi992025k. [DOI] [PubMed] [Google Scholar]
- 3.Dickinson BC, Chang CJ. Nat. Chem. Biol. 2011;7:504–511. doi: 10.1038/nchembio.607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nagashima S, Nakasako M, Dohmae N, Tsujimura M, Takio K, Odaka M, Yohda M, Kamiya N, Endo I. Nat. Struct. Mol. Biol. 1998;5:347–351. doi: 10.1038/nsb0598-347. [DOI] [PubMed] [Google Scholar]
- 5.Arakawa T, Kawano Y, Katayama Y, Nakayama H, Dohmae N, Yohda M, Odaka M. J. Am. Chem. Soc. 2009;131:14838–14843. doi: 10.1021/ja903979s. [DOI] [PubMed] [Google Scholar]
- 6.Arakawa T, Kawano Y, Kataoka S, Katayama Y, Kamiya N, Yohda M, Odaka M. J. Mol. Biol. 2007;366:1497–1509. doi: 10.1016/j.jmb.2006.12.011. [DOI] [PubMed] [Google Scholar]
- 7.Katayama Y, Hashimoto K, Nakayama H, Mino H, Nojiri M, Ono T, Nyunoya H, Yohda M, Takio K, Odaka M. J. Am. Chem. Soc. 2006;128:728–729. doi: 10.1021/ja057010q. [DOI] [PubMed] [Google Scholar]
- 8.Huang WJ, Jia J, Cummings J, Nelson M, Schneider G, Lindqvist Y. Structure. 1997;5:691–699. doi: 10.1016/s0969-2126(97)00223-2. [DOI] [PubMed] [Google Scholar]
- 9.Miyanaga A, Fushinobu S, Ito K, Wakagi T. Biochem. Biophys. Res. Commun. 2001;288:1169–1174. doi: 10.1006/bbrc.2001.5897. [DOI] [PubMed] [Google Scholar]
- 10.Nakasako M, Odaka M, Yohda M, Dohmae N, Takio K, Kamiya N, Endo I. Biochemistry. 1999;38:9887–9898. doi: 10.1021/bi982753s. [DOI] [PubMed] [Google Scholar]
- 11.Song L, Wang M, Shi J, Xue Z, Wang M-X, Qian S. Biochem. Biophys. Res. Commun. 2007;362:319–324. doi: 10.1016/j.bbrc.2007.07.184. [DOI] [PubMed] [Google Scholar]
- 12.Murakami T, Nojiri M, Nakayama H, Dohmae N, Takio K, Odaka M, Endo I, Nagamune T, Yohda M. Protein Sci. 2000;9:1024–1030. doi: 10.1110/ps.9.5.1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Endo I, Odaka M, Yohda M. Trends Biotechnol. 1999;17:244–248. doi: 10.1016/s0167-7799(99)01303-7. [DOI] [PubMed] [Google Scholar]
- 14.Zhou Z, Hashimoto Y, Kobayashi M. Actinomycetologica. 2005;19:18–26. [Google Scholar]
- 15.Kobayashi M, Nagasawa T, Yamada H. Trends Biotechnol. 1992;10:402–408. doi: 10.1016/0167-7799(92)90283-2. [DOI] [PubMed] [Google Scholar]
- 16.Noveron JC, Olmstead MM, Mascharak PK. J. Am. Chem. Soc. 1999;121:3553–3554. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- 17.Kovacs JA. Chem. Rev. 2004;104:825–848. doi: 10.1021/cr020619e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kung I, Schweitzer D, Shearer J, Taylor WD, Jackson HL, Lovell S, Kovacs JA. J. Am. Chem. Soc. 2000;122:8299–8300. [Google Scholar]
- 19.Shearer J, Kung IY, Lovell S, Kaminsky W, Kovacs JA. J. Am. Chem. Soc. 2001;123:463–468. doi: 10.1021/ja002642s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Galvez C, Ho DG, Azod A, Selke M. J. Am. Chem. Soc. 2001;123:3381–3382. doi: 10.1021/ja003993+. [DOI] [PubMed] [Google Scholar]
- 21.Rat M, Alves de Sousa R, Vaissermann J, Leduc P, Mansuy D, Artaud I. J. Inorg. Biochem. 2001;84:207–213. doi: 10.1016/s0162-0134(01)00181-7. [DOI] [PubMed] [Google Scholar]
- 22.Shearer J, Callan PE, Amie J. Inorg. Chem. 2010;49:9064–9077. doi: 10.1021/ic101765h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Masitas CA, Kumar M, Mashuta MS, Kozlowski PM, Grapperhaus CA. Inorg. Chem. 2010;49:10875–10881. doi: 10.1021/ic101221z. [DOI] [PubMed] [Google Scholar]
- 24.Farmer PJ, Solouki T, Mills DK, Soma T, Russell DH, Reibenspies JH, Darensbourg MY. J. Am. Chem. Soc. 1992;114:4601–4605. [Google Scholar]
- 25.Mirza SA, Pressler MA, Kumar M, Day RO, Maroney MJ. Inorg. Chem. 1993;32:977–987. [Google Scholar]
- 26.Farmer PJ, Verpeaux J-N, Amatore C, Darensbourg MY, Musie G. J. Am. Chem. Soc. 1994;116:9355–9356. [Google Scholar]
- 27.Noveron JC, Olmstead MM, Mascharak PK. J. Am. Chem. Soc. 2001;123:3247–3259. doi: 10.1021/ja001253v. [DOI] [PubMed] [Google Scholar]
- 28.Kaasjager VE, Bouwman E, Gorter S, Reedijk J, Grapperhaus CA, Reibenspies JH, Smee JJ, Darensbourg MY, Derecskei-Kovacs A, Thomson LM. Inorg. Chem. 2002;41:1837–1844. doi: 10.1021/ic010784x. [DOI] [PubMed] [Google Scholar]
- 29.Lee C-M, Hsieh C-H, Dutta A, Lee G-H, Liaw W-F. J. Am. Chem. Soc. 2003;125:11492–11493. doi: 10.1021/ja035292t. [DOI] [PubMed] [Google Scholar]
- 30.Lee C-M, Chen C-H, Chen H-W, Hsu J-L, Lee G-H, Liaw W-F. Inorg. Chem. 2005;44:6670–6679. doi: 10.1021/ic050108l. [DOI] [PubMed] [Google Scholar]
- 31.Chohan BS, Maroney MJ. Inorg. Chem. 2006;45:1906–1908. doi: 10.1021/ic051982h. [DOI] [PubMed] [Google Scholar]
- 32.Grapperhaus CA, Darensbourg MY, Sumner LW, Russell DH. J. Am. Chem. Soc. 1996;118:1791–1792. [Google Scholar]
- 33.Galardon E, Giorgi M, Artaud I. Chem. Commun. 2004:286–287. doi: 10.1039/b312318a. [DOI] [PubMed] [Google Scholar]
- 34.Bourles E, de Sousa RA, Galardon E, Giorgi M, Artaud I. Angew. Chem. Int. Edit. 2005;44:6162–6165. doi: 10.1002/anie.200500814. [DOI] [PubMed] [Google Scholar]
- 35.Zhou Z, Hashimoto Y, Shiraki K, Kobayashi M. Proc. Natl. Acad. Sci. USA. 2008;105:14849–14854. doi: 10.1073/pnas.0803428105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou ZM, Hashimoto Y, Kobayashi M. J. Biol. Chem. 2009;284:14930–14938. doi: 10.1074/jbc.M808464200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou Z, Hashimoto Y, Cui T, Washizawa Y, Mino H, Kobayashi M. Biochemistry. 2010;49:9638–9648. doi: 10.1021/bi100651v. [DOI] [PubMed] [Google Scholar]
- 38.Sloan CP, Krueger JH. Inorg. Chem. 1975;14:1481–1485. [Google Scholar]
- 39.Lange BA, Libson K, Deutsch E, Elder RC. Inorg. Chem. 1976;15:2985–2989. [Google Scholar]
- 40.Higashi LS, Lundeen M, Hilti E, Seff K. Inorg. Chem. 1977;16:310–313. [Google Scholar]
- 41.Lundeen M, Firor RL, Seff K. Inorg. Chem. 1978;17:701–706. doi: 10.1021/ic50184a007. [DOI] [PubMed] [Google Scholar]
- 42.Elder RC, Heeg MJ, Payne MD, Trkula M, Deutsch E. Inorg. Chem. 1978;17:431–440. [Google Scholar]
- 43.Kubota M, Rothrock RK, Kernan MR, Haven RB. Inorg. Chem. 1982;21:2491–2493. [Google Scholar]
- 44.Bertz SH, Dabbagh G. Inorg. Chem. 1985;24:3488–3490. [Google Scholar]
- 45.Stoll S, Schweiger A. J. Magn. Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 46.Flores M, Isaacson R, Abresch E, Calvo R, Lubitz W, Feher G. Biophys. J. 2007;92:671–682. doi: 10.1529/biophysj.106.092460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Evans DF. J. Chem. Soc. 1959:2003–2005. [Google Scholar]
- 48.Grant DH. J. Chem. Educ. 1995;72:39. [Google Scholar]
- 49.Shearer J, Dehestani A, Abanda F. Inorg. Chem. 2008;47:2649–2660. doi: 10.1021/ic7019878. [DOI] [PubMed] [Google Scholar]
- 50.Shearer J, Scarrow RS. EXAFS123. University of Nevada; Reno: 2011. [Google Scholar]
- 51.IgorPro . Wavemetrics, Inc.; Lake Oswego, OR: 2011. [Google Scholar]
- 52.Ankudinov AL, Ravel B, Rehr JJ, Conradson SD. Phys. Rev. B. 1998;58:7565–7576. [Google Scholar]
- 53.Dutta A, Hamilton GA, Hartnett HE, Jones AK. Inorg. Chem. 2012;51:9580–9588. doi: 10.1021/ic2026818. [DOI] [PubMed] [Google Scholar]
- 54.Neupane KP, Gearty K, Francis A, Shearer J. J. Am. Chem. Soc. 2007;129:14605–14618. doi: 10.1021/ja0731625. [DOI] [PubMed] [Google Scholar]
- 55.Ellman GL. Arch. Biochem. Biophys. 1958;74:443–450. doi: 10.1016/0003-9861(58)90014-6. [DOI] [PubMed] [Google Scholar]
- 56.Choudhury SB, Lee J-W, Davidson G, Yim Y-I, Bose K, Sharma ML, Kang S-O, Cabelli DE, Maroney MJ. Biochemistry. 1999;38:3744–3752. doi: 10.1021/bi982537j. [DOI] [PubMed] [Google Scholar]
- 57.Wuerges J, Lee J-W, Yim Y-I, Yim H-S, Kang S-O, Carugo KD. Proc. Natl. Acad. Sci. USA. 2004;101:8569–8574. doi: 10.1073/pnas.0308514101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barondeau DP, Kassmann CJ, Bruns CK, Tainer JA, Getzoff ED. Biochemistry. 2004;43:8038–8047. doi: 10.1021/bi0496081. [DOI] [PubMed] [Google Scholar]
- 59.Lever ABP. Inorganic Electronic Spectroscopy. 3rd ed. Elsevier; 1986. [Google Scholar]
- 60.Petros AK, Shaner SE, Costello AL, Tierney DL, Gibney BR. Inorg. Chem. 2004;43:4793–4795. doi: 10.1021/ic0497679. [DOI] [PubMed] [Google Scholar]
- 61.Petros AK, Reddi AR, Kennedy ML, Hyslop AG, Gibney BR. Inorg. Chem. 2006;45:9941–9958. doi: 10.1021/ic052190q. [DOI] [PubMed] [Google Scholar]
- 62.Drum DE, Vallee BL. Biochem. Biophys. Res. Commun. 1970;41:33–39. doi: 10.1016/0006-291x(70)90464-x. [DOI] [PubMed] [Google Scholar]
- 63.May SW, Kuo J-Y. Biochemistry. 1978;17:3333–3338. doi: 10.1021/bi00609a025. [DOI] [PubMed] [Google Scholar]
- 64.Vasak M, Kaegi JHR, Holmquist B, Vallee BL. Biochemistry. 1981;20:6659–6664. doi: 10.1021/bi00526a021. [DOI] [PubMed] [Google Scholar]
- 65.VanZile ML, Cosper NJ, Scott RA, Giedroc DP. Biochemistry. 2000;39:11818–11829. doi: 10.1021/bi001140o. [DOI] [PubMed] [Google Scholar]
- 66.Nakashini K, Berova N, Woody RW. Circular Dichroism: Principles and Applications. 1st ed. Wiley-VCH publishers; Darmstadt: 1994. [Google Scholar]
- 67.Condrate RA, Nakamoto K. J. Chem. Phys. 1965;42:2590–2598. doi: 10.1063/1.1696337. [DOI] [PubMed] [Google Scholar]
- 68.Kincaid JR, Nakamoto K. Spectrochim. Acta, Part A. 1976;32:277–283. [Google Scholar]
- 69.Rizzi AC, Brondino CD, Calvo R, Baggio R, Garland MT, Rapp RE. Inorg. Chem. 2003;42:4409–4416. doi: 10.1021/ic026111b. [DOI] [PubMed] [Google Scholar]
- 70.Pietrzyk P, Srebro M, Radoń M, Sojka Z, Michalak A. J. Phys. Chem. A. 2011;115:2316–2324. doi: 10.1021/jp109524t. [DOI] [PubMed] [Google Scholar]
- 71.Dey A, Chow M, Taniguchi K, Lugo Mas P, Davin S, Maeda M, Kovacs JA, Odaka M, Hodgson KO, Hedman B, Solomon EI. J. Am. Chem. Soc. 2006;128:533–541. doi: 10.1021/ja0549695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rompel A, Cinco RM, Latimer MJ, McDermott AE, Guiles RD, Quintanilha A, Krauss RM, Sauer K, Yachandra VK, Klein MP. Proc. Natl. Acad. Sci. USA. 1998;95:6122–6127. doi: 10.1073/pnas.95.11.6122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Angelosante J, Schopp L, Lewis B, Vitalo A, Titus D, Swanson R, Stanley A, Abolins B, Frome M, Cooper L, Tierney D, Moore C, Rheingold A, Daley CA. J. Biol. Inorg. Chem. 2011;16:937–947. doi: 10.1007/s00775-011-0794-7. [DOI] [PubMed] [Google Scholar]
- 74.Shigehiro S, Nakasako M, Dohmae N, Tsujimura M, Tokoi K, Odaka M, Yohda M, Kamiya N, Endo I. Nat. Struct. Biol. 1998;5:347–351. doi: 10.1038/nsb0598-347. [DOI] [PubMed] [Google Scholar]
- 75.Komeda H, Kobayashi M, Shimizu S. Proc. Natl. Acad. Sci. USA. 1997;94:36–41. doi: 10.1073/pnas.94.1.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Okamoto S, Eltis LD. Metallomics. 2011;3:963–970. doi: 10.1039/c1mt00056j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.