

Background: NADPH oxidases (Nox) are involved in pathogenesis of inflammatory and fibrotic diseases.
Results: High-throughput screening methodology is developed for discovery and determination of Nox inhibitors and mechanism.
Conclusion: Rapid and specific monitoring of superoxide and hydrogen peroxide is possible, enabling high-throughput screening of Nox isoform-specific inhibitors.
Significance: The proposed strategy will enable more rigorous research on the chemical biology of Nox enzymes.
Keywords: Enzyme Inhibitor, Fluorescence, High-performance Liquid Chromatography (HPLC), High-throughput Screening (HTS), Hydrogen Peroxide, NADPH Oxidase, Reactive Oxygen Species (ROS), Superoxide Ion
Abstract
Recent progress characterizing the reaction mechanism(s) of fluorescent probes with reactive oxygen species has made it possible to rigorously analyze these reactive species in biological systems. We have developed rapid high throughput-compatible assays for monitoring cellular production of superoxide radical anion and hydrogen peroxide using hydropropidine and coumarin boronic acid probes, respectively. Coupling plate reader-based fluorescence measurements with HPLC-based simultaneous monitoring of superoxide radical anion and hydrogen peroxide provides the basis for the screening protocol for NADPH oxidase (Nox) inhibitors. Using this newly developed approach along with the medium-throughput plate reader-based oximetry and EPR spin trapping as confirmatory assays, it is now eminently feasible to rapidly and reliably identify Nox enzyme inhibitors with a markedly lower rate of false positives. These methodological advances provide an opportunity to discover selective inhibitors of Nox isozymes, through enhanced conceptual understanding of their basic mechanisms of action.
Introduction
The Nox3 family of enzymes (Nox1 to -5, Duox1 and -2) and mitochondrial electron transport enzymes are among the major sources of cellular reactive oxygen species (ROS), including superoxide radical anion (O2⨪), hydrogen peroxide (H2O2), and related oxidants (1, 2). However, unlike mitochondrial enzymes from which ROS are generated as an “accidental” byproduct of their primary catalytic function, Nox enzymes (Nox1 to -5, Duox1 and -2) are “professional” ROS generators, with no other known reported function (3, 4). Nox enzyme-generated ROS are reported to play a prominent role in both inflammatory and fibrotic diseases (5, 6). Nox-related inflammatory diseases include acute respiratory disease syndrome, chronic obstructive pulmonary disease, neurodegenerative diseases (including Alzheimer disease, Parkinson disease, and ischemic stroke), and reperfusion injury occurring during transplantation (7). Fibrotic diseases include liver fibrosis, idiopathic pulmonary fibrosis, and diabetic kidney disease (8, 9). Nox2 has been implicated in inflammatory diseases, and Nox4 has been implicated in fibrotic diseases (8, 9). Existing therapies for both classes of diseases include steroids and other anti-inflammatory approaches but are largely ineffective (10). Hence, there is a need for the development of novel drugs based on new targets (10). Nox enzymes are a promising drug target, based on mounting evidence in humans and in experimental animal models (10–16).
Despite decades of research, only a limited number of Nox inhibitors are currently available, few if any of which are isoform-specific; researchers are therefore actively seeking new small molecules as isoform-selective Nox inhibitors (17–20). The limited progress thus far is due in part to limitations of the assays that are currently in use for detection and quantitation of ROS (21). Nox activity is assessed by measuring ROS generation, but the lack of robust methods for monitoring specific individual species of reactive oxygen (or indeed to even distinguish between reactive oxygen and reactive nitrogen species) has hampered screening for Nox inhibitors. Primary screening assays for monitoring Nox activity often include the use of nonspecific and artifact-prone probes (e.g. lucigenin, luminol, L-012) that self-generate O2⨪ via redox cycling of intermediates (22, 23), resulting in an unusually higher rate of false positives and potentially missing weaker but selective “hits” that are lost in the “noise.” We propose that the use of more specific probes, including site- and species-specific probes will enable more efficient high-throughput screening of Nox isoenzyme-specific inhibitors. The use of ROS-specific probes whose redox chemistry is better understood with respect to reaction kinetics, stoichiometry, and product formation is therefore critically important (24–26).
Recent progress made with regard to development of new fluorescent probes for ROS and understanding their chemistry has enabled us to detect superoxide and hydrogen peroxide generated in cellular/subcellular compartments (27–31). Recently, we demonstrated the global profiling of oxidizing, nitrosating, and nitrating species in activated macrophages using a 96-well plate format (32). Here, we report the rapid, high throughput-compatible analyses of O2⨪ and H2O2 and screening of inhibitors of NADPH oxidases. The overall workflow of the screening protocol for inhibitors of NADPH oxidases and detection of ROS is shown in Fig. 1. Specifically, we used hydropropidine (HPr+), a cell-impermeable analog of hydroethidine (HE), for detecting extracellular O2⨪ and coumarin-7-boronic acid (CBA) for peroxidase-independent detection of H2O2 in the primary assays (33, 34). In orthogonal assays, we used HPLC-based detection of 2-hydroxyethidium, a specific product of the reaction of hydroethidine with O2⨪, and peroxidase-catalyzed oxidation of Amplex Red to resorufin for H2O2 measurement (35). The modes and specific parameters of detection are listed in Table 1. In addition to the assays listed above, a 96-well plate-based oximetry is used as a confirmatory assay, providing a direct “probe-free” measurement of NADPH oxidase activity in a medium-throughput manner. We expect that the methodology described will provide a basic set of tools for rapid, reliable, and selective monitoring of O2⨪ and H2O2 dynamics in cellular and cell-free systems.
FIGURE 1.

Probe chemistry and assay design. a, probes and products formed in primary assays. b, probes and products formed in orthogonal assays. c, workflow scheme for screening of Nox inhibitors.
TABLE 1.
Fluorescence settings used in primary and orthogonal assays for O2⨪ and H2O2
| Probe | Species detected | Product detected | Fluorescence detection settings |
|
|---|---|---|---|---|
| Emission λmax | Excitation λmax | |||
| nm | ||||
| Primary assays | ||||
| HPr+ + DNA | O2⨪ | 2-OH-Pr2+ bound to DNA | 485 | 574 |
| CBA | H2O2 | COH | 355 | 460 |
| Orthogonal assays | ||||
| HE | O2⨪ | 2-OH-E+ detected by HPLC | 370 | 565 |
| Amplex® Red + HRP | H2O2 | Resorufin | 535 | 595 |
EXPERIMENTAL PROCEDURES
Materials
Hydropropidine (HPr+) was synthesized, according to a previously published procedure (33). The stock solution was prepared in deoxygenated water at a final concentration of 1 mm, aliquoted, and stored at −80 °C. DNA (from salmon testes) was obtained from Sigma-Aldrich and dissolved at a 2 mg/ml concentration in Tris buffer (10 mm) containing EDTA (1 mm) and stored at 4 °C. CBA was synthesized according to a published procedure (34). The stock solution of CBA at 0.1 m concentration was prepared in DMSO and stored at −20 °C. Amplex Red (10-acetyl-3,7-dihydroxyphenoxazine) was from Cayman, and the stock solution at 50 mm concentration in DMSO was stored at −20 °C. Horseradish peroxidase at a concentration of 1 kilounit/ml (HRP, type VI, from Sigma) was prepared in phosphate buffer (50 mm, pH 7.4) containing DTPA (100 μm) and stored at 4 °C. HE was from Invitrogen, and the stock solution at 20 mm concentration in deoxygenated DMSO was aliquoted and stored at −80 °C. Superoxide dismutase (SOD), catalase (CAT), diphenyleneiodonium (DPI), 6-hydroxydopamine (6-OH-DA), phenylarsine oxide, apocynin, ebselen, and neopterin were obtained from Sigma-Aldrich. Diapocynin was synthesized, as described recently (36). VAS2870 was purchased from Enzo. Compounds 13–44 were synthesized at Emory University. Stock solutions of inhibitors were prepared in DMSO at a concentration of 10 mm, with the exception of DPI (1 mm), apocynin (1 m), and diapocynin (50 mm). Authentic standard of 7-hydroxycoumarin (COH) was from Sigma-Aldrich, and 2-hydroxyethidium (2-OH-E+) was synthesized as described elsewhere (37). All other reagents were obtained from Sigma-Aldrich.
Cellular Models of Nox2, Nox4, and Nox5
Human promyelocytic leukemia HL60 cells (Sigma) differentiated into neutrophils by incubation for 4–5 days with all-trans-retinoic acid (ATRA; 1 μm) were used as a source of Nox2. HL60 cells were grown in RPMI 1640 medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Sigma), penicillin (100 units/ml), and streptomycin (100 μg/ml) and differentiated in the same medium but in the presence of ATRA. For comparison, non-differentiated cells were used as control (Fig. 2). Nox2 activation was achieved by treatment of differentiated HL60 cells with phorbol myristate acetate (PMA; 1 μm). To study Nox4 isoenzyme, we used HEK293 cells overexpressing Nox4 (HEK-Nox4), obtained from Dr. Ralf Brandes (Goethe-University, Frankfurt, Germany). Nox4 activity was deducted from the comparison of probe oxidation by HEK-Nox4 and HEK293 (non-transfected) cells. For Nox5 activity measurements, we used HEK293 cells overexpressing Nox5 (HEK-Nox5), obtained from Dr. David Fulton (Georgia Regents University, Augusta, GA). To stimulate Nox5 activity, cells were treated with ionomycin (1 μm). HEK293, HEK-Nox4, and HEK-Nox5 were grown in DMEM (Invitrogen) supplemented with 5% FBS, 0.1% Mito+ (BD Biosciences), and ciproflaxin (10 μg/ml).
FIGURE 2.

dHL60 model for monitoring the activity of Nox2 NADPH oxidase. a, comparison of the mitochondrial (OCR) and glycolytic (extracellular acidification rate (ECAR)) status of non-differentiated (ndHL60) and differentiated (dHL60) cells. b, HPr+/DNA-based titration with PMA; ndHL60 versus dHL60. c, CBA-based titration with PMA; ndHL60 versus dHL60. d, HE/HPLC-based titration with PMA; dHL60. e, Amplex Red/HRP-based titration with PMA in dHL60. f, expression of protein Nox2 and p47phox in non-differentiated HL60 cells (lane 2) and HL60 cells differentiated with 1.3% DMSO (lane 3) or with 1 μm ATRA (lane 4). Molecular mass markers are shown in lane 1. g, profile of HE oxidation products at high cell density (1 × 106 cells/ml) after incubation of 20 μm HE with dHL60 in HBSS for 1 h. h, kinetics of oxidation of HE (20 μm) in suspension of dHL60 in HBSS at a cell density of 1 × 105 cells/ml. i, same as g, but the probe HPr+ (20 μm) was used. b, c, and e, rate of probe oxidation by dHL60 in the absence of PMA was taken as 1. a.u., arbitrary units. Error bars, S.D.
Plate Reader-based Monitoring of Superoxide Radical Anion and Hydrogen Peroxide
Differentiated HL60 cells were suspended in HBSS (Invitrogen) containing glucose (5.5 mm), CaCl2 (1.3 mm), MgCl2 (0.5 mm), MgSO4 (0.4 mm), KCl (5.3 mm), KH2PO4 (0.4 mm), NaHCO3 (4.2 mm), NaCl (0.14 m), and Na2HPO4 (0.3 mm) and supplemented with HEPES (25 mm) and DTPA (100 μm). Cells were counted and diluted to a final concentration of 1 × 105 cells/ml. Next, cells were incubated at 37 °C in a CO2-free incubator for 30 min in deep well 96-well plates with and without the inhibitors. During the incubation, 384-well plates (Costar, clear-bottom black 384-well plates) were preloaded with freshly prepared probe solutions (5× concentrated, 10 μl/well) in HBSS and with 0.1% BSA in HBSS (odd-numbered columns, control) or 5 μm PMA, 0.1% BSA in HBSS (even-numbered columns). Probe solutions included HPr+ (100 μm) plus DNA (0.5 mg/ml) for superoxide detection and CBA (500 μm) or Amplex Red (250 μm) plus HRP (0.5–5 units/ml) for H2O2 detection. After incubation with inhibitors, the HL60 cell suspension was transferred into prepared 384-well plates (30 μl/well), and plates were sealed with sealing film to minimize water evaporation and placed in the plate readers prewarmed at 37 °C. The changes in fluorescence intensity were monitored over a 2-h period. Beckman Coulter DTX880 and BMG Labtech FLUOstar Omega plate readers were used, and fluorescence intensities were measured from the bottom of the plates. The detection settings for each of the probes are shown in Table 1.
Simultaneous Detection of Superoxide Radical Anion and Hydrogen Peroxide; Rapid Isolation of Products by HPLC
Cells were incubated with inhibitors as described for the plate reader-based assays. Two 96-well plates were preloaded (40 μl/well) with a freshly prepared solution of HE (100 μm) plus CBA (500 μm) in HBSS containing HEPES (25 mm), DTPA (100 μm), and BSA (0.1 mg/ml) and an additional two plates with similar solutions but containing PMA (5 μm). After incubation with inhibitors, the cell suspension was transferred into the plates (160 μl/well), and the mixtures were incubated for 1 h at 37 °C in a CO2-free incubator. After incubation, the solutions (180-μl aliquots) were transferred into four 96-well plates preloaded with the mixture of SOD (0.5 mg/ml) and CAT (1 kilounit/ml). Plates were centrifuged (250 × g for 2 min at 25 °C), and the 90-μl aliquots of the supernatants were transferred from four 96-well plates into one 384-well plate. The plate was sealed and placed in the HPLC autosampler (thermostatted at 25 °C) for HPLC analysis. HPLC analysis was performed using an Agilent 1100 system equipped with absorption and fluorescence detection. For rapid separation of CBA, HE, 2-OH-E+, and COH, isocratic elution on a Kinetex Phenyl-Hexyl column (Phenomenex; 50 mm × 4.6 mm, 2.6 μm) was used. The mobile phase consisted of water (70% by volume), acetonitrile (30% by volume), and trifluoroacetic acid (0.1% by volume), and the flow rate was 2.0 ml/min. CBA (retention time, 25 s) was quantitated based on the absorption trace recorded at 290 nm. HE, COH, and 2-OH-E+ (retention times of 20, 30, and 48 s, respectively) were quantitated based on the fluorescence trace (excitation at 370 nm, emission at 565 nm). The fluorescence parameters were selected to avoid switching the fluorescence parameters during a short HPLC run and to minimize the peak intensity of ethidium cation (retention time of 53 s). HE was also quantitated based on the absorption trace recorded at 370 nm. The same method was used for rapid HPLC monitoring of oxidation of HE or CBA when used alone.
Complete Profiling of HE and HPr+ Oxidation Products by HPLC
Full profiling of the products of HE and HPr+ oxidation by differentiated HL60 cells was performed according to published methods (32, 33). Briefly, after incubation, the cell supernatant was injected on a Kinetex C18 column (Phenomenex, 100 mm × 4.6 mm, 2.6 μm) equilibrated with a water/acetonitrile mixture (9:1 for HPr+ or 8:2 for HE), and the probe and products were separated by an increase in acetonitrile content at a flow rate of 1.5 ml/min. An absorption detector was used to monitor probes and the dimeric oxidation products, whereas the fluorescence detector was used to monitor and quantitate 2-OH-E+ and E+ or 2-hydroxypropidium and propidium.
Complete Profiling of HE Oxidation Products and CBA by HPLC and LC-MS
For simultaneous monitoring of CBA oxidation and complete profiling of HE oxidation products, LC-MS/MS or HPLC with absorption and fluorescence detectors was used. In the LC-MS/MS method, the Shimadzu Nexera UHPLC system equipped with an LCMS 8030 triple quadrupole was used. The compounds were separated on a Supelco Titan C18 column (Sigma-Aldrich; 100 mm × 2.1 mm, 1.9 μm) using a mobile phase consisting of water and acetonitrile in the presence of formic acid (0.1%). The elution of analytes was achieved by a linear increase in the concentration of acetonitrile from 15 to 50% over 7 min at a flow rate of 0.5 ml/min. For selective detection, the multiple-reaction monitoring mode was used with the following transitions: 316.1 > 287.1 (HE), 330.1 > 255.0 (2-OH-E+), 314.1 > 284.0 (E+), 313.10 > 298.9 (E+-E+), 235.1 > 189.05 (CBA), and 160.9 > 133.0 (COH). HE and its oxidation products were analyzed in positive ion mode, whereas CBA and COH were detected in negative ion mode. CBA was detected as an adduct of formate anion to the probe. Alternatively, HPLC with absorption and fluorescence detection was used, as described above for complete profiling of HE oxidation products, with the following modifications: (a) the column used was Synergi Polar-RP (Phenomenex; 100 mm × 2.0 mm, 2.5 μm); (b) the gradient was obtained by an increase in acetonitrile fraction from 30 to 100% over a time period of 5.5 min at a flow rate of 0.45 ml/min; (c) fluorescence parameters for HE and COH detection (excitation at 370 nm, emission at 440 nm) were set during the first 2.5 min of the run, and fluorescence parameters for 2-OH-E+ and E+ (excitation at 490 nm, emission at 567 nm) were set after the first 2.5 min.
Oxygen Consumption Measurements
Oxygen consumption by HL60 cells was monitored using a Seahorse XF96 extracellular flux analyzer by modification of previously published protocols established for adherent cells (38, 39). Cell suspension was prepared in phenol red-free RPMI medium (without bicarbonate) and aliquoted (80 μl/well) into a 96-well plate to obtain a final cell amount of 2 × 104 cells/well. Cells were spun down, and an additional amount of RPMI medium (100 μl/well) was added. Oxygen measurements were started, and at the specified time points, the solutions of inhibitors and/or activator (PMA) were subsequently added. Alternatively, cells preincubated with inhibitors were transferred into measurement plates and tested for response to PMA. That way, the same cell suspensions could be used for plate reader-based ROS measurements and Seahorse XF96-based monitoring of oxygen consumption rates, for direct comparison. The solutions for injections were prepared 10× concentrated, and the volumes injected were 20, 22.2, 24.7, and 27.4 μl for first, second, third, and fourth injection, respectively. PMA injection causes a 4-fold increase in oxygen consumption rate (OCR), confirming the possibility of medium throughput (96-well plate-based) monitoring of NADPH oxidase activity by direct oxygen consumption measurements. A mixture of rotenone (1 μm) and antimycin A (10 μm) was injected when the contribution of mitochondrial respiration to the total OCR was to be tested. In contrast to primary neutrophils, differentiated HL60 cells exhibited a significant mitochondrial respiration rate (Fig. 2a), which enabled us to monitor the effects of inhibitors not only on the activity of NADPH oxidase but also on mitochondrial respiration, as exemplified by the inhibitory effect of DPI on basal respiration.
Cytotoxicity Assay
The short term toxicity of the inhibitors was tested by measurement of the effect of inhibitors on intracellular ATP levels in control and PMA-stimulated cells. This was to establish whether the observed effects of inhibitors were due to Nox inhibition or to interference with cell function. Intracellular ATP level was measured using a kit (Sigma) based on ATP-dependent luciferase-mediated oxidation of luciferin with light emission, as described elsewhere (38). White 96-well plates (Greiner) were used, and light intensity was measured with a Beckman Coulter DTX880 plate reader. Of note, compounds 40, 41, and 42 displayed strong short term toxicity, with concomitant membrane permeabilization as confirmed by Sytox dye uptake measurements (data not shown). Therefore, the observed apparent Nox2-inhibitory effect of those compounds was attributed to their toxicity.
Spin Trapping
The production of superoxide radical anion was monitored from differentiated HL60 cells (1 × 105) incubated for 1 h at 25 °C with 50 mm DEPMPO with or without PMA (10 μm) in HBSS containing HEPES (25 mm) and DTPA (100 μm). After incubation, the cell suspension was transferred into a 50-μl capillary tube, and the EPR spectrum was collected with a Bruker EMX spectrometer working in X-band. The spectrometer parameters were as follows: modulation frequency, 100 kHz; modulation amplitude, 1.0 gauss; receiver gain, 1 × 106; microwave power, 20 milliwatts; scan range, 140 gauss; center field, 3505 gauss; conversion time, 40.96 ms; time constant, 1.28 ms; sweep time, 41.94 s. The spectra shown are the averages of five scans.
Nitro Blue Tetrazolium (NBT) Staining
N27 cells were grown in RPMI 1640 medium supplemented with 10% FBS, penicillin (100 units/ml), and streptomycin (100 μg/ml) and treated for 6 h with 6-OH-DA (100 μm). The aqueous solution of 6-OH-DA (0.1 m) was prepared freshly under anaerobic conditions immediately before the experiment and was protected from light. After a 6-h period, the aqueous solution of NBT (10 mg/ml) was added to a final concentration of 0.3 mg/ml and incubated for 1 h. After incubation, cells were washed twice with ice-cold PBS and harvested, and cell pellets were frozen in liquid nitrogen. For measurement of the extent of formazan formation (the reduction product of NBT), cell pellets were vortexed for 1 h with 240 μl of an aqueous solution of potassium hydroxide (KOH; 2 m), followed by the addition of 280 μl of DMSO and additional vortexing for 30 min. Absorption spectra were collected, and absorbance at 685 nm was used to measure the yield of the formazan.
RESULTS
Monitoring ROS from NADPH Oxidase Activity
Human promyelocytic leukemia HL60 cells differentiated into neutrophils with ATRA were used as a source of endogenous Nox2 (40). The expression of Nox2 in the differentiated cells was confirmed by Western blotting of both membrane-bound gp91phox and cytosolic p47phox subunits (Fig. 2). Both DMSO (1.3%) and ATRA (1 μm) were able to induce cell differentiation, and we chose the ATRA model for further experiments. The differentiated cells (dHL60) were compared with non-differentiated cells (ndHL60) in their response to PMA, an activator of protein kinase C, leading to the phosphorylation of the p47phox cytosolic subunit, which, in turn, binds to p22phox membrane protein. After the assembly of all cytosolic and membrane components, NADPH is oxidized, and electrons are transferred to oxygen, generating O2⨪. As O2⨪ is released, it oxidizes the HPr+ probe, forming a fluorescent product, 2-hydroxypropidium (2-OH-Pr2+). The fluorescence yield was further increased by the addition of DNA to the medium, thereby increasing the sensitivity of the O2⨪ assay (Figs. 2b and 3a). In the orthogonal assay, 2-hydroxyethidium was measured using a rapid HPLC method (Figs. 2d and 3d).
FIGURE 3.

Monitoring Nox2 activity of dHL60 cells. a, effect of SOD (0.1 mg/ml) and CAT (1 kilounit/ml) on the time-dependent oxidation of HPr+ induced by stimulation of dHL60 with PMA (1 μm). b, same as a, but CBA was used instead of HPr+. c, same as a, but using Amplex Red/HRP probe. d, HPLC traces of the media from incubation of dHL60 with PMA (1 μm) for 90 min in the presence of HE (20 μm). e–h, same as a–d, but the effect of DPI and VAS2870 inhibitors (10 μm each) was tested. i, OCR measurements; response to PMA of ndHL60 versus dHL60 cells. j, same as i, but the effect of DPI (10 μm) on basal and PMA-induced OCR in dHL60 cells was tested. k, same as j, but the effect of VAS2870 (10 μm) was tested. l, same as j, but the effect of rotenone (1 μm) + antimycin (10 μm) was tested. a.u., arbitrary units. Error bars, S.D.
Both Nox4 and Duox1/2 isoenzymes reportedly generate H2O2 as a major product directly from a two-electron reduction of oxygen. In the primary assay, H2O2 is detected using the CBA probe that is converted to a blue fluorescent COH product (Figs. 2b and 3b). HRP/H2O2-induced oxidation of Amplex Red, forming a red fluorescent product (resorufin), was employed in the orthogonal assay (Figs. 2e and 3c). To further confirm the identity of the oxidizing species, SOD and CAT, specific scavengers of O2⨪ and H2O2, respectively, were used (Fig. 3, a–d). Fluorescent products formed from HPr+ and HE probes were completely inhibited by SOD, but not by CAT, confirming the identity of the oxidant as O2⨪. The oxidation of CBA to COH was inhibited by CAT, but not by SOD, indicating the oxidant as H2O2. Unambiguous identification of ROS produced by dHL60 cells as O2⨪ and H2O2 helped to establish the cellular source of ROS. PMA stimulated production of O2⨪ and H2O2 in differentiated but not in non-differentiated HL60 cells (Fig. 2, b and c). This, together with increased Nox2 expression during cell differentiation (Fig. 2f), implicates Nox2 as a primary source of ROS. The oxidation of cell-impermeable HPr+ probe is consistent with a Nox2 mechanism that functions in the extracellular milieu. The enzymatic source of ROS was further confirmed using Nox2 inhibitors, DPI and VAS2870. Both inhibitors significantly attenuated the oxidation of the probes, consistent with Nox2 as the major source of ROS (Fig. 3, e–h).
HPLC analysis of HE-derived oxidation products indicates formation of O2⨪ product, 2-hydroxyethidium, as well as ethidium and dimeric products (Fig. 2g). Extensive research on oxidation chemistry of HE suggests that although ethidium can be formed via one- or two-electron oxidation pathways, the dimeric products arise from one-electron oxidation of the probe (41, 42). Analysis of HE oxidation in PMA-stimulated dHL60 cells at high cell density (106 cell/ml) shows both superoxide-specific (2-OH-E+) and one-electron oxidation-specific dimeric product (E+-E+) (Fig. 2g). SOD and CAT inhibited 2-OH-E+ and E+-E+ formation, respectively, whereas, Nox2 inhibitor, VAS2870, inhibited both 2-OH-E+ and E+-E+. These data are consistent with 2-OH-E+ being the specific marker of O2⨪ and E+-E+ dimer as a marker of peroxidatic activity. To minimize the peroxidatic activity and selectively measure O2⨪, we decreased the cell density and observed significantly lower nonspecific probe oxidation, with 2-OH-E+ or 2-OH-Pr2+ as the major products using HE and HPr+ probes, respectively (Fig. 2, h and i). Therefore, we used the cell density of 1 × 105 cells/ml in subsequent experiments.
Next, we tested whether oxygen consumption measurements can be used in a medium-throughput manner to monitor the activity of Nox2 without an exogenous probe. The Seahorse XF96 extracellular flux analyzer, which allows simultaneous monitoring of OCRs in all wells of the 96-well plates, was used. The addition of PMA induced a significant increase in OCR in differentiated (dHL60) but not in non-differentiated cells (Fig. 3i). Stimulation of OCR was prevented by Nox2 inhibitors, DPI and VAS2870, added prior to PMA (Fig. 3, j and k). Interestingly, both inhibitors also decreased the basal OCR, although DPI inhibited OCR to a greater extent than VAS2870. This may be attributed to inhibition of mitochondrial respiration because basal oxygen consumption is completely blocked by the mitochondrial inhibitors, rotenone plus antimycin A (Fig. 3l). In contrast to DPI and VAS2870, rotenone plus antimycin A did not affect oxygen consumption stimulated by PMA, indicating that DPI and VAS2870 blocked Nox2 activation independent of their effect on mitochondrial respiration. The oxygen consumption data further validate the use of the dHL60 model for screening and identifying potential inhibitors of Nox2.
Simultaneous Monitoring of O2⨪ and H2O2
The development of a rapid HPLC method for detecting 2-OH-E+ and COH enables simultaneous monitoring of O2⨪ and H2O2 generated from the same incubation mixture. The HPLC method allows the separation and quantitation of HE, 2-OH-E+, and COH, using a single fluorescence detection method (λex = 370 nm, λem = 565 nm) (Fig. 4). Additionally, the use of an absorption detector makes it feasible to separate and quantify the HE and CBA consumption rate, thereby confirming the overall mechanism. We used an enzymatic system containing hypoxanthine and xanthine oxidase, which generates both O2⨪ and H2O2, to validate the assay. Fig. 4 shows a very good chromatographic separation between the probes (HE and CBA) and products (2-OH-E+ and COH) when present together. Also, the effects of SOD and catalase are the same, whether the probes are used singly or together. SOD inhibits conversion of HE into 2-OH-E+, whereas catalase inhibits oxidation of CBA to COH. Similar conclusions can be drawn from an analogous experiment but with PMA-stimulated dHL60 cells (see below).
FIGURE 4.

Simultaneous monitoring of superoxide radical anion and hydrogen peroxide formed in a hypoxanthine plus xanthine oxidase system, using high-throughput HPLC method. a, concentration of HE, 2-OH-E+, CBA, and COH from incubation of 20 μm HE and 100 μm CBA in the presence and absence of hypoxanthine (HX; 200 μm) and xanthine oxidase (XO; 0.07 milliunits/ml). b, same as a, but HPLC chromatograms of samples analyzed after incubation for 2 h. Line 1, HE alone; line 2, HE + CBA; line 3, HE + CBA + XO; line 4, HE + CBA + XO + HX. c and d, same as a and b, but full system (HX + XO) was used and the effect of SOD (0.1 mg/ml) or CAT (0.1 kilounit/ml) was tested. Line 1, HE + CBA + XO + HX; line 2, with SOD; line 3, with CAT. e and f, same as a and b, but full system (HX + XO) was used, and the oxidation of probes alone was compared with co-incubation with both probes. Line 1, HE + CBA + XO + HX; line 2, HE + XO + HX; line 3, CBA + XO + HX.
The HPLC-based method was used to rapidly and simultaneously monitor O2⨪ and H2O2 formation in cellular systems expressing Nox2 and Nox4 (Fig. 5, a–d). Both O2⨪ and H2O2 have been detected in dHL60 cells stimulated with PMA. To further confirm their identity, we tested the effect of SOD and catalase and observed that the signal was decreased to basal levels observed from unstimulated cells lacking the Nox activity. Nox4 is a constitutively active protein, with its cellular activity regulated primarily at the transcriptional level. Oxidants produced by Nox4 were monitored by comparing the profiles of oxidation of HE and CBA in the wild-type HEK293 cells and in HEK293 cells overexpressing Nox4 (HEK-Nox4 cells). As shown in Fig. 5, c and d, the presence of Nox4 has virtually no effect on the yield of the superoxide product, 2-OH-E+, but significantly increases the catalase-sensitive oxidation of CBA probe to COH. This indicates that overexpressing Nox4 protein in HEK293 cells induced H2O2 production but not O2⨪. Because there are reports on increased intracellular HE-derived red fluorescence, which was attributed to Nox4-derived O2⨪ (43–46), we also profiled intracellular HE oxidation products under same conditions (Fig. 6). After incubation of cells with HE and CBA, we observed only very little increase in 2-OH-E+ level, when comparing HEK293 and HEK-Nox4 cells, with significant increase in E+ and E+-E+ products, indicating nonspecific oxidation of the probe. This indicates that Nox4-derived H2O2 induces intracellular one-electron oxidation of HE, with the formation of red fluorescent E+ product. The observed small increase in 2-OH-E+ level may therefore be due to increased steady-state level of HE-derived radical in HEK-Nox4 cells because we have recently shown that one-electron oxidation of HE may increase the yield of 2-OH-E+ at constant flux of O2⨪ (41). Interestingly, in the presence of DPI, the yield of 2-OH-E+ remained unchanged, whereas there was a significant increase in the yield of both E+ and E+-E+ (Fig. 6). This indicates that 2-OH-E+ formation was Nox4-independent and that DPI may exhibit pro-oxidative effects in both HEK293 and HEK-Nox4 cells. Simultaneous analysis of the extent of CBA oxidation in the medium indicated increased, DPI-inhibitable formation of COH in HEK-Nox4 cells, consistent with the results described above. We also tested if rapid simultaneous monitoring of O2⨪ and H2O2 could be extended to investigate another Nox family member, Nox5 (Fig. 5, e and f), whose activity is regulated by Ca2+ and phosphorylation status. Ionomycin increased the intracellular calcium level, thereby stimulating Nox5 activity in HEK293 cells overexpressing Nox5 protein (HEK-Nox5 cells). Activation of Nox5 increased both 2-OH-E+ and COH levels, which were inhibited by SOD and catalase.
FIGURE 5.
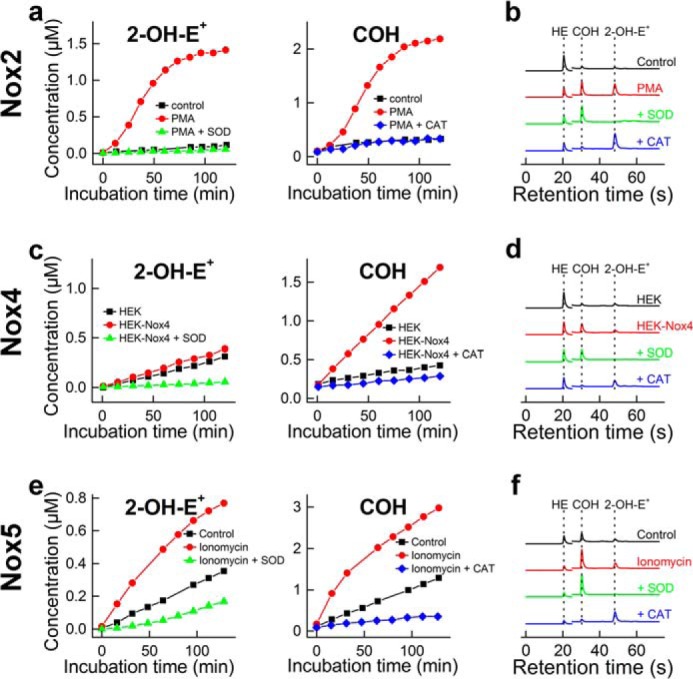
Simultaneous monitoring of superoxide radical anion and hydrogen peroxide in cells enriched with Nox2, Nox4, and Nox5, using a high-throughput HPLC method. a, the concentration of 2-OH-E+ and COH detected in the media of dHL60 cells stimulated with PMA (1 μm) in the presence of 20 μm HE and 100 μm CBA and in the presence and absence of SOD (0.1 mg/ml) or CAT (0.1 kilounit/ml). b, same as a, but HPLC traces of samples analyzed after 2 h incubation are shown. c and d, same as a and b, but HEK and HEK-Nox4 cells were used. e and f, same as a and b, but HEK-Nox5 cells were used. Ionomycin (1 μm) was used to stimulate production of O2⨪ and H2O2.
FIGURE 6.

Profiling of HE and CBA oxidation in HEK293 and HEK-Nox4 cells. HEK293 and HEK-Nox4 were incubated for 1 h with 20 μm HE and 100 μm CBA in the absence and presence of DPI (2 μm), and the cell lysates and media were analyzed by LC-MS/MS. a, profiles of HE oxidation products, as measured in cell lysates. b, analysis of the extent of the conversion of CBA to COH in cell media. Error bars, S.D.
High-throughput Compatibility of the Assays
Successful implementation of a high-throughput screening method is dependent both on the speed and the dynamic range of the probe in response to stimuli and inhibitors such that easy discrimination between positive and negative “hits” can be made, enabling the determination of the IC50 values. The most common parameter used to describe the quality of an assay is Z′ value (47), defined by the equation,
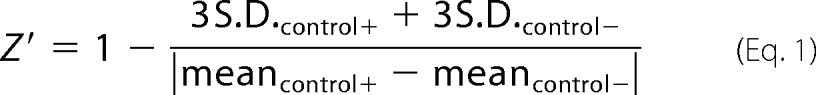 |
where S.D.control+ and S.D.control− represent S.D. values of the signals from positive and negative controls, respectively, and meancontrol+ and meancontrol− represent corresponding values of the means. The data for the present assays are included in Table 2. As can be seen, all assays are judged to be “excellent,” with sufficiently large separation bands between positive and negative samples, according to the published definition (47). By comparing the kinetic and end point modes of analysis of the plate reader-based data, we concluded that the assays could be used in end point-type measurement mode without sacrificing the assay quality. The use of end point measurement not only increases the throughput but also eliminates the possibility of photochemical transformation of the fluorogenic probes during continuous light exposure (35, 48).
TABLE 2.
Z′ values of the primary and orthogonal assays
| Assay/Probe | Detection mode | Type of analysis | Z′ |
|---|---|---|---|
| Primary assays | |||
| HPr+ + DNA | Plate reader | Kinetic | 0.60 ± 0.17 |
| Plate reader | End point | 0.55 ± 0.11 | |
| CBA | Plate reader | Kinetic | 0.67 ± 0.15 |
| Plate reader | End point | 0.68 ± 0.14 | |
| Orthogonal assays | |||
| HE | HPLC | End point | 0.75 ± 0.07 |
| Amplex Red + HRP | Plate reader | Kinetic | 0.73 ± 0.11 |
| Plate reader | End point | 0.74 ± 0.11 | |
Primary Assays for O2⨪ and H2O2
To test and validate the proposed workflow for screening of Nox inhibitors shown in Fig. 1, we screened a group of compounds synthesized in Dr. Lambeth's laboratory. Fig. 7 shows the compounds tested, including antioxidant enzymes (SOD (3) and catalase (4)), DPI (5), and other reported or potential inhibitors of Nox enzymes (6–44). Of those, compounds 13-44 were selected based on independent high-throughput screening using the less selective probe, L-012 (23). We have independently confirmed that of those 22 compounds, 20 exhibited a significant inhibitory activity on the luminescence signal from L-012 probe in the Nox2 cellular model (data not shown). The current primary assays included hydropropidine-based detection of O2⨪ and coumarin boronate-based detection of H2O2 in 384-well plates. The plate layout is shown in Fig. 8a. The time course of probe oxidation by dHL60 cells treated with and without PMA is shown in Fig. 8b (O2⨪ detection) and Fig. 8c (H2O2 detection). Each window represents a single treatment (control, antioxidant, or inhibitor), with the number corresponding to the structure/identity provided in Fig. 7. The kinetic traces presented show a good separation of fluorescence signals between non-treated (black traces) and PMA-treated (red traces) cells, in agreement with the Z′ values (Table 2). Additionally, DPI (5), a well known flavoprotein inhibitor, blocked the fluorescence increase from both HPr+ and CBA probes, whereas SOD (3) and catalase (4) blocked selectively HPr+- and CBA-derived signal, respectively. For quantitative analyses, we determined the slopes of the fluorescence increase from the linear part of the dependence of fluorescence intensity versus time and set the criterion for positive hits as at least 50% inhibition at 10 μm concentration. The results (presented as a percentage of the slope of untreated cells) for each treatment are shown in Fig. 8, d and e. These data indicate that, of the 40 small molecules tested here, we identified 11 positive hits using HPr+ probe and 13 positive hits using CBA. The difference in the number of positive hits is due to H2O2 scavenging activity of compound 9 (4-acetylphenylboronic acid), which does not exhibit any activity in the superoxide assay, and the modest inhibitory activity of compound 17. Overall, there was excellent agreement between the results of both assays using two different detection probes.
FIGURE 7.

Structures of the compounds tested as potential Nox2 inhibitors.
FIGURE 8.

Primary screening of Nox2 inhibitors. a, 384-well plate layout. The numbers correspond to the inhibitors and antioxidants, as defined in Fig. 7. b, effect of inhibitors on the time course of HPr+ oxidation by dHL60 cells without PMA (black symbols) or treated with 1 μm PMA (red symbols). c, same as b, but CBA probe was used. d, quantitative analysis of the effects of inhibitors on the rate of PMA-stimulated oxidation of HPr+. 100% corresponds to the rate of HPr+ oxidation by PMA-stimulated cells in the absence of inhibitors. Positive hits are indicated by green color. e, same as d but with CBA probe. a.u., arbitrary units. Error bars, S.D.
Orthogonal Assays for O2⨪ and H2O2
As shown in Fig. 1, for each of the primary assays, we used an orthogonal assay, based on a completely different mechanism or detection mode. For superoxide detection, we used the HPLC-based monitoring of 2-hydroxyethidium. The specific detection of the superoxide/HE diagnostic marker product by HPLC has several advantages, as discussed elsewhere (24). With respect to H2O2 detection, peroxidase-catalyzed oxidation of Amplex Red by H2O2 is based on a one-electron oxidation of the probe, via formation of an intermediate probe radical.
This mechanism is different from the direct oxidation of boronate-based probes by H2O2. The HPLC-based monitoring of 2-OH-E+ in a 384-well plate (Fig. 9, a and c) yielded almost the same positive hits as determined using HPr+, but with compounds 13 and 14 as negatives. With the Amplex Red-based assay (Fig. 9, b and d), the number of identified positive hits was 15; of them, four compounds displayed an inhibitory activity closer to the threshold set. The remaining 11 hits were in perfect agreement with the results of the other assays. Due to the relatively complex mechanism of oxidation and the requirement of peroxidase activity, the Amplex Red-based assay yields more positive hits, thereby producing numerous false positives and precluding this assay as the best choice for a primary assay. It serves, however, as a good orthogonal assay, validating the inhibitory activity of “hits” from the primary assay for H2O2.
FIGURE 9.

Orthogonal screening for Nox2 inhibitors. The numbers correspond to the inhibitors and antioxidants, as defined in Fig. 7. a, the effect of inhibitors on the conversion of HE into 2-OH-E+ by dHL60 cells without PMA (black traces) or treated with 1 μm PMA (red traces). The traces represent HPLC chromatograms zoomed at the time range of 2-OH-E+ elution. b, effect of inhibitors on the time course of Amplex Red oxidation by dHL60 cells without PMA (black symbols) or treated with 1 μm PMA (red symbols) in the presence of HRP (1 unit/ml); c, quantitative analysis of the effects of inhibitors on the yield of 2-OH-E+. 100% corresponds to the yield of 2-OH-E+ formed by PMA-stimulated cells in the absence of inhibitors. d, similar to c, but the rate of PMA-stimulated oxidation of Amplex Red in the presence of HRP was analyzed. 100% corresponds to the rate of Amplex Red oxidation by PMA-stimulated cells in the absence of inhibitors. Positive hits are indicated in green. a.u., arbitrary units. Error bars, S.D.
Screening for False Positives
The positive hits from the primary and the orthogonal assays were further tested for their ability to inhibit the signal in the Nox-independent enzymatic source of O2⨪ and H2O2 generated from xanthine oxidase-catalyzed oxidation of hypoxanthine (Fig. 10a). Of the seven positive hits tested, DPI was the most potent inhibitor, in agreement with its non-selective but potent inhibitory activity toward a variety of flavoproteins. Additionally, whereas ebselen (10) and VAS2870 (11) displayed some inhibitory activity on the fluorescence signal, compounds 16, 42, and 43 displayed only weak or no inhibitory effects. We have chosen compound 43 for further evaluation, because it displayed no interference in any of the assays tested and no short term toxicity in cells treated up to 100 μm concentration (see below).
FIGURE 10.
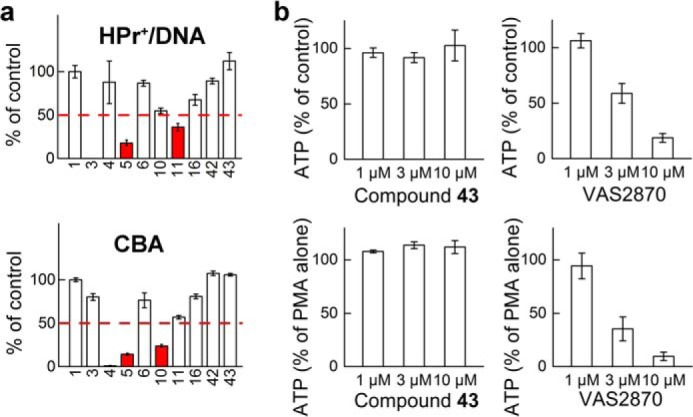
Analyses of false positives and cytotoxicity of the hits. a, analysis of the effects of inhibitors on the rate of oxidation of HPr+ (top) and CBA (bottom) probes in a Nox-independent system. Top, HPr+ (20 μm) was incubated in the presence of DNA (0.1 mg/ml) with hypoxanthine (100 μm) and xanthine oxidase (0.1 milliunit/ml), and fluorescence was monitored over time. Numbers below the x axis indicate the presence of inhibitor/scavenger, as defined in Fig. 7. Potential false positives are indicated by red color. Bottom, same as above, but CBA oxidation was followed. Instead of HPr+ and DNA, the solutions contained 100 μm CBA probe. b, comparison of short term cytotoxicity of compound 43 and VAS2870. Cells (dHL60) were preincubated for 30 min with inhibitors (left panels, compound 43; right panels, VAS2870), followed by the addition of DMSO (control samples; top panels) or PMA (1 μm; bottom panels) and additional incubation for 1 h. Toxicity was estimated based on the effect of the compounds on cellular ATP levels. Error bars, S.D.
Confirmatory and Cytotoxicity Assays for Potential Inhibitors of Nox Activity
The positive hits were further evaluated using the cytotoxicity assay (Fig. 10b) followed by a set of confirmatory assays (Fig. 11). Intracellular ATP level was measured as an indicator of acute toxicity of the compounds used in the presence of PMA stimulation. When tested at 10 μm concentration, most of the compounds tested exhibited some degree of toxicity, with compound 43 as one of the few exceptions (Fig. 10b). Of note, VAS2870, which has been recently reported to exhibit off-target effects (49), showed dose-dependent toxicity. Therefore, as mentioned above, we chose compound 43 as a positive hit for further evaluation in confirmatory assays. We compared the effects of the compound 43 with the known Nox2 inhibitors, DPI (5) and VAS2870 (11) (Fig. 11). The first confirmatory assay is based on direct monitoring of oxygen consumption by activated dHL60 cells. As can be seen in Fig. 11a, all three compounds inhibited the response of dHL60 cells to PMA. Of note, both DPI and VAS2870 and not compound 43 inhibited basal mitochondrial respiration. The EPR spin trapping of Nox-derived O2⨪ with DEPMPO was used as another confirmatory assay. Upon trapping O2⨪, DEPMPO spin trap formed a relatively stable superoxide adduct, with characteristic EPR spectrum (50). As shown in Fig. 11b, all three inhibitors tested, as well as SOD but not catalase, completely blocked the production of the DEPMPO-•OOH radical adduct. Finally, we tested the dose response for all three inhibitors to determine their IC50 values. To this end, we used the HPLC-based simultaneous monitoring of O2⨪ and H2O2 as described above. The response of both 2-OH-E+ and COH to varying concentrations of inhibitors was similar for the given inhibitor (Fig. 11c), with IC50 values of 2.3, 0.13, and 0.13 μm for compound 43, VAS2870 and DPI, respectively. Thus, compound 43 exerted inhibitory action below its potential toxicity. Also VAS2870, which was toxic to dHL60 cells at the concentration of 10 μm, exhibited a strong inhibitory effect at less than 1 μm, with no short term toxicity (Fig. 10b). Compound 43 was observed to not affect Nox4 activity at concentrations up to 50 μm (data not shown).
FIGURE 11.

Confirmatory assays for compound 43, VAS2870, and DPI. a, measurement of OCRs by dHL60 cells using a 96-well extracellular flux analyzer, Seahorse XF96. b, effect of compound 43 (50 μm), VAS2870 (1 μm), DPI (1 μm), SOD (0.1 mg/ml), and CAT (0.1 kilounit/ml) on the yield of spin adduct formed by trapping dHL60-derived O2⨪ by DEPMPO (50 mm). c, concentration dependence of the effect of compound 43, VAS2870, and DPI on PMA-stimulated probe oxidation by dHL60 cells. HE oxidation to 2-OH-E+ and CBA oxidation to COH were monitored simultaneously by HPLC. Error bars, S.D.
Lack of Evidence for O2⨪ Formation from N27 Cells Treated with 6-Hydroxydopamine; Nox1 Case Study
Previously, it was reported that treatment of N27 cells with 6-OH-DA (100 μm) for 6 h induces Nox1 protein expression accompanied by an increase in Nox1 activity (i.e. O2⨪ formation), as measured by the reduction of NBT dye (51). However, we reported recently that 6-OH-DA failed to stimulate O2⨪ formation in N27 cells (39). In an effort to reconcile these differences, we investigated the effect of 6-OH-DA in N27 cells using NBT and HE. As shown in Fig. 12, we monitored the distribution of different oxidation products of HE, as measured by HPLC. As can be seen in Fig. 12a, incubation of N27 cells with 6-OH-DA increased the ability of cells to reduce NBT, corresponding to formation of formazan (increase in the intensity of the absorption band between 500 and 800 nm). This was in agreement with the previous report (51). However, when these experiments were carried out using HE, we failed to observe any increase in the yield of the O2⨪-specific product, 2-OH-E+ (Fig. 12b). Although 6-OH-DA stimulated HE oxidation, the major product was ethidium cation (E+). Similarly, no increase in 2-OH-E+ was observed in extracellular medium, with E+ responsible for the majority of HE consumption (data not shown). These results reveal the pitfalls of using nonspecific, artifact-prone probes (e.g. NBT) and further emphasize the need for HPLC-based assays of specific products formed from superoxide reaction with the probe (HE) in determining the activity of Nox enzymes.
FIGURE 12.
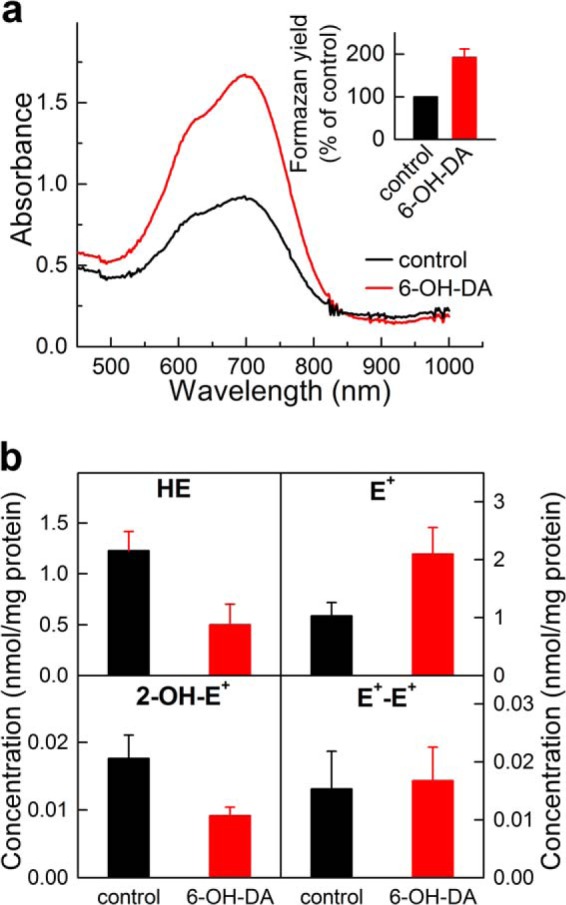
Monitoring superoxide radical anion in N27 cells after treatment with 6-hydroxydopamine. N27 cells were treated with 6-OH-DA (100 μm) for 6 h, followed by a 1-h treatment with NBT (0.3 mg/ml) (a) or 10 μm HE (b). a, UV-visible absorption spectra of the cell extracts after treatment with NBT. The inset shows the relative amount of the formazan formed, as determined based on the absorbance measured at 685 nm. b, profiles of HE oxidation determined in the cell lysates. Data shown in a and b are averages of three independent experiments. Error bars, S.D.
DISCUSSION
Despite being implicated in various disease conditions, high throughput-compatible and reliable detection of O2⨪ and H2O2 has been a challenge. Major drawbacks associated with commonly used probes for ROS include the lack of specificity and the reactivity of the probe-derived radical species with oxygen to artifactually produce O2⨪ and H2O2. Currently available high-throughput screening approaches use the artifact-prone probes, luminol or L-012, and HRP enzyme in the primary assay (23). In this assay, the luminescence emitted from an unstable product of O2⨪ and luminol or L-012 radical is measured. Unfortunately, the luminol radical (and L-012 radical) itself is capable of artifactually generating O2⨪ via a redox-cycling mechanism in the presence of molecular oxygen (21, 23). As the luminol radical or L-012 radical is derived from HRP/H2O2-dependent peroxidatic oxidation of luminol or L-012, any peroxidase inhibitor (or competing substrate) will inhibit luminescence and be identified as a potential hit for Nox inhibition. This assay is therefore subject to a high rate of false positives and in our opinion should not be used as a primary assay specific for O2⨪ (23). In this work, we used hydropropidine as a cell-impermeable analog of hydroethidine. Although both HE and HPr+ can be oxidized by several oxidants, the extracellular targeting of the probe (i.e. HPr+) vastly decreases the contribution of superoxide-independent oxidation. Additionally, HPLC-based detection of superoxide-specific product, 2-hydroxyethidium or 2-hydroxypropidium, further improves the assay specificity. The intermediate radical species, HE radical cation, is not a reducing species and therefore does not react with oxygen to produce O2⨪. With respect to H2O2 detection, the use of a boronate-based assay allows monitoring of H2O2 in a direct reaction with the probe, without the involvement of the probe radical. In comparison with HPr+- and CBA-based primary assays, the L-012 assay yielded significantly more hits. Using the probes and methodology described here, we estimate that the false positive rate should be decreased by more than 75%. Also, assay specificity might allow identification of weaker inhibitors that can be subsequently improved by medicinal chemistry.
Development of inhibitors of NADPH oxidase is an active area of research (7, 10, 16, 52). However, the lack of availability of an ideal screening methodology has impeded progress. We propose here the complete workflow for screening of potential inhibitors of NADPH oxidases, with monitoring of both O2⨪ and H2O2, thus enabling screening for inhibitors of various Nox isoforms. Because boronate-based probes will allow monitoring of not only hydrogen peroxide, but also peroxynitrite and hypohalous acids, the use of specific enzymatic inhibitors and/or scavengers is recommended to establish the identity of the species detected. Using the dHL60 cells, we did not observe the inhibitory effects of the inhibitor of nitric oxide synthase, l-NG-nitroarginine methyl ester (compound 2), but the oxidation was completely inhibited by the H2O2 scavenger, catalase, confirming the identity of the oxidant as H2O2. The proposed approach for rapid monitoring of the production of O2⨪ and H2O2 can readily be extended to other cellular models/sources of ROS production.
The Z′ values calculated for O2⨪ and H2O2 detection assays in a 384-well format are within the acceptable range for development and optimization of cell-based high-throughput screening assays. The enhanced assay performance is due to increased specificity, sensitivity, and stability of probes (shelf half-life of several months at −20 °C) and rigorously developed detection approaches. With automation, the Z′ values of the assays will increase due to elimination of inaccuracy in traditional pipetting. The high-throughput analyses of ROS described in this study will enable the screening and identification of promising Nox inhibitors with a much lower rate of false positive “hits” from different chemical libraries.
Although the present study is focused on developing a strategy for high-throughput screening of inhibitors of NADPH oxidases, the chemical probes and methodological advances described can be applied in basic research centered on Nox enzymes and other cellular sources of O2⨪ and H2O2. This is exemplified by the comparative analysis of the dynamics of generation and identity of the oxidizing species produced by three Nox isoforms: Nox2, Nox4, and Nox5 (Fig. 5). We have shown that whereas Nox2 and Nox5 yield both O2⨪ and H2O2, Nox4 produces H2O2 as the only detectable product in HEK-Nox4 cellular model, in agreement with earlier reports (19). The present data also indicate that HE-derived red fluorescence in Nox4-expressing HEK cells is due to nonspecific probe oxidation rather than superoxide-specific product (Fig. 6). Thus, we reiterate that HE-derived intracellular red fluorescence cannot be equated to O2⨪ formation (53) and that there is no experimental proof so far for Nox4-derived O2⨪ in cells. An additional HPLC-based assay should be performed in Nox4-containing systems. The comparison of NBT reduction results with profiling of HE oxidation products in N27 dopaminergic cells treated with 6-hydroxydopamine exemplifies a need for reliable Nox activity assays, which do not suffer from redox cycling of probe-derived intermediates and from a lack of specificity of the products. Recent identification of 2-chloroethidium as the specific product formed from HE under chlorinating conditions (54) opens up another opportunity for simultaneous monitoring of different reactive species as well as Nox2 and MPO activity using a hydroethidine assay coupled with rapid HPLC analysis.
In summary, we conclude that rapid and rigorous detection and quantitation of O2⨪ and H2O2 will lead to better understanding of the chemical biology of O2⨪/H2O2-producing enzymes (e.g. Nox isoforms) and will also help in the discovery of specific inhibitors of Nox isoforms.
Acknowledgments
We thank Dr. Ralf Brandes (Goethe-University, Frankfurt, Germany) for providing wild type HEK293 and HEK-Nox4 cells, Dr. David Fulton (Georgia Regents University, Augusta, GA) for providing HEK-Nox5 cells, and Dr. Micaël Hardy (Université Aix-Marseille, Marseille, France) for providing the spin trap, DEPMPO.
This work was supported, in whole or in part, by National Institutes of Health Grants R01 HL063119 and R01 HL073056 (to B. K.). This work was also supported by the Harry R. and Angeline E. Quadracci endowment (to B. K.).
- Nox
- NADPH oxidase(s)
- ROS
- reactive oxygen species
- HPr+
- hydropropidine
- HE
- hydroethidine
- CBA
- coumarin-7-boronic acid
- DTPA
- diethylenetriaminepentaacetic acid
- SOD
- superoxide dismutase
- CAT
- catalase
- DPI
- diphenyleneiodonium
- 6-OH-DA
- 6-hydroxydopamine
- COH
- 7-hydroxycoumarin
- 2-OH-E+
- 2-hydroxyethidium
- ATRA
- all-trans-retinoic acid
- PMA
- phorbol 12-myristate 13-acetate
- HBSS
- Hanks' balanced salt solution
- E+
- ethidium
- DEPMPO
- 5-(diethoxyphosphoryl)-5-methyl-1-pyrroline-N-oxide
- dHL60
- HL60 cells differentiated with all-trans-retinoic acid
- ndHL60
- non-differentiated HL60 cells
- OCR
- oxygen consumption rate
- NBT
- nitro blue tetrazolium
- HX
- hypoxanthine
- XO
- xanthine oxidase.
REFERENCES
- 1. Finkel T. (2011) Signal transduction by reactive oxygen species. J. Cell Biol. 194, 7–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambeth J. D. (2004) NOX enzymes and the biology of reactive oxygen. Nat. Rev. Immunol. 4, 181–189 [DOI] [PubMed] [Google Scholar]
- 3. Leto T. L., Morand S., Hurt D., Ueyama T. (2009) Targeting and regulation of reactive oxygen species generation by Nox family NADPH oxidases. Antioxid. Redox Signal. 11, 2607–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Streeter J., Thiel W., Brieger K., Miller F. J., Jr. (2013) Opportunity nox: the future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc. Ther. 31, 125–137 [DOI] [PubMed] [Google Scholar]
- 5. Bulua A. C., Simon A., Maddipati R., Pelletier M., Park H., Kim K. Y., Sack M. N., Kastner D. L., Siegel R. M. (2011) Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in TNFR1-associated periodic syndrome (TRAPS). J. Exp. Med. 208, 519–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hecker L., Cheng J., Thannickal V. J. (2012) Targeting NOX enzymes in pulmonary fibrosis. Cell Mol. Life Sci. 69, 2365–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Al Ghouleh I., Khoo N. K., Knaus U. G., Griendling K. K., Touyz R. M., Thannickal V. J., Barchowsky A., Nauseef W. M., Kelley E. E., Bauer P. M., Darley-Usmar V., Shiva S., Cifuentes-Pagano E., Freeman B. A., Gladwin M. T., Pagano P. J. (2011) Oxidases and peroxidases in cardiovascular and lung disease: new concepts in reactive oxygen species signaling. Free Radic. Biol. Med. 51, 1271–1288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Laleu B., Gaggini F., Orchard M., Fioraso-Cartier L., Cagnon L., Houngninou-Molango S., Gradia A., Duboux G., Merlot C., Heitz F., Szyndralewiez C., Page P. (2010) First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis. J. Med. Chem. 53, 7715–7730 [DOI] [PubMed] [Google Scholar]
- 9. Pendyala S., Gorshkova I. A., Usatyuk P. V., He D., Pennathur A., Lambeth J. D., Thannickal V. J., Natarajan V. (2009) Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid. Redox Signal. 11, 747–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lambeth J. D., Krause K. H., Clark R. A. (2008) NOX enzymes as novel targets for drug development. Semin. Immunopathol. 30, 339–363 [DOI] [PubMed] [Google Scholar]
- 11. Guzik T. J., Harrison D. G. (2006) Vascular NADPH oxidases as drug targets for novel antioxidant strategies. Drug Discov. Today 11, 524–533 [DOI] [PubMed] [Google Scholar]
- 12. Jaquet V., Bedard K. (2009) Editorial: Genetic mapping: the path of discovery for novel functions of the NOX NADPH oxidases. J. Leukoc. Biol. 86, 461–463 [DOI] [PubMed] [Google Scholar]
- 13. Jaquet V., Scapozza L., Clark R. A., Krause K. H., Lambeth J. D. (2009) Small-molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid. Redox Signal. 11, 2535–2552 [DOI] [PubMed] [Google Scholar]
- 14. Krause K. H., Lambeth D., Krönke M. (2012) NOX enzymes as drug targets. Cell Mol. Life Sci. 69, 2279–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rey F. E., Cifuentes M. E., Kiarash A., Quinn M. T., Pagano P. J. (2001) Novel competitive inhibitor of NAD(P)H oxidase assembly attenuates vascular O2⨪ and systolic blood pressure in mice. Circ. Res. 89, 408–414 [DOI] [PubMed] [Google Scholar]
- 16. Williams H. C., Griendling K. K. (2007) NADPH oxidase inhibitors: new antihypertensive agents? J. Cardiovasc. Pharmacol. 50, 9–16 [DOI] [PubMed] [Google Scholar]
- 17. Cifuentes-Pagano E., Csanyi G., Pagano P. J. (2012) NADPH oxidase inhibitors: a decade of discovery from Nox2ds to HTS. Cell Mol. Life Sci. 69, 2315–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rivera J., Sobey C. G., Walduck A. K., Drummond G. R. (2010) Nox isoforms in vascular pathophysiology: insights from transgenic and knockout mouse models. Redox. Rep. 15, 50–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Serrander L., Cartier L., Bedard K., Banfi B., Lardy B., Plastre O., Sienkiewicz A., Fórró L., Schlegel W., Krause K. H. (2007) NOX4 activity is determined by mRNA levels and reveals a unique pattern of ROS generation. Biochem. J. 406, 105–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cifuentes-Pagano E., Saha J., Csányi G., Ghouleh I. A., Sahoo S., Rodríguez A., Wipf P., Pagano P. J., Skoda E. M. (2013) Bridged tetrahydroisoquinolines as selective NADPH oxidase 2 (Nox2) inhibitors. Medchemcomm. 4, 1085–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Faulkner K., Fridovich I. (1993) Luminol and lucigenin as detectors for O2⨪. Free Radic. Biol. Med. 15, 447–451 [DOI] [PubMed] [Google Scholar]
- 22. Liochev S. I., Fridovich I. (1997) Lucigenin (bis-N-methylacridinium) as a mediator of superoxide anion production. Arch. Biochem. Biophys. 337, 115–120 [DOI] [PubMed] [Google Scholar]
- 23. Zielonka J., Lambeth J. D., Kalyanaraman B. (2013) On the use of L-012, a luminol-based chemiluminescent probe, for detecting superoxide and identifying inhibitors of NADPH oxidase: a reevaluation. Free Radic. Biol. Med. 65, 1310–1314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalyanaraman B., Dranka B. P., Hardy M., Michalski R., Zielonka J. (2014) HPLC-based monitoring of products formed from hydroethidine-based fluorogenic probes: the ultimate approach for intra- and extracellular superoxide detection. Biochim. Biophys. Acta 1840, 739–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Winterbourn C. C. (2008) Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 4, 278–286 [DOI] [PubMed] [Google Scholar]
- 26. Koto T., Michalski R., Zielonka J., Joseph J., Kalyanaraman B. (2014) Detection and identification of oxidants formed during •NO/O2⨪ reaction: a multi-well plate CW-EPR spectroscopy combined with HPLC analyses. Free Radic. Res. 48, 478–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kalyanaraman B. (2011) Oxidative chemistry of fluorescent dyes: implications in the detection of reactive oxygen and nitrogen species. Biochem. Soc. Trans. 39, 1221–1225 [DOI] [PubMed] [Google Scholar]
- 28. Kalyanaraman B., Darley-Usmar V., Davies K. J., Dennery P. A., Forman H. J., Grisham M. B., Mann G. E., Moore K., Roberts L. J., 2nd, Ischiropoulos H. (2012) Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 52, 1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zielonka J., Vasquez-Vivar J., Kalyanaraman B. (2008) Detection of 2-hydroxyethidium in cellular systems: a unique marker product of superoxide and hydroethidine. Nat. Protoc. 3, 8–21 [DOI] [PubMed] [Google Scholar]
- 30. Zielonka J., Hardy M., Kalyanaraman B. (2009) HPLC study of oxidation products of hydroethidine in chemical and biological systems: ramifications in superoxide measurements. Free Radic. Biol. Med. 46, 329–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zielonka J., Sikora A., Hardy M., Joseph J., Dranka B. P., Kalyanaraman B. (2012) Boronate probes as diagnostic tools for real time monitoring of peroxynitrite and hydroperoxides. Chem. Res. Toxicol. 25, 1793–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zielonka J., Zielonka M., Sikora A., Adamus J., Joseph J., Hardy M., Ouari O., Dranka B. P., Kalyanaraman B. (2012) Global profiling of reactive oxygen and nitrogen species in biological systems: high-throughput real-time analyses. J. Biol. Chem. 287, 2984–2995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Michalski R., Zielonka J., Hardy M., Joseph J., Kalyanaraman B. (2013) Hydropropidine: a novel, cell-impermeant fluorogenic probe for detecting extracellular superoxide. Free Radic. Biol. Med. 54, 135–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zielonka J., Sikora A., Joseph J., Kalyanaraman B. (2010) Peroxynitrite is the major species formed from different flux ratios of co-generated nitric oxide and superoxide: direct reaction with boronate-based fluorescent probe. J. Biol. Chem. 285, 14210–14216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhao B., Summers F. A., Mason R. P. (2012) Photooxidation of Amplex Red to resorufin: implications of exposing the Amplex Red assay to light. Free Radic. Biol. Med. 53, 1080–1087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dranka B. P., Gifford A., Ghosh A., Zielonka J., Joseph J., Kanthasamy A. G., Kalyanaraman B. (2013) Diapocynin prevents early Parkinson's disease symptoms in the leucine-rich repeat kinase 2 (LRRK2R(1)(4)(4)(1)G) transgenic mouse. Neurosci. Lett. 549, 57–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zielonka J., Zhao H., Xu Y., Kalyanaraman B. (2005) Mechanistic similarities between oxidation of hydroethidine by Fremy's salt and superoxide: stopped-flow optical and EPR studies. Free Radic. Biol. Med. 39, 853–863 [DOI] [PubMed] [Google Scholar]
- 38. Cheng G., Zielonka J., Dranka B. P., McAllister D., Mackinnon A. C., Jr., Joseph J., Kalyanaraman B. (2012) Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 72, 2634–2644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Dranka B. P., Zielonka J., Kanthasamy A. G., Kalyanaraman B. (2012) Alterations in bioenergetic function induced by Parkinson's disease mimetic compounds: lack of correlation with superoxide generation. J. Neurochem. 122, 941–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Teufelhofer O., Weiss R. M., Parzefall W., Schulte-Hermann R., Micksche M., Berger W., Elbling L. (2003) Promyelocytic HL60 cells express NADPH oxidase and are excellent targets in a rapid spectrophotometric microplate assay for extracellular superoxide. Toxicol. Sci. 76, 376–383 [DOI] [PubMed] [Google Scholar]
- 41. Michalski R., Michalowski B., Sikora A., Zielonka J., Kalyanaraman B. (2014) On the use of fluorescence lifetime imaging and dihydroethidium to detect superoxide in intact animals and ex vivo tissues: a reassessment. Free Radic. Biol. Med. 67, 278–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zielonka J., Srinivasan S., Hardy M., Ouari O., Lopez M., Vasquez-Vivar J., Avadhani N. G., Kalyanaraman B. (2008) Cytochrome c-mediated oxidation of hydroethidine and mito-hydroethidine in mitochondria: identification of homo- and heterodimers. Free Radic. Biol. Med. 44, 835–846 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McCann S. K., Dusting G. J., Roulston C. L. (2008) Early increase of Nox4 NADPH oxidase and superoxide generation following endothelin-1-induced stroke in conscious rats. J. Neurosci. Res. 86, 2524–2534 [DOI] [PubMed] [Google Scholar]
- 44. Topchiy E., Panzhinskiy E., Griffin W. S., Barger S. W., Das M., Zawada W. M. (2013) Nox4-generated superoxide drives angiotensin II-induced neural stem cell proliferation. Dev. Neurosci. 35, 293–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Varga Z. V., Kupai K., Szűcs G., Gáspár R., Pálóczi J., Faragó N., Zvara A., Puskás L. G., Rázga Z., Tiszlavicz L., Bencsik P., Görbe A., Csonka C., Ferdinandy P., Csont T. (2013) MicroRNA-25-dependent up-regulation of NADPH oxidase 4 (NOX4) mediates hypercholesterolemia-induced oxidative/nitrative stress and subsequent dysfunction in the heart. J. Mol. Cell Cardiol. 62, 111–121 [DOI] [PubMed] [Google Scholar]
- 46. Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M. D., Sadoshima J. (2010) NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proc. Natl. Acad. Sci. U.S.A. 107, 15565–15570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang J. H., Chung T. D., Oldenburg K. R. (1999) A simple statistical parameter for use in evaluation and validation of high throughput screening assays. J. Biomol. Screen. 4, 67–73 [DOI] [PubMed] [Google Scholar]
- 48. Zielonka J., Vasquez-Vivar J., Kalyanaraman B. (2006) The confounding effects of light, sonication, and Mn(III)TBAP on quantitation of superoxide using hydroethidine. Free Radic. Biol. Med. 41, 1050–1057 [DOI] [PubMed] [Google Scholar]
- 49. Sun Q. A., Hess D. T., Wang B., Miyagi M., Stamler J. S. (2012) Off-target thiol alkylation by the NADPH oxidase inhibitor 3-benzyl-7-(2-benzoxazolyl)thio-1,2,3-triazolo[4,5-d]pyrimidine (VAS2870). Free Radic. Biol. Med. 52, 1897–1902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sikora A., Zielonka J., Lopez M., Dybala-Defratyka A., Joseph J., Marcinek A., Kalyanaraman B. (2011) Reaction between peroxynitrite and boronates: EPR spin-trapping, HPLC analyses, and quantum mechanical study of the free radical pathway. Chem. Res. Toxicol. 24, 687–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Choi D. H., Cristóvão A. C., Guhathakurta S., Lee J., Joh T. H., Beal M. F., Kim Y. S. (2012) NADPH oxidase 1-mediated oxidative stress leads to dopamine neuron death in Parkinson's disease. Antioxid. Redox Signal. 16, 1033–1045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gianni D., Taulet N., Zhang H., DerMardirossian C., Kister J., Martinez L., Roush W. R., Brown S. J., Bokoch G. M., Rosen H. (2010) A novel and specific NADPH oxidase-1 (Nox1) small-molecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells. ACS Chem. Biol. 5, 981–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zielonka J., Kalyanaraman B. (2010) Hydroethidine- and MitoSOX-derived red fluorescence is not a reliable indicator of intracellular superoxide formation: another inconvenient truth. Free Radic. Biol. Med. 48, 983–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Maghzal G. J., Cergol K. M., Shengule S. R., Suarna C., Newington D., Kettle A. J., Payne R. J., Stocker R. (2014) Assessment of myeloperoxidase activity by the conversion of hydroethidine to 2-chloroethidium. J. Biol. Chem. 289, 5580–5595 [DOI] [PMC free article] [PubMed] [Google Scholar]