

Background: FBXL5, the F-box protein subunit of an SCF-type ubiquitin-ligase complex, is a regulator of mammalian iron homeostasis.
Results: The HECT-type E3 ligase HERC2 binds to FBXL5 and regulates its stability.
Conclusion: HERC2 controls iron metabolism by promoting ubiquitin-dependent degradation of FBXL5.
Significance: Our results provide new mechanistic insight into the proteolytic control of iron metabolism.
Keywords: E3 Ubiquitin Ligase, Iron Metabolism, Protein Degradation, Proteomics, Ubiquitylation (Ubiquitination)
Abstract
FBXL5 (F-box and leucine-rich repeat protein 5) is the F-box protein subunit of, and therefore responsible for substrate recognition by, the SCFFBXL5 ubiquitin-ligase complex, which targets iron regulatory protein 2 (IRP2) for proteasomal degradation. IRP2 plays a central role in the maintenance of cellular iron homeostasis in mammals through posttranscriptional regulation of proteins that contribute to control of the intracellular iron concentration. The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo, given that mice lacking FBXL5 die during early embryogenesis as a result of unrestrained IRP2 activity and oxidative stress attributable to excessive iron accumulation. Despite its pivotal role in the control of iron homeostasis, however, little is known of the upstream regulation of FBXL5 activity. We now show that FBXL5 undergoes constitutive ubiquitin-dependent degradation at the steady state. With the use of a proteomics approach to the discovery of proteins that regulate the stability of FBXL5, we identified the large HECT-type ubiquitin ligase HERC2 (HECT and RLD domain containing E3 ubiquitin protein ligase 2) as an FBXL5-associated protein. Inhibition of the HERC2-FBXL5 interaction or depletion of endogenous HERC2 by RNA interference resulted in the stabilization of FBXL5 and a consequent increase in its abundance. Such accumulation of FBXL5 in turn led to a decrease in the intracellular content of ferrous iron. Our results thus suggest that HERC2 regulates the basal turnover of FBXL5, and that this ubiquitin-dependent degradation pathway contributes to the control of mammalian iron metabolism.
Introduction
Iron is an essential nutrient for almost all living organisms, with iron deficiency resulting in the impairment of various cellular processes including oxygen transport, energy production, and DNA repair (1). On the other hand, iron is highly reactive and can be deleterious to cells if present in excess (2). Given that both iron deficiency and iron overload are harmful to cells, cellular iron homeostasis is subject to strict regulation (3).
In mammalian cells, iron homeostasis is regulated through coordination of iron uptake, storage, export, and utilization (4). Coordinated expression of proteins mediating these processes is regulated at the posttranscriptional level by iron regulatory protein 1 (IRP1)2 and IRP2. These two orthologous RNA-binding proteins interact with conserved cis-regulatory hairpin structures known as iron-responsive elements (IREs) during iron-limiting conditions to regulate the translation and stability of mRNAs for proteins that contribute to iron homeostasis (5). As a consequence of these interactions, IRP1 and IRP2 increase the size of the bioavailable (ferrous, Fe2+) iron pool during iron-limiting conditions (6). When bioavailable iron levels are high, this activity of IRP1/2 is lost as a result of the assembly of a [4Fe-4S] cluster within IRP1 (holo-IRP1) or of the degradation of both apo-IRP1 (IRP1 without the [4Fe-4S] cluster) and IRP2 by the ubiquitin-proteasome system (7).
The stability of apo-IRP1 and IRP2 is dependent on the action of an iron- and oxygen-regulated SCF-type ubiquitin ligase, SCFFBXL5 (8, 9). FBXL5 (F-box and leucine-rich repeat protein 5) is a member of the F-box family of adaptor proteins that confer substrate specificity on SCF-type ubiquitin ligases (E3s), which consist of the RING-finger protein RBX1, the scaffold protein CUL1, and the adaptor protein SKP1 in addition to an F-box protein (10–12). Under iron-replete conditions, FBXL5 recruits apo-IRP1 and IRP2 to the SCF complex, resulting in their ubiquitylation. In contrast, under iron-limiting conditions, FBXL5 itself is targeted for ubiquitylation and degradation by the proteasome, resulting in the stabilization of apo-IRP1 and IRP2 and their interaction with IREs.
Indicative of the pivotal role of FBXL5 in the control of mammalian iron homeostasis, we have recently shown that mice lacking FBXL5 die early in embryogenesis as a result of deleterious effects of the accumulation of free iron (13). Additional ablation of IRP2, which dominates control of iron homeostasis in vivo, rescued this embryonic mortality of FBXL5-null mice, suggesting that strict regulation of IRP2 by FBXL5 is required for the maintenance of iron homeostasis as well as for viability during development. In addition, liver-specific ablation of FBXL5 was found to result in death from acute liver failure when mice were fed a high-iron diet. The FBXL5-IRP2 axis is thus integral to control of iron metabolism in vivo. Despite recognition of its pivotal role in the regulation of iron homeostasis, however, little is known of the upstream mechanisms responsible for the control of FBXL5 activity.
A regulatory element that renders the stability of FBXL5 sensitive to iron resides within the hemerythrin-like domain at the NH2 terminus of the protein (14). Hemerythrin is a member of a family of iron- and oxygen-binding proteins in bacteria and invertebrates. A previous study suggested that a degron present within the hemerythrin-like domain of FBXL5 becomes preferentially accessible to an as yet unknown E3 in the absence of iron binding to the domain, resulting in ubiquitin-dependent degradation of FBXL5 under iron-limiting conditions (15). The hemerythrin-like domain of FBXL5 is thus considered to be pivotal to the iron-dependent stability of the protein. It remains unclear how FBXL5 is regulated at the steady state, however.
We have now found that FBXL5 undergoes constitutive proteasomal degradation at the steady state, and that a mutant form of FBXL5 that lacks the hemerythrin-like domain is similarly degraded, suggesting the existence of a mechanism for regulation of FBXL5 stability that is independent of this domain. With the use of a proteomics approach, we identified the large HECT-type ubiquitin ligase HERC2 (HECT and RLD domain containing E3 ubiquitin protein ligase 2) as an FBXL5-interacting protein. Depletion of endogenous HERC2 by RNAi stabilized FBXL5 and thereby increased its abundance, resulting in dysregulation of its downstream targets. Our results indicate that HERC2 regulates the basal turnover of FBXL5 and thereby modulates iron metabolism.
EXPERIMENTAL PROCEDURES
Plasmids
Complementary DNAs encoding WT or mutant forms of human FBXL5; mouse SKP2, FBXW5, or FBXW7α; or fragments of human HERC2, each with an NH2-terminal FLAG tag, were subcloned into pcDNA3 (Invitrogen). A cDNA encoding human ubiquitin, tagged at its NH2 terminus with the HA epitope, was subcloned into pCGN (16).
Antibodies
Antibodies to FBXL5 (N0036) were obtained from NeoClone Biotechnology International, HERC2 (ab85832) were from Abcam, IRP1 (sc-14216) and IRP2 (sc-33682) were from Santa Cruz Biotechnology, transferrin receptor 1 (13-6800) from Invitrogen, SKP1 (610530) and HSP90 (610419) were from BD Biosciences, CUL1 (71-8700) was from Zymed Laboratories Inc., and the FLAG epitope (M2) was from Sigma.
Cell Culture and Transfection
HEK293T cells and HeLa cells were cultured under an atmosphere of 5% CO2 at 37 °C in DMEM (Invitrogen) supplemented with 10% FBS (Invitrogen), 1 mm sodium pyruvate, penicillin (100 units/ml), streptomycin (100 mg/ml), 2 mm l-glutamine, and nonessential amino acids (10 μl/ml; Invitrogen). Transfection was performed with the FuGENE HD reagent (Roche Applied Science). The empty pcDNA3 plasmid was included to ensure that cells were transfected with equal amounts of total DNA.
Preparation of Lentiviral Vectors
Lentiviral vectors were prepared as described (17). Gene silencing by RNAi was performed with the lentivirus-based shRNA expression vector CSII-U6tet (18). The shRNA-encoding DNA oligonucleotide inserts were generated with the use of the Insert Design Tool for pSilencer vectors (Applied Biosystems). The target sequences were 5′-GGCCTTGCCTCCAGAAACA-3′ (HERC2-1) and 5′-GGAAAGCACTGGATTCGTT-3′ (HERC2-2) for HERC2 and 5′-GCATACAGCTCTGCAGTTTCC-3′ for FBXL5. An shRNA specific for LacZ (5′-AAGGCCAGACGCGAATTAT-3′), which did not match any existing sequence in the human database, was used as a control.
Tetracycline-inducible shRNA Expression System
HEK293T and HeLa cells were infected with a lentivirus encoding TetR (CSII-EF-TetR-IRES-puro) (18) for 2 days. The cells were then incubated with puromycin (5 μg/ml) for selection and amplification of puromycin-resistant cells. The TetR-expressing cells were then infected with lentiviruses encoding shRNAs. For induction of shRNA expression, the cells were incubated with doxycycline (1 μg/ml) for the indicated times, with replenishment of the drug after 48 h.
Immunoprecipitation and Immunoblot Analysis
HEK293T cells expressing FLAG-tagged proteins were lysed with a buffer comprising 50 mm Tris-HCl (pH 7.5), 150 mm NaCl, 0.5% Triton X-100, aprotinin (10 μg/ml), leupeptin (10 μg/ml), 1 mm PMSF, 400 μm Na3VO4, 400 μm EDTA, 10 mm NaF, and 10 mm sodium pyrophosphate. The lysates were centrifuged at 20,000 × g for 10 min at 4 °C to remove debris, and the resulting supernatants were adjusted with lysis buffer to achieve a protein concentration of 3 mg/ml. The supernatants were then incubated for 30 min at 4 °C with agarose beads conjugated with M2 antibodies to FLAG (Sigma). The resulting immunoprecipitates were washed three times with ice-cold lysis buffer, fractionated by SDS-PAGE, and subjected to immunoblot analysis with primary antibodies and HRP-conjugated secondary antibodies to mouse (Promega), rabbit (Promega), or goat (Santa Cruz Biotechnology) IgG. Immune complexes were detected with an ECL system (Thermo Scientific). For detection of the interaction between endogenous HERC2 and FBXL5, HEK293T cells were incubated for 4 h in the presence of the proteasome inhibitor MG132 (5 μm; Peptide Institute) and then subjected to extraction as described above. The resulting supernatants were subjected to immunoprecipitation for 1 h at 4 °C with antibodies to HERC2 (or control rabbit IgG) and protein A-Sepharose 4 Fast Flow (GE Healthcare). The immunoprecipitates were washed three times with lysis buffer and then subjected to immunoblot analysis, as described above.
Immunoaffinity Purification and Protein Identification by LC-MS/MS Analysis
A cDNA for FLAG-tagged FBXL5 was subcloned into pMX-puro-CMV (19). The resulting vector was introduced together with pCL-Ampho (Imgenex) into HEK293T cells with the use of FuGENE 6 (Promega). The recombinant retroviruses thereby generated were used to infect HEK293T cells, which were then subjected to selection in medium containing puromycin (5 μg/ml). The resulting cells (1 × 108) stably expressing FLAG-FBXL5 were incubated for 6 h in the presence of MG132 (10 μm) and then lysed in 8 ml of a lysis buffer containing 20 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, 1% digitonin, 10 mm NaF, 10 mm Na4P2O7, 0.4 mm Na3VO4, 0.4 mm EDTA, leupeptin (20 μg/ml), aprotinin (10 μg/ml), and 1 mm PMSF. The lysate was centrifuged at 20,000 × g for 20 min at 4 °C to remove debris, and the resulting supernatant was adjusted with lysis buffer to achieve a protein concentration of 3 mg/ml before incubation for 50 min at 4 °C with agarose beads conjugated with M2 antibodies to FLAG (Sigma). The beads were then washed three times with 4 ml of a solution containing 10 mm HEPES-NaOH (pH 7.5), 150 mm NaCl, and 0.1% Triton X-100, after which bead-bound proteins were eluted with 400 μl of FLAG peptide solution (500 μg/ml; Sigma), precipitated with ice-cold 20% TCA, and washed with acetone. The concentrated proteins were dissolved in SDS sample buffer, fractionated by SDS-PAGE on a 10.5% gel, and stained with silver. Individual lanes of the stained gel were sliced into 18 pieces, and proteins within these pieces were subjected to in-gel digestion with trypsin. The resulting peptides were subjected to LC-MS/MS analysis with an ion-trap mass spectrometer (LTQ-XL, Thermo Finnigan), and the obtained data sets were analyzed on the basis of spectral counting as described previously (19).
RT and Real-time PCR Analysis
Total RNA (1 μg) isolated from cells with the use of Isogen (Nippon Gene) was subjected to RT and real-time PCR analysis with primers (forward and reverse, respectively) specific for human FBXL5 (5′-GCCTATGGAATCATGCTGAAGAGC-3′ and 5′-GCATTACCTCAGGAGGAAGATGGG-3′) and human GAPDH (5′-GCAAATTCCATGGCACCGT-3′ and 5′-TCGCCCCACTTGATTTTGG-3′), as described previously (20). The amount of FBXL5 mRNA was normalized by the corresponding amount of GAPDH mRNA.
Determination of Ferrous Iron
The concentration of Fe2+ was determined with the phenanthroline method as described previously (21), with slight modifications. In brief, shRNA-expressing HEK293T cells (1 × 108) were harvested with trypsin, washed five times with 1 ml of PBS (Invitrogen), and subjected to ultrasonic treatment in 500 μl of a solution (pH 4.6) containing 500 μm 1,10-phenanthroline (Sigma) and 100 mm sodium acetate (Sigma). The samples were then centrifuged at 100,000 × g for 30 min at 4 °C, and the absorbance of the resulting supernatants was measured immediately at 510 nm for the spectrophotometric detection of Fe2+. A certified iron standard (FeSO4; Sigma) was used to determine Fe2+ levels. A standard curve for Fe2+ concentrations between 0.2 and 2 μg/ml revealed a linearity of response with a slope of ∼1. Samples were diluted appropriately to fall within the linear range. Cellular Fe2+ content was expressed as a percentage for control cells.
RESULTS
FBXL5 Undergoes Proteasomal Degradation in a Manner Independent of Its Hemerythrin-like Domain
To explore the mechanisms by which the cellular abundance of FBXL5 is controlled, we examined the stability of FBXL5 at steady state in HEK293T cells cultured in the presence of the protein synthesis inhibitor cycloheximide and in the absence or presence of the proteasome inhibitor MG132. FBXL5 was rapidly degraded, with a half-life of <1 h, in the absence of MG132, whereas this rapid turnover was almost completely blocked by the proteasome inhibitor (Fig. 1A). These results suggested that FBXL5 is constitutively degraded and that this basal degradation is dependent largely on the proteasome.
FIGURE 1.

FBXL5 undergoes constitutive proteasomal degradation in a manner independent of its hemerythrin-like domain. A, HEK293T cells incubated with cycloheximide (100 μg/ml) for the indicated times in the absence or presence of the proteasome inhibitor MG132 (10 μm) were subjected to immunoblot (IB) analysis with antibodies to FBXL5 and HSP90 (loading control). The percentage of FBXL5 remaining after the various incubation times was quantitated by image analysis. B, HEK293T cells incubated for 16 h in the absence or presence of ferric ammonium citrate (FAC; 100 μg/ml) or the ferric-iron chelator desferrioxamine (DFO; 100 μm) were subjected to immunoblot analysis with antibodies to FBXL5 and HSP90. C, HEK293T cells transfected with expression vectors for FLAG-FBXL5(1–161) or FLAG-FBXL5(Δ1–161) were incubated for 6 h in the absence or presence of FAC (100 μg/ml) or DFO (100 μm) and then subjected to immunoblot analysis with antibodies to FLAG and to HSP90. D, HEK293T cells transfected with an expression vector for FLAG-FBXL5(Δ1–161) were exposed to cycloheximide (100 μg/ml) for the indicated times in the absence or presence of MG132 (10 μm) and then subjected to immunoblot analysis with antibodies to FLAG and HSP90. The percentage of FLAG-FBXL5(Δ1–161) remaining after the various incubation times was quantitated by image analysis.
Consistent with previous observations suggesting that FBXL5 is targeted for proteasomal degradation in an iron-dependent manner (14), we found that the level of FBXL5 was greatly diminished when iron was limiting and markedly increased under iron-replete conditions (Fig. 1B). A fragment of FBXL5 comprising residues 1–161 also showed similar iron-dependent changes in stability, whereas an FBXL5 mutant lacking this region did not (Fig. 1C), suggesting that a regulatory element that renders FBXL5 stability sensitive to iron resides within its hemerythrin-like domain (amino acids 1–161). To investigate the requirement for the hemerythrin-like domain in the basal degradation of FBXL5, we measured the stability of the FBXL5(Δ1–161) mutant in HEK293T cells cultured in the presence of cycloheximide and in the absence or presence of MG132. As with the WT protein, FBXL5(Δ1–161) was rapidly degraded, with a half-life of <1 h, in the absence of MG132, whereas this rapid turnover was markedly attenuated by the proteasome inhibitor (Fig. 1D). These results suggested that an FBXL5 mutant that lacks the hemerythrin-like domain still undergoes proteasome-dependent degradation at the steady state, and that this domain is therefore dispensable for the basal degradation of FBXL5.
Identification of HERC2 as an FBXL5-associated Protein by a Proteomics Approach
To elucidate the mechanism underlying the basal turnover of FBXL5, we adopted a proteomics approach to identify molecules that associate with this protein. Lysates of HEK293T cells stably infected with a retroviral expression vector for FLAG epitope-tagged FBXL5 were subjected to immunoprecipitation with antibodies to FLAG, and the resulting immunoprecipitated proteins were eluted from the antibody-coated beads with the FLAG peptide and subjected to SDS-PAGE followed by silver staining (Fig. 2A). LC-MS/MS analysis of peptides derived from the fractionated proteins identified CUL1 and SKP1 (both of which are components of the SCFFBXL5 E3 complex) as well as IRP1 and IRP2, both of which were previously identified as substrates for FBXL5 (Fig. 2B) (8, 9). These results thus validated our approach and revealed its high reproducibility. Among the other identified FBXL5-interacting proteins, we focused on HERC2, given that it possesses a COOH-terminal HECT domain, a hallmark of a subfamily of E3 ligases (22).
FIGURE 2.

Identification of HERC2 as a protein that interacts with FBXL5. A, HEK293T cells stably expressing FLAG-tagged FBXL5 were incubated for 6 h in the presence of MG132 (10 μm) and then subjected to immunoaffinity purification with agarose-conjugated antibodies to FLAG. The purified proteins were fractionated by SDS-PAGE and stained with silver. The positions of bands corresponding to various purified proteins are indicated. B, the proteins found to interact with FLAG-FBXL5 in A were identified by LC-MS/MS analysis. For clarity, only HERC2 and previously identified FBXL5-interacting proteins are listed. The peptide count as well as the percentage sequence coverage for each protein are also shown. C, HEK293T cells transfected (or not) with expression vectors for FLAG-SKP2, FLAG-tagged WT, or mutant (PE/AA) forms of FBXL5, FLAG-FBXW5, or FLAG-FBXW7α were subjected to immunoprecipitation (IP) with antibodies to FLAG, and the resulting precipitates as well as a portion of the original cell lysates (Input) were subjected to immunoblot (IB) analysis with antibodies to the indicated proteins. D, HEK293T cells were incubated for 4 h in the presence of MG132 (5 μm) and then subjected to immunoprecipitation with antibodies to HERC2 (or with normal control IgG), and the resulting precipitates as well as a portion of the original cell lysates (Input) were subjected to immunoblot analysis with antibodies to FBXL5 and HERC2. The asterisk indicates a nonspecific band.
To confirm the interaction between FBXL5 and HERC2 in mammalian cells, we transfected HEK293T cells with expression vectors for FLAG-tagged F-box proteins including SKP2, FBXL5, FBXW5, and FBXW7α and subjected cell lysates to immunoprecipitation with antibodies to FLAG. Immunoblot analysis of the resulting precipitates with antibodies to HERC2 revealed that endogenous HERC2 interacted with FLAG-FBXL5, but not with FLAG-SKP2, FLAG-FBXW5, or FLAG-FBXW7α (Fig. 2C). To determine whether formation of an SCF complex by FBXL5 is required for its association with HERC2, we replaced two amino acids (Pro209 and Glu211) in the F-box domain of FBXL5 that are essential for its binding to CUL1 with alanine (19). This FBXL5(PE/AA) mutant failed to bind endogenous CUL1 but retained the ability to associate with endogenous HERC2 (Fig. 2C). These results thus suggested that FLAG-FBXL5 interacts specifically with endogenous HERC2 in a manner independent of SCF complex formation.
We also performed similar experiments to detect the interaction between endogenous proteins. Immunoprecipitates prepared from HEK293T cell extracts with antibodies to HERC2 were subjected to immunoblot analysis with antibodies to FBXL5. Endogenous FBXL5 was co-immunoprecipitated with endogenous HERC2 (Fig. 2D), suggesting that HERC2 interacts with FBXL5 under physiological conditions.
Molecular Dissection of the HERC2-FBXL5 Interaction
We next investigated which region of FBXL5 is required for binding to HERC2 by generating a series of FLAG-tagged deletion mutants of FBXL5 (Fig. 3A) and examining their ability to associate with endogenous HERC2 with a co-immunoprecipitation assay in HEK293T cells. Whereas full-length FBXL5 and all three mutants that contained amino acids 257–297 interacted with HERC2, all the other mutants that lacked this region failed to do so (Fig. 3, B and C), suggesting that residues 257–297 are required for the interaction of FBXL5 with HERC2. Given that this region of FBXL5 is adjacent to the F-box domain, we examined the association of FBXL5(Δ257–297) with CUL1 and SKP1. This mutant retained the ability to interact with both CUL1 and SKP1 (Fig. 3C). On the other hand, an FBXL5 mutant lacking the F-box domain (Δ195–256) was found to bind to HERC2 but not to CUL1 or SKP1. Furthermore, full-length FBXL5 was found to associate with both CUL1-SKP1 and HERC2, consistent with our observation that FBXL5 interacts with HERC2 in a manner independent of the SCF complex formation.
FIGURE 3.

Delineation of the region of FBXL5 responsible for the interaction with HERC2. A, domain organization of full-length (FL) human FBXL5 and structure of deletion mutants thereof. A summary of the ability of the mutants to bind HERC2 as determined in B and C is shown on the right. B and C, HEK293T cells transfected (or not) with expression vectors for FLAG-tagged full-length or mutant forms of FBXL5 were subjected to immunoprecipitation with antibodies to FLAG, and the resulting precipitates as well as a portion of the original cell lysates were subjected to immunoblot (IB) analysis with antibodies to the indicated proteins.
We also determined the region of HERC2 required for binding to FBXL5. Various fragments of HERC2 fused with the FLAG epitope were tested for the ability to interact with endogenous FBXL5 with a co-immunoprecipitation assay in HEK293T cells (Fig. 4). Whereas both fragments that contained amino acids 2930–3327 (a region that includes the RCC1–2 domain) interacted with FBXL5, all the other mutants that lacked this region failed to do so, suggesting that residues 2930–3327 are sufficient for the interaction of HERC2 with FBXL5. Our observations thus indicated that a region of HERC2 that includes the RCC1–2 domain (amino acids 2930–3327) interacts with a region adjacent to the F-box domain of FBXL5 (amino acids 257–297).
FIGURE 4.
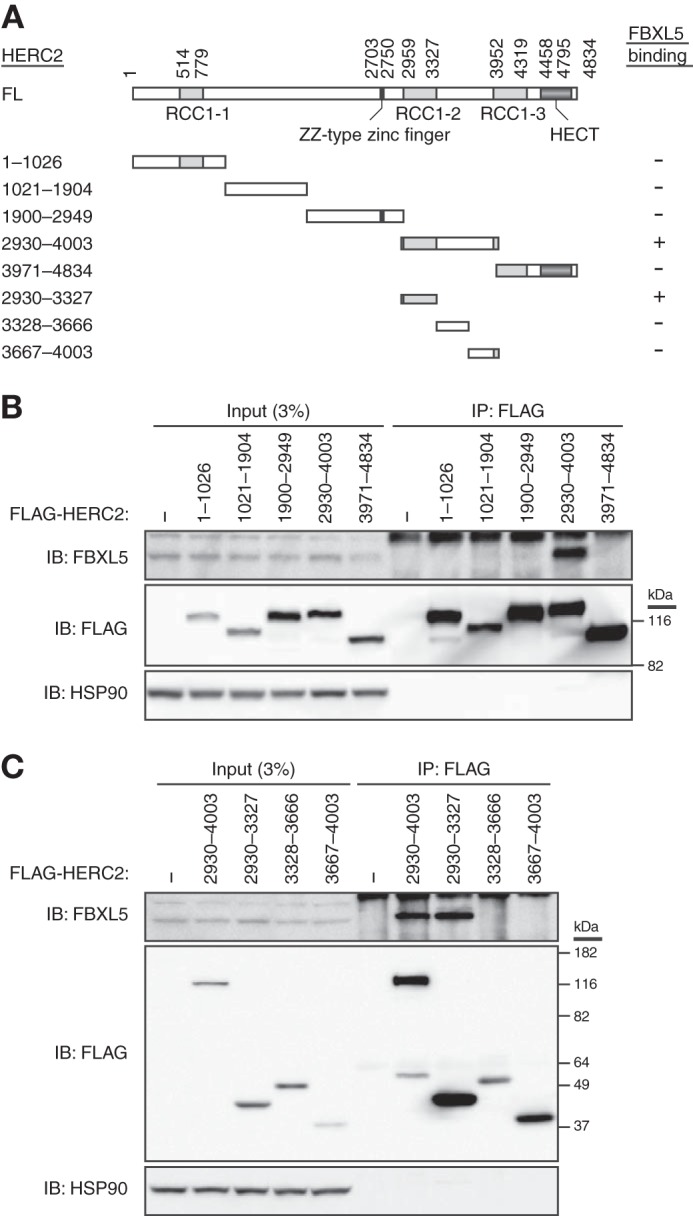
Delineation of the region of HERC2 responsible for the interaction with FBXL5. A, domain organization of human HERC2 and structure of deletion mutants thereof. A summary of the ability of the mutants to bind FBXL5 as determined in B and C is shown on the right. B and C, HEK293T cells transfected (or not) with expression vectors for FLAG-tagged mutants of HERC2 were subjected to immunoprecipitation with antibodies to FLAG, and the resulting precipitates as well as a portion of the original cell lysates were subjected to immunoblot (IB) analysis with antibodies to the indicated proteins.
HERC2 Mediates Ubiquitin-dependent Degradation of FBXL5
Given that HERC2 contains a HECT domain, a hallmark of a subfamily of E3 ubiquitin ligases, we hypothesized that interaction of HERC2 with FBXL5 results in the ubiquitin-dependent proteolysis of the latter protein. To test this possibility, we first transfected HEK293T cells with various amounts of an expression vector for the FBXL5 binding domain of HERC2 (amino acids 2930–3327), which would be expected to inhibit the interaction between endogenous HERC2 and FBXL5. Immunoblot analysis with antibodies to FBXL5 revealed that expression of FLAG-HERC2(2930–3327) in HEK293T cells increased the abundance of endogenous FBXL5 in a concentration-dependent manner (Fig. 5A). We next measured the stability of an FBXL5 mutant that lacks the HERC2 binding domain (amino acids 257–297). Cycloheximide chase analysis revealed that half-life of the FBXL5(Δ257–297) mutant in HEK293T cells was substantially longer than that of the WT protein (Fig. 5B). These results suggested that HERC2 binding destabilizes FBXL5 at the steady state. To investigate further the mechanism of basal FBXL5 degradation, we measured the stability of the FBXL5(PE/AA) mutant (which does not assemble into an SCF complex), given that the F-box subunits of SCF E3 complexes are often subjected to autoubiquitylation and degradation (23). The half-life of FBXL5(PE/AA) did not differ from that of WT FBXL5 (Fig. 5B), suggesting that the basal turnover of FBXL5 is regulated in a manner independent of its assembly into an SCF complex. Collectively, our observations suggested that the stability of FBXL5 is regulated by HERC2, but not through autoubiquitylation resulting from formation of the SCFFBXL5 complex.
FIGURE 5.

Inhibition of HERC2-FBXL5 interaction stabilizes FBXL5. A, HEK293T cells transfected (or not) with various amounts (1×, 2×, 4×) of an expression vector for FLAG-HERC2(2930–3327) were subjected to immunoblot analysis with antibodies to the indicated proteins. B, HEK293T cells transfected with expression vectors for FLAG-FBXL5(WT), FLAG-FBXL5(PE/AA), or FLAG-FBXL5(Δ257–297) were exposed to cycloheximide (100 μg/ml) for the indicated times and then subjected to immunoblot analysis with antibodies to FLAG and HSP90. The percentage of the FLAG-tagged proteins remaining after the various incubation times was quantitated by image analysis.
To confirm that HERC2 indeed regulates the stability of FBXL5, we examined the effect of shRNA-mediated depletion of endogenous HERC2 on the steady state level of endogenous FBXL5. Depletion of HERC2 in HEK293T cells resulted in the accumulation of FBXL5 in proportion to the extent of depletion (Fig. 6A), without a corresponding effect on the amount of FBXL5 mRNA (Fig. 6B). These results suggested that endogenous HERC2 regulates the abundance of FBXL5 at a posttranscriptional level.
FIGURE 6.

Depletion of endogenous HERC2 stabilizes FBXL5. A, HEK293T cells infected (or not) with lentiviral vectors encoding shRNAs specific for LacZ (control) or HERC2 (HERC2–1 or HERC2–2) for 48 h were subjected to immunoblot analysis with antibodies to the indicated proteins. B, total RNA extracted from cells infected as in A was subjected to RT and real-time PCR analysis of FBXL5 mRNA. Data are mean ± S.D. of triplicates from a representative experiment. C, tetracycline repressor (TetR)-expressing HEK293T cells were infected with lentiviral vectors encoding shRNAs specific for LacZ (control) or HERC2, incubated with doxycycline (1 μg/ml) for the indicated times, and then subjected to immunoblot (IB) analysis with antibodies to the indicated proteins. D, HEK293T cells infected as in C were incubated with doxycycline for 48 h and then exposed to cycloheximide (100 μg/ml) for the indicated times before immunoblot analysis with antibodies to the indicated proteins. The percentage of FBXL5 remaining after the various incubation times was quantitated by image analysis. E, HEK293T cells infected and treated with doxycycline as in D were transfected with expression vectors for HA-ubiquitin and for either FLAG-FBXL5(WT) or FLAG-FBXL5(PE/AA). The cells were then incubated for 4 h in the presence of MG132 (10 μm) prior to immunoprecipitation with antibodies to FLAG. The resulting precipitates were subjected to immunoblot analysis with antibodies to the indicated proteins. The positions of bands corresponding to polyubiquitylated ((Ub)n) forms of FLAG-FBXL5 proteins are indicated. F, TetR-expressing HeLa cells were infected with lentiviral vectors encoding shRNAs specific for LacZ (control) or HERC2, incubated with doxycycline for 48 h, and then exposed to cycloheximide for the indicated times before immunoblot analysis with antibodies to the indicated proteins. The percentage of FBXL5 remaining after the various incubation times was quantitated by image analysis.
For reproducible expression of shRNA, we took advantage of HEK293T cells in which a tetracycline-inducible construct encoding HERC2 shRNA was stably integrated. Immunoblot analysis revealed a substantial decrease in the abundance of HERC2 in such cells as early as 48 h after the onset of doxycycline treatment (Fig. 6C). We therefore cultured these cells with doxycycline for 48 h to deplete HERC2 and then subjected the cells to cycloheximide chase analysis to measure the stability of FBXL5 (Fig. 6D). The half-life of FBXL5 was markedly increased in cells depleted of HERC2 compared with that in control cells. Furthermore, to evaluate the effect of HERC2 depletion on the ubiquitylation of FBXL5, we transfected HERC2-depleted HEK293T cells with expression vectors for HA-tagged ubiquitin and FLAG-tagged WT or mutant (PE/AA) forms of FBXL5 and then subjected lysates of the transfected cells to immunoprecipitation with antibodies to FLAG. Immunoblot analysis of the resulting precipitates revealed that depletion of endogenous HERC2 resulted in a marked decrease in the ubiquitylation levels of both FLAG-FBXL5(WT) and FLAG-FBXL5(PE/AA) (Fig. 6E). The extent of ubiquitylation of FLAG-FBXL5(PE/AA) did not differ from that of FLAG-FBXL5(WT) in control cells, suggesting that basal ubiquitylation of FBXL5 is mediated in a manner independent of its assembly into an SCF complex, again arguing against an autoubiquitylation model for control of basal FBXL5 degradation. Collectively, our observations suggested that HERC2 serves as an E3 ligase for FBXL5 and promotes its basal turnover.
We also examined the stability of FBXL5 in the absence or presence of HERC2 in HeLa cells. As observed with HEK293T cells, the half-life of FBXL5 was markedly greater in HERC2-depleted HeLa cells than in the corresponding control cells (Fig. 6F), suggesting that the role of HERC2 in regulation of the basal turnover of FBXL5 is not specific to one cell line.
HERC2 Modulates Iron Metabolism by Regulating the Basal Turnover of FBXL5
Given that HERC2 was shown to serve as an E3 ligase for FBXL5 at the steady state, we next examined whether HERC2 also mediates iron-dependent changes in FBXL5 stability. We cultured control or HERC2-depleted HEK293T cells under iron-replete conditions and then subjected them to iron limitation for various times before immunoblot analysis (Fig. 7A). Although HERC2 depletion resulted in a marked increase in the abundance of FBXL5 before iron chelation, the subsequent rate of degradation of FBXL5 in cells depleted of HERC2 was almost identical to that in control cells, suggesting that HERC2 is dispensable for iron-dependent changes in the stability of FBXL5. Our observations thus indicated that HERC2 regulates the basal turnover of FBXL5 rather than its iron-dependent degradation.
FIGURE 7.

HERC2 controls iron metabolism by regulating the basal degradation of FBXL5. A, TetR-expressing HEK293T cells were infected with lentiviral vectors encoding LacZ (control) or HERC2 shRNAs, treated with doxycycline (1 μg/ml) for 48 h, incubated with ferric ammonium citrate (FAC) (100 μg/ml) for 16 h, and then exposed to desferrioxamine (DFO) (100 μm) for the indicated times before immunoblot (IB) analysis with antibodies to the indicated proteins. The percentage of FBXL5 remaining after the various incubation times was quantitated by image analysis. B, TetR-expressing HEK293T or HeLa cells infected with lentiviral vectors encoding LacZ (control) or HERC2 shRNAs were treated with doxycycline for 48 h and then subjected to immunoblot analysis with antibodies to the indicated proteins. C, TetR-expressing HEK293T cells infected with lentiviral vectors encoding LacZ (control), HERC2, or FBXL5 shRNAs, as indicated, were treated with doxycycline for 48 h, incubated in the absence or presence of FAC (100 μg/ml) or DFO (100 μm) for an additional 12 h, and then subjected to immunoblot analysis with antibodies to the indicated proteins. D, ferrous iron content of HEK293T cells infected and treated with doxycycline as in C. Data are mean ± S.D. from three independent experiments. ***, p < 0.005 (two-tailed Student's t test).
To evaluate the role of HERC2 in regulation of intracellular iron homeostasis, we examined the effects of HERC2 depletion on the expression of proteins related to such homeostasis (Fig. 7B). For both HEK293T and HeLa cells, the increase in the abundance of FBXL5 in cells depleted of HERC2 was associated with a decrease in the amounts of IRP1 and IRP2, which in turn was accompanied by a small but reproducible decrease in the abundance of transferrin receptor 1, the mRNA that is stabilized by IRP1/2 binding (24). On the other hand, the response of these various iron homeostasis-related proteins to changes in iron availability was maintained in the HERC2-depleted cells (Fig. 7C), consistent with the finding that HERC2 is dispensable for iron-dependent degradation of FBXL5. Furthermore, the additional depletion of FBXL5 greatly attenuated the effects of HERC2 depletion (Fig. 7C), suggesting that the latter effects are mediated by up-regulation of FBXL5 expression. Our results thus indicated that the accumulation of FBXL5 in HERC2-depleted HEK293T and HeLa cells results in dysregulation of the expression of proteins related to iron homeostasis.
Finally, we examined how such FBXL5 accumulation resulting from HERC2 depletion affects cellular iron homeostasis. The level of ferrous iron (Fe2+) in HERC2-depleted HEK293T cells was significantly lower than that in control cells (Fig. 7D), suggesting that accumulated FBXL5 prevented Fe2+ loading, likely as a result of reduced IRP1/2 expression, in the former cells. We confirmed that additional depletion of FBXL5 greatly attenuated this effect of HERC2 depletion, suggesting that the effect is indeed mediated by up-regulation of FBXL5 expression (Fig. 7D). Collectively, our observations thus indicated that HERC2 modulates iron metabolism by regulating the amount of FBXL5.
DISCUSSION
Iron homeostasis in mammals is strictly controlled to ensure provision of a proper amount of iron to cells. Regulation of IRP1/2 stability by the ubiquitin-proteasome pathway is critical for cells to maintain an appropriate ferrous iron (Fe2+) content, and thereby to avoid deleterious iron deficiency and prevent iron excess (25). FBXL5 plays a pivotal role in the control of iron metabolism by promoting the ubiquitin-dependent degradation of IRP1/2 under iron-replete conditions (13). Furthermore, a decline in cellular iron availability and consequent loss of direct iron binding to the hemerythrin-like domain of FBXL5 results in the ubiquitylation of FBXL5 itself by an as yet unknown E3 followed by its degradation (8, 9).
Recent molecular approaches have provided insight into the iron-dependent regulation of FBXL5 stability. The hemerythrin-like domain at the NH2 terminus of FBXL5 is essential and sufficient for iron sensitivity of stability (14). Mutation of the iron-ligating residues in this domain results in constitutive destabilization of FBXL5, suggesting that iron binding is essential for protein stabilization. Moreover, residues 77–81 within the hemerythrin-like domain appear to constitute part of a degron responsible for iron-dependent degradation of FBXL5 (15). These observations thus suggest a model in which the hemerythrin-like domain acts as an iron-binding switch that regulates accessibility of the degron to an as yet unknown E3.
We now propose an additional mechanism for ubiquitin-dependent regulation of FBXL5 stability, providing further evidence for the prominent role of the ubiquitin-proteasome system in the control of iron homeostasis. We have shown that FBXL5 is constitutively degraded at the steady state in a manner independent of its hemerythrin-like domain. Our biochemical results suggest that the large HECT-type ubiquitin ligase HERC2 is responsible for the basal degradation of FBXL5. HERC2 interacts with a region of FBXL5 immediately downstream of the F-box domain, and it regulates the basal turnover of FBXL5 rather than its iron-dependent degradation. Our findings further indicate that HERC2 sets the steady state level of FBXL5 and thereby modulates iron metabolism.
The human HERC2 gene comprises 93 exons and encodes a 4834-amino acid protein with a predicted molecular size of 528 kDa (26). It is a highly mutable gene located at a deletion breakpoint hot spot on human chromosome 15q11-q13 (27). A homozygous missense mutation of HERC2 is associated with a global developmental delay and autism spectrum disorder (28). Although the function of HERC2 had long remained unknown, the protein was recently shown to contribute to the response of cells to DNA damage. HERC2 is thus recruited to sites of DNA double strand breaks and facilitates assembly of the ubiquitin-conjugating enzyme UBC13 with RNF8 (RING finger protein 8), thereby promoting ubiquitin-dependent retention of repair factors (29). HERC2 also shuttles between the nucleus and the cytoplasm, acting as an E3 ubiquitin ligase that targets xeroderma pigmentosum A in the circadian control of nucleotide excision repair (30) as well as BRCA1 (breast cancer 1) in control of the cell cycle (31). These observations suggest that HERC2 might regulate multiple aspects of the DNA damage response by contributing to the ubiquitylation and degradation of key protein participants (32).
DNA repair proteins commonly require iron as a cofactor (33), with this requirement likely endowing iron with protective effects against DNA damage (34). We have now uncovered a function of HERC2 that relates to iron metabolism. Cells exposed to DNA damage might require iron to facilitate the production of DNA repair proteins. In this regard, it is of interest that HERC2 also sets the cellular iron concentration by regulating the abundance of FBXL5.
Collectively, our observations suggest the existence of at least two distinct pathways for the ubiquitin-dependent regulation of FBXL5 stability, with these two pathways being responsible for the basal degradation and the iron-dependent degradation of the protein. These pathways might contribute differentially to FBXL5 degradation in a context-dependent manner. Further molecular analysis of the regulation of FBXL5, including identification of the as yet unknown E3 for the iron-dependent degradation pathway, should provide new insight into the control of iron metabolism mediated by ubiquitin-dependent proteolysis.
Acknowledgments
We thank H. Miyoshi and S. Yonehara for lentiviral vectors, T. Kitamura for pMX-puro, M. Oda and E. Koba for technical assistance, and T. Asano for discussion.
Footnotes
- IRP
- iron regulatory protein
- IRE
- iron-responsive element
- FBXL5
- F-box and leucine-rich repeat protein 5
- SCF
- SKP1–CUL1–F-box protein
- HERC2
- HECT and RLD domain containing E3 ubiquitin protein ligase 2
- TetR
- tetracycline repressor.
REFERENCES
- 1. Andrews N. C., Schmidt P. J. (2007) Iron homeostasis. Annu. Rev. Physiol. 69, 69–85 [DOI] [PubMed] [Google Scholar]
- 2. Prohaska R., Sibon O. C., Rudnicki D. D., Danek A., Hayflick S. J., Verhaag E. M., Vonk J. J., Margolis R. L., Walker R. H. (2012) Brain, blood, and iron: perspectives on the roles of erythrocytes and iron in neurodegeneration. Neurobiol. Dis. 46, 607–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. De Domenico I., McVey Ward D., Kaplan J. (2008) Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat. Rev. Mol. Cell Biol. 9, 72–81 [DOI] [PubMed] [Google Scholar]
- 4. Hentze M. W., Muckenthaler M. U., Galy B., Camaschella C. (2010) Two to tango: regulation of mammalian iron metabolism. Cell 142, 24–38 [DOI] [PubMed] [Google Scholar]
- 5. Rouault T. A. (2006) The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2, 406–414 [DOI] [PubMed] [Google Scholar]
- 6. Wang J., Pantopoulos K. (2011) Regulation of cellular iron metabolism. Biochem. J. 434, 365–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Evstatiev R., Gasche C. (2012) Iron sensing and signalling. Gut 61, 933–952 [DOI] [PubMed] [Google Scholar]
- 8. Salahudeen A. A., Thompson J. W., Ruiz J. C., Ma H. W., Kinch L. N., Li Q., Grishin N. V., Bruick R. K. (2009) An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 326, 722–726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Vashisht A. A., Zumbrennen K. B., Huang X., Powers D. N., Durazo A., Sun D., Bhaskaran N., Persson A., Uhlen M., Sangfelt O., Spruck C., Leibold E. A., Wohlschlegel J. A. (2009) Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 326, 718–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jin J., Cardozo T., Lovering R. C., Elledge S. J., Pagano M., Harper J. W. (2004) Systematic analysis and nomenclature of mammalian F-box proteins. Genes Dev. 18, 2573–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nakayama K. I., Nakayama K. (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369–381 [DOI] [PubMed] [Google Scholar]
- 12. Skaar J. R., Pagan J. K., Pagano M. (2013) Mechanisms and function of substrate recruitment by F-box proteins. Nat. Rev. Mol. Cell. Biol. 14, 369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moroishi T., Nishiyama M., Takeda Y., Iwai K., Nakayama K. I. (2011) The FBXL5-IRP2 axis is integral to control of iron metabolism in vivo. Cell Metab. 14, 339–351 [DOI] [PubMed] [Google Scholar]
- 14. Chollangi S., Thompson J. W., Ruiz J. C., Gardner K. H., Bruick R. K. (2012) Hemerythrin-like domain within F-box and leucine-rich repeat protein 5 (FBXL5) communicates cellular iron and oxygen availability by distinct mechanisms. J. Biol. Chem. 287, 23710–23717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Thompson J. W., Salahudeen A. A., Chollangi S., Ruiz J. C., Brautigam C. A., Makris T. M., Lipscomb J. D., Tomchick D. R., Bruick R. K. (2012) Structural and molecular characterization of iron-sensing hemerythrin-like domain within F-box and leucine-rich repeat protein 5 (FBXL5). J. Biol. Chem. 287, 7357–7365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kitagawa M., Hatakeyama S., Shirane M., Matsumoto M., Ishida N., Hattori K., Nakamichi I., Kikuchi A., Nakayama K., Nakayama K. (1999) An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of β-catenin. EMBO J. 18, 2401–2410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tahara-Hanaoka S., Sudo K., Ema H., Miyoshi H., Nakauchi H. (2002) Lentiviral vector-mediated transduction of murine CD34(−) hematopoietic stem cells. Exp. Hematol. 30, 11–17 [DOI] [PubMed] [Google Scholar]
- 18. Kobayashi Y., Yonehara S. (2009) Novel cell death by downregulation of eEF1A1 expression in tetraploids. Cell Death Differ. 16, 139–150 [DOI] [PubMed] [Google Scholar]
- 19. Yumimoto K., Matsumoto M., Oyamada K., Moroishi T., Nakayama K. I. (2012) Comprehensive identification of substrates for F-box proteins by differential proteomics analysis. J. Proteome Res. 11, 3175–3185 [DOI] [PubMed] [Google Scholar]
- 20. Nishiyama M., Skoultchi A. I., Nakayama K. I. (2012) Histone H1 recruitment by CHD8 is essential for suppression of the Wnt-β-catenin signaling pathway. Mol. Cell. Biol. 32, 501–512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mahler H. R., Elowe D. G. (1954) Studies on metalloflavoproteins. II. The role of iron in diphosphopyridine nucleotide cytochrome c reductase. J. Biol. Chem. 210, 165–179 [PubMed] [Google Scholar]
- 22. Rotin D., Kumar S. (2009) Physiological functions of the HECT family of ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 10, 398–409 [DOI] [PubMed] [Google Scholar]
- 23. Ang X. L., Wade Harper J. (2005) SCF-mediated protein degradation and cell cycle control. Oncogene 24, 2860–2870 [DOI] [PubMed] [Google Scholar]
- 24. Piccinelli P., Samuelsson T. (2007) Evolution of the iron-responsive element. RNA 13, 952–966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Thompson J. W., Bruick R. K. (2012) Protein degradation and iron homeostasis. Biochim. Biophys. Acta 1823, 1484–1490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hochrainer K., Mayer H., Baranyi U., Binder B., Lipp J., Kroismayr R. (2005) The human HERC family of ubiquitin ligases: novel members, genomic organization, expression profiling, and evolutionary aspects. Genomics 85, 153–164 [DOI] [PubMed] [Google Scholar]
- 27. Ji Y., Rebert N. A., Joslin J. M., Higgins M. J., Schultz R. A., Nicholls R. D. (2000) Structure of the highly conserved HERC2 gene and of multiple partially duplicated paralogs in human. Genome Res. 10, 319–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Puffenberger E. G., Jinks R. N., Wang H., Xin B., Fiorentini C., Sherman E. A., Degrazio D., Shaw C., Sougnez C., Cibulskis K., Gabriel S., Kelley R. I., Morton D. H., Strauss K. A. (2012) A homozygous missense mutation in HERC2 associated with global developmental delay and autism spectrum disorder. Hum. Mutat. 33, 1639–1646 [DOI] [PubMed] [Google Scholar]
- 29. Bekker-Jensen S., Rendtlew Danielsen J., Fugger K., Gromova I., Nerstedt A., Lukas C., Bartek J., Lukas J., Mailand N. (2010) HERC2 coordinates ubiquitin-dependent assembly of DNA repair factors on damaged chromosomes. Nat. Cell. Biol. 12, 80–86; sup pp 1–12 [DOI] [PubMed] [Google Scholar]
- 30. Kang T. H., Lindsey-Boltz L. A., Reardon J. T., Sancar A. (2010) Circadian control of XPA and excision repair of cisplatin-DNA damage by cryptochrome and HERC2 ubiquitin ligase. Proc. Natl. Acad. Sci. U.S.A. 107, 4890–4895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wu W., Sato K., Koike A., Nishikawa H., Koizumi H., Venkitaraman A. R., Ohta T. (2010) HERC2 is an E3 ligase that targets BRCA1 for degradation. Cancer Res. 70, 6384–6392 [DOI] [PubMed] [Google Scholar]
- 32. Ulrich H. D., Walden H. (2010) Ubiquitin signalling in DNA replication and repair. Nat. Rev. Mol. Cell Biol. 11, 479–489 [DOI] [PubMed] [Google Scholar]
- 33. Lill R., Mühlenhoff U. (2008) Maturation of iron-sulfur proteins in eukaryotes: mechanisms, connected processes, and diseases. Annu. Rev. Biochem. 77, 669–700 [DOI] [PubMed] [Google Scholar]
- 34. Berthelet S., Usher J., Shulist K., Hamza A., Maltez N., Johnston A., Fong Y., Harris L. J., Baetz K. (2010) Functional genomics analysis of the Saccharomyces cerevisiae iron responsive transcription factor Aft1 reveals iron-independent functions. Genetics 185, 1111–1128 [DOI] [PMC free article] [PubMed] [Google Scholar]