
Figure 8.
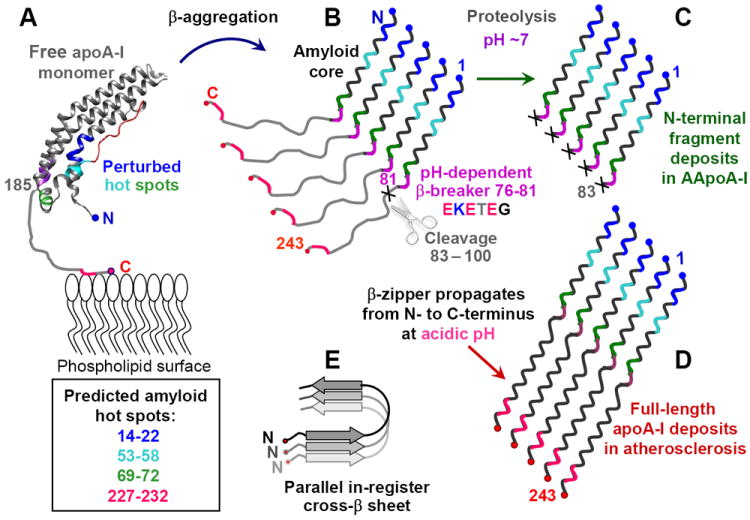
Hypothetical pathway of apoA-I misfolding in familial and in acquired amyloidosis. (A) Free apoA-I monomer, generated de-novo or dissociated from HDL, forms a four-helix bundle containing residues 1-184 (helix ribbon) followed by the partially unstructured C-terminal residues 185-243 (thin line) containing the primary lipid binding site. Four predicted amyloid hot spots are color-coded. AApoAI mutation in familial amyloidosis or M86 oxidation in acquired amyloidosis may perturb native structure in these hot spots and initiate β-aggregation. This leads to formation of the protease-resistant amyloid core in the first 75 residues containing N-terminal amyloid hot spots (in blue, teal and green) (B). In the parallel in-register β-sheet predicted for apoA-I, each hot spot (color-coded) stacks in register against its counterparts from the adjacent molecules. At pH~7, the β-breaking EKETEG residues 76-81 probably impede the amyloid core propagation towards the C-terminus and hamper the in-register stacking of the similarly charged groups. As a result, amyloid core stops at residues 76-81, leading to protein cleavage shortly thereafter. This explains why amyloid core in the patient-derived AApoAI deposits contains residue segments 1-83 to 1-100 (C). (D) At acidic pH, partial protonation of the adjacent Glu is expected to diminish the β-breaking potential of the EKETEG motif. This facilitates the β-sheet propagation towards the C-terminus and formation of fibrils containing full-length apoA-I, such as those found in atherosclerotic plaques. In the resulting parallel in-register β-sheet structure, different hot spots within the molecule act in synergy (D), while similar secondary structural elements from different molecules (in different shades of gray, E) are stacked in register.