

Abstract
Neuroferritinopathy or hereditary ferritinopathy is an inherited neurodegenerative disease caused by mutations in ferritin light chain (FTL) gene. The clinical features of the disease are highly variable, and include a movement disorder, behavioral abnormalities, and cognitive impairment. Neuropathologically, the disease is characterized by abnormal iron and ferritin deposition in the central nervous system. We report a family in which neuroferritinopathy begins with chronic headaches, later developing progressive orolingual and arm dystonia, dysarthria, cerebellar ataxia, pyramidal tract signs, and psychiatric symptoms. In the absence of classic clinical symptoms, the initial diagnosis of the disease was based on magnetic resonance imaging studies. Biochemical studies on the proband showed normal serum ferritin levels, but remarkably low cerebrospinal fluid (CSF) ferritin levels. A novel FTL mutation was identified in the proband. Our findings expand the genetic and clinical diversity of neuroferritinopathy and suggest CSF ferritin levels as a novel potential biochemical marker for the diagnosis of neuroferritinopathy.
Keywords: Ferritin, neurodegeneration, hereditary ferritinopathy, iron, brain
1. INTRODUCTION
Neuroferritinopathy or hereditary ferritinopathy (Muhoberac, 2013) is an autosomal dominant movement disorder caused by mutations in the ferritin light chain gene (FTL) on chromosome 19q13.3. Neuropathologically, the disease is characterized by the abnormal depositions of iron and ferritin in the brain, particularly in the basal ganglia. Thus far, six different mutations in exon four of the FTL gene have been reported, all affecting the FTL polypeptide C-terminus (Curtis, 2001; Vidal, 2004; Mancuso, 2005; Ohta, 2008; Devos, 2009; Kubota, 2009). Clinically, the disease presents as a middle-age–onset chorea and dystonia. Clinical presentation may also include extrapyramidal and pyramidal tract signs as well as cerebellar ataxia, dysautonomia, cognitive decline, and psychiatric symptoms; however, the clinical presentation is highly variable both within and between families (Wills, 2002; Chinnery, 2003; Mir, 2005; Chinnery, 2007; Cassidy, 2011; Crompton, 2005; Ory-Magne, 2009). Several in vitro and in vivo studies (reviewed in Muhoberac, 2013) have implicated at least two key toxic mechanisms in the pathogenesis of the disease: abnormal iron metabolism and generation of free radicals, and abnormal ferritin aggregation. These two mechanisms may be acting together to lead to neurodegeneration and thus to the progression of the disease. The mutant FTL polypeptide can cause deregulation of cellular iron metabolism (ferritin loss of function), oxidative stress, and overproduction of ferritin polypeptides (a positive feedback loop), while excess iron and ferritin could trigger the formation of ferritin aggregates, which may physically interfere with normal cellular functions (gain of a toxic function) (Vidal, 2011).
Herein, we report the identification of a novel mutation in the FTL gene in a Japanese family with neuroferritinopathy, and highlight the utility of T2-weighted magnetic resonance imaging and biochemical studies as potential biomarkers for the diagnosis of the disease.
2. SUBJECTS AND METHODS
2.1. Case Report
The proband, a 44-year-old, right-handed Japanese female, presented initially with chronic headaches at the age of 42. She was allergic to milk, wheat, and eggs. There was no history of anoxia at birth or carbon monoxide poisoning and no family history of neurodegenerative disorders or consanguineous marriage (Fig. 1A). At the age of 43, she demonstrated psychiatric disturbances such as emotional lability, and was diagnosed with panic disorder at a mental health clinic. At the age of 44, she experienced difficulty in speaking and walking as well as clumsiness in her left arm. Neurological examination showed emotional incontinence, mild cognitive decline (Full scale Intelligence Quotient = 83, Verbal Intelligence Quotient = 84, Performance Intelligence Quotient = 84), slurred speech, bilateral hyperextensibility and hypotonus, left-sided cerebellar ataxia, hyperreflexia, and extensor plantar response. Cranial nerve examination showed slow eye saccades and involuntary movement of tongue. Rigidity, spasticity, tremor, dystonia, chorea, and parkinsonism were not observed initially. Her gait was unsteady (not wide-based); she exhibited some difficulty during tandem gait. Gait disturbance gradually progressed, and her gait became increasingly unsteady with a tendency to fall. Limb weakness, sensory disturbance, and bladder or rectal disturbances were not observed. Orthostatic hypotension was detected by the head-up tilt test (blood pressure: 121/79 mmHg in the supine, 90/63 mmHg in the standing position with dizziness). Several months after the initial presentation, oromandibular, orolingual, and left dominant arm dystonia, tongue dyskinesia, tongue wiggling movements, tongue biting, and dysphagia developed. Her serum ferritin levels were 20 ng/mL (normal range, 5–204 ng/mL). Serum levels of iron, copper, ceruloplasmin, hemoglobin, and vitamin E were normal. Routine hematological studies and thyroid function studies were also normal. No tumor markers or autoimmune antibodies such as anti-glutamic acid decarboxylase and anti-gliadin were detected. An abdominal computed tomography (CT)/MRI showed the presence of a left ovarian cyst; however, tests for paraneoplastic antibodies such as anti-Hu and anti-Yo in serum and CSF were negative. Analysis of CSF revealed a remarkably low ferritin level (<1.00 ng/mL, normal range 6.68 ± 0.93 ng/mL). Several medications, including trihexyphenidyl, benzodiazepine, valproate, and muscle relaxants, were attempted; however, they did not improve the patient’s symptoms. Herbal medicine (Shakuyaku-kanzo-to) relieved the left finger hyperextensibility due to dystonia.
Figure 1.
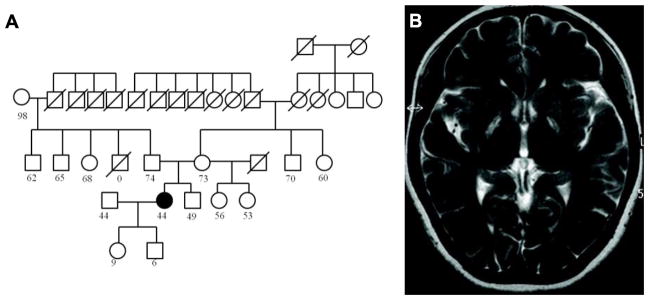
A. Pedigree of the family. The square symbols represent males and the circle symbols represent females. The filled symbol represents the proband. The symbol “/” represents deceased individuals. The numbers under the symbols represent current age. B. MRI of the proband at the age of 42, before onset of neurological symptoms.
2.2. Methods
MRI, laboratory tests including measurement of serum ferritin levels and genetic analysis were performed on the proband and her family (parents and brother). CSF ferritin levels were also measured on the proband. Brain MRI was performed using a 1.5-Tesla system (GyroscanAchieva; Philips Medical Systems, Best, The Netherlands). T1-weighted (TR = 600 ms, TE = 12 mc), T2-weighted (TR = 4423 ms, TE = 100 ms), and T2*-weighted (TR = 640 ms, TE = 23 ms) sequences were acquired in the transverse plane. In the proband, susceptibility-weighted imaging (SWI) based on 3-dimensional T1-weighted fast field echo (3DT1FFE) and 123I-iodoamphetamine (IMP) single photon emission tomography (IMP-SPECT) were also performed.
After informed consent was obtained, genomic DNA was extracted from a 500 μl saliva aliquot collected with the Oragene Discover Collection Kit (DNAgenotek, Ottawa, Canada) using prepIT-C2D Genomic DNA MiniPrep Kit (Oragene) in accordance with the manufacturer’s instructions. PCR amplification was performed on 0.15 ug of genomic DNA to amplify all four exons of the FTL gene using the oligonucleotide primer pairs: Exon 1(352bp) F: 5′-ACGTCCCCTCGCAGTTCGGCGG-3′ and R: 5′-GGAGGTGCGCAGCTGGAGG-3′; Exon 2(327bp)F: 5′-GGTAAACAGAGGGCGGAGTC-3′ and R: 5′-ACCGAACTCAATCTCCCAGA-3′; and Exon 3 & 4(660 bp) F: 5′-TGTAGGTTTAGTTCTATGTG-3′ and R: 5′-AAGCCCTATTACTTTGCAAG-3′; at 2 mmol each oligonucleotide, 200 μmol dNTPs, and 1.5 mM MgCl2 in a 50 μl reaction, and cycled for 35 cycles of 94°C for 30 s, 45°C for 45 s, and 72°C for 45 s. DNA fragments were separated on a 1% agarose gel, visualized by ethidium bromide staining, and the corresponding bands excised and purified using the GeneJET Gel Extraction Kit (Thermo Scientific, Lithuania). DNA sequencing was performed in both directions as described (Vidal, 2004) using a CEQ 8000 GeXP Genetic Analysis System and Software (Beckman Coulter). Amplification products of exons 3 and 4 were also subcloned into pCR 2.1 vector (Invitrogen, Carlsbad, CA) and transformed into One Shot® TOP10 Chemically Competent E. coli cells (Invitrogen) according to the manufacturer’s protocol. Recombinant plasmid DNA was isolated from 10 clones of different PCR reactions and sequenced in both directions as described (Vidal, 2004).
3. RESULTS
3.1. Imagining Studies
Brain MRI of the proband showed symmetrical hyperintense areas surrounded by hypointense areas in the bilateral posterior globus pallidus and putamen at age 42 (Fig. 1B). At age 43, Brain CT revealed symmetrical low density areas in the bilateral basal ganglia (Fig. 2A). T1-wighted MRI images demonstrated cortical atrophy of the cerebrum and cerebellum. T2-weighted MRI revealed symmetrical hyperintense areas surrounded by hypointense areas in the bilateral posterior globus pallidus and putamen, whereas T2*-weighted and SWI-MRI revealed hypointense areas in the globus pallidus, putamen, thalamus, red nucleus, dentate nucleus, and cerebral cortex (Fig. 2 and 3). No spinal cord lesions were observed. 123I-iodoamphetamine single photon emission tomography (IMP-SPECT) revealed bilateral mild hypoperfusion of the cerebellum and right hypoperfusion of the basal ganglia (Fig. 4). Cardiac 123I-metaiodobenzylguanidine (MIBG) uptake was not decreased (H/M ratios for early phase 2.70 and delayed phase 2.78).
Figure 2.

CT and MRI scans of the proband at the age of 44. A. Axial head CT shows symmetrical hypodense lesions in the basal ganglia, bilaterally. B. T1 weighted axial MRI image shows symmetrical hypointense areas in the basal ganglia, revealing the degenerative and partially cystic nature of the lesions. C. T2 weighted axial MRI image showing symmetrical hyperintense areas encircled by hypointense areas in the bilateral posterior globus pallidus and putamen. D–H. T2* weighted axial MRI images showing hypointense areas in dentate nucleus (D), red nucleus (E), globus pallidus, putamen, caudate, thalamus (F, G), and cerebral cortex (H), suggesting increased deposition of ferritin in multiple nuclei. I. T2* weighted coronal MRI image showing a hypointense rim and a hyperintense center reminiscent of the “eye of the tiger” sign in the basal ganglia.
Figure 3.

Axial susceptibility-weighted images (SWI) show hypointense areas in the globus pallidus, putamen, caudate, thalamus, red nucleus, dentate nucleus, and motor cortex.
Figure 4.
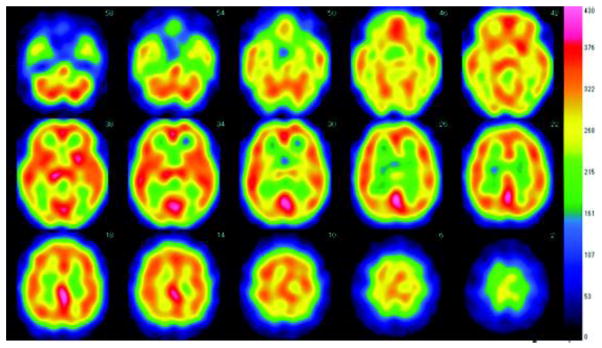
123I-iodoamphetamine single photon emission tomography (IMP-SPECT) shows hypoperfusion in the bilateral cerebellum and right basal ganglia.
3.2 Ferritin measurements
Serum ferritin values in the proband, her parents and brother were in the normal range (5–204 ng/mL). CSF ferritin levels in the proband were remarkably low (<1.00 ng/mL, normal range 6.68 ± 0.93 ng/mL).
3.3. Genetic Analysis
The sequence of exons 1, 2 and 3 of the FTL gene and adjacent intronic sequences were found to be normal. Gel migration analysis of a PCR product comprising exons 3 and 4 of FTL revealed the presence of two amplification products of 660 and 676 bp in the proband, whereas the control sample showed only the 660 bp product (Fig. 5A). DNA sequencing of exon 4 of the FTL gene in the proband revealed a novel mutation in one allele of the FTL gene, c.468_483dupTGGCCCGGAGGCTGGG (Fig. 5B). The position of the mutation was near to that of previously reported Japanese (Ohta, 2008) and Italian (Storti, 2013) mutation, c.469_484dupGGCCCGGAGGCTGGGC, shifted one nucleotide to the 5′ of the previously reported mutation sequence. To confirm the presence of the mutation found in the proband, the PCR products of the amplification of exons 3 and 4 were subcloned and recombinant plasmid DNA isolated and sequenced in both directions. Sequence analysis confirmed the presence of the 16 bp insertion in the mutant allele (Fig. 5B). As a result, the predicted mutant polypeptide (p.Leu162TrpfsX24) and the previously reported mutant polypeptide (p.Leu162ArgfsX24) (Ohta, 2008; Storti, 2013) have a different amino acid at codon 162 (tryptophan in p.Leu162TrpfsX24 and arginine in p.Leu162ArgfsX24) but identical sequence and length of the C-terminus of the mutant protein (Fig. 5C). The mutation was not observed in the proband’s parents or her brother, who had normal serum ferritin levels and unremarkable MRI. Genetic analyses for mutations associated with spinocerebellar degeneration, such as spinocerebellar ataxias (SCA1, SCA2, SCA3, SCA6, SCA7, SCA8, SCA10, SCA12, SCA17), and dentatorubral–pallidoluysian atrophy, were negative. No mutations were observed in the pantothenate kinase 2 (PANK2) gene associated with pantothenate kinase-associated neurodegeneration (PKAN).
Figure 5.

Genetic analyses of the FTL gene in the proband. A. Analysis of PCR products containing exons 3 and 4 of the FTL gene by agarose gels show that the proband (P) has an additional band (676 bp) in addition to the normal amplification product of 660 bp observed in control individuals. B. Direct sequencing of genomic DNA from the proband (top) and cloned DNA containing the mutant allele (bottom) clearly showing the beginning (thymine) of the 16-nucleotide duplication (c.468_483dupTGGCCCGGAGGCTGGG). Wild-type nucleotide sequence is indicated on top and the duplicated sequence on bottom. C. Wild-type sequence (top) and mutant sequences (bottom) of the FTL gene showing the 16-bp duplication. The duplicated nucleotide sequences are underlined. The p.Leu162ArgfsX24 was previously reported in Japanese (Ohta, 2008) and Italian (Storti, 2013) affected individuals. The predicted C-terminal amino acid sequences are indicated. Amino acid 162 is in bold.
4. DISCUSSION
We report a Japanese female with neuroferritinopathy harboring a novel mutation in exon 4 of the FTL gene. The proband presented by age 42 with chronic headaches, developing during the next two years progressive orolingual and arm dystonia, dysarthria, cerebellar ataxia, pyramidal tract signs, and psychiatric symptoms. Previously reported individuals with neuroferritinopathy show dystonia and dysarthria although our proband did not present initially with dystonia, she gradually exhibited progressive orolingual and limb dystonia over several months. It has been previously reported that the clinical presentation of the disease may be highly variable not only between families but also within a family (Wills, 2002; Chinnery, 2003; Mir, 2005; Chinnery, 2007; Cassidy, 2011; Crompton, 2005; Ory-Magne, 2009; Vidal, 2011), suggesting that a heterogeneous presentation may be a characteristic of neuroferritinopathy. Although dystonia and dysarthria are the main manifestations of this condition, the disease cannot be diagnosed only on the basis of clinical symptoms alone because many other clinical features such as pyramidal signs, cerebellar ataxia, dysautonomia, cognitive impairment, and psychiatric symptoms may be observed in affected individuals.
The MRI findings in patients with neuroferritinopathy change with disease progression according to clinical stage. In the early clinical stage, hypointense lesions in the basal ganglia are observed on T2*-weighted images and SWI, particularly in the globus pallidus and putamen (Lehn, 2012). With disease progression, the T2* signal loss extends to the dentate nucleus, red nucleus, substantia nigra, thalamus, caudate nucleus, and cerebral cortex (Ohta, 2012). In later stages, hyperintense lesions in the basal ganglia reflecting neuronal loss and gliosis due to iron deposition and ferritin accumulation can be seen on T2-weighted images. In our case, hypointense lesions on T2* images indicative of widespread abnormal iron deposition were observed in theglobus pallidus, putamen, caudate, thalamus, red nucleus, dentate nucleus, and motor cortex. In addition, T2-weighted imaging revealed mild cerebral and cerebellar cortical atrophy and symmetrical hyperintense areas surrounded by hypointense areas in the bilateral posterior globus pallidus and putamen. The present individual has not revealed the bilateral large cavities observed in advanced-stage disease. And interestingly, in our case, brain MRI showed the hypointensity in the basal ganglia on T2-weighted MRI images before symptom onset (Fig. 1B). A previous study of young carriers of the c.460InsA mutation (6–36 years old), showed the presence of hypointense signals on T2*-weighted images indicative of iron deposition (Keogh, 2012), strongly suggesting that iron deposition may actually begin several decades before the appearance of clinical symptoms. Coronal T2*-weighted images showed a combination of a hypointense rim and hyperintense center in the basal ganglia called the “eye of the tiger” sign, which reflects iron deposition in the basal ganglia on both T2- and T2*-weighted MRI. This sign has been considered to be pathognomonic of PKAN, but it has also been observed in other neurodegenerative diseases such as cortical–basal ganglionic degeneration, multiple system atrophy, progressive supranuclear palsy, pure akinesia with gait freezing (Erro, 2011), and neuroferritinopathy (McNeill, 2008). These studies point out a substantial degree of overlap between neuroferritinopathy and other NBIA diseases in terms of presentation on T2*-weighted images and SWI. Thus, the diagnosis of neuroferritinopathy may not be made solely on the basis of neuroimaging. Brain perfusion studies in the proband (IMP-SPECT) showed asymmetric hypoperfusion of the cerebellum and the basal ganglia with predominance on the right side. This finding is likely to reflect the laterality of neurological symptoms.
Low serum ferritin levels were initially reported in individuals with the c.460InsA mutation (Curtis, 2001); however, low serum ferritin levels are not always observed in individuals with neuroferritinopathy (Curtis, 2001; Wills, 2002; Mancuso, 2005; Mir, 2005; Chinnery, 2007; Ohta, 2008; Ory-Magne, 2009; Devos, 2009; Ondo, 2010; Storti, 2013). Serum ferritin levels in our premenopausal female patient were 20 ng/mL (normal range, 5–204 ng/mL), and thus not found to be significantly decreased compared to normal range. Interestingly, CSF ferritin levels in our patient (<1.00 ng/mL) were significantly lower than normal levels in control individuals (6.68 ± 0.93 ng/mL), and also lower than ferritin levels in patients with restless leg syndrome (below 3.5 to 4.1ng/mL), a disease associated with lower CSF ferritin levels (Earley, 2000; Mizuno, 2005). Further studies will demonstrate whether the measurement of CSF ferritin levels could be used as a novel biomarker for neuroferritinopathy.
Patients carrying the mutation c.469_484dupGGCCCGGAGGCTGGGC have been described in Japan (Ohta, 2008) and Italy (Storti, 2013). This mutation is similar to the one presented here, with one nucleotide difference that causes a single amino acid change between both mutant ferritins at codon 162 (Fig. 5C); however, we have observed significant degree of variability in the presentation of the disease between the affected individuals from the Japanese family (tremor was the main characteristic symptom) (Ohta, 2008), the Italian patient (behavioral changes and tremor) (Storti, 2013) and our case. This difference may not be explained at the protein level, since the general structure of the spherical protein shell seems to be maintained in mutant ferritin as was seen by X-ray crystallography (Baraibar, 2010). All known mutations associated with the disease may have a similar effect: the disruption of the 4-fold pores, making the pores unstable and leaky, and the gradual aggregation of ferritin and iron into a precipitate mediated by the unraveled and extended mutant C-termini (reviewed by Muhoberac, 2013). Further research will be needed to understanding the effect of amino acid variations between mutant C-terminal sequences in the pathogenic process of the disease and the role of genetic and environmental factors in the disease.
In conclusion, our findings expand the genetic and phenotypic diversity of neuroferritinopathy. The observed clinical features of the disease in this family further imply the importance of testing for FTL mutations in patients with distinctive MRI findings. Further studies will determine the clinical significance of low levels of CSF ferritin and the potential value of determining CSF ferritin levels for the diagnosis of the disease.
Highlights.
We provide novel evidence of the genetic and phenotypic diversity of neuroferritinopathy
Discovered a novel FTL mutation associated with neuroferritinopathy
We presented data on the atypical presentation of the disease
We propose CSF ferritin levels as a novel biochemical marker for diagnosis
Acknowledgments
We gratefully acknowledge Dr. Emiko Ohta and Dr. Yamawaki Takemori for useful discussions as well as Dr. Hiroshi Ichinose for analysis of the PANK2 gene. Dr. Suketaka Momoshima gave us insightful comments of neuroimaging analysis. We also acknowledge Sachiko Imai for technical help.
Footnotes
Conflict of Interest: None
Full Financial Disclosures of all Authors
This study was supported by grants from the National Institute on Neurological Disorders and Stroke NS050227 and NS063056 (RV), and by grants for Research on Measures for Intractable Diseases (H24-nanchi-ippan-063) and the Comprehensive Brain Science Network (MT).
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baraibar MA, et al. Unraveling of the E helices and disruption of 4-fold pores are associated with iron mishandling in a mutant ferritin causing neurodegeneration. J Biol Chem. 2010;285:1950–1956. doi: 10.1074/jbc.M109.042986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassidy AJ, et al. The man who could not walk backward: an unusual presentation of neuroferritinopathy. Mov Disord; 2011;26:362–364. doi: 10.1002/mds.23415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, et al. Neuroferritinopathy in a French family with late onset dominant dystonia. J Med Genet. 2003;40:e69. doi: 10.1136/jmg.40.5.e69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnery PF, et al. Clinical features and natural history of neuroferritinopathy caused by the FTL1 460InsA mutation. Brain; 2007;130:110–119. doi: 10.1093/brain/awl319. [DOI] [PubMed] [Google Scholar]
- Crompton DE, et al. Spectrum of movement disorders in neuroferritinopathy. Mov Disord; 2005;20:95–99. doi: 10.1002/mds.20284. [DOI] [PubMed] [Google Scholar]
- Curtis AR, et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat Genet; 2001;28:350–354. doi: 10.1038/ng571. [DOI] [PubMed] [Google Scholar]
- Devos D, et al. Clinical features and natural history of neuroferritinopathy caused by the 458dupA FTL mutation. Brain. 2009;132:e109. doi: 10.1093/brain/awn274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley CJ, et al. Abnormalities in CSF concentrations of ferritin and transferrin in restless legs syndrome. Neurology; 2000;54:1698–1700. doi: 10.1212/wnl.54.8.1698. [DOI] [PubMed] [Google Scholar]
- Erro R, et al. The “eye of the tiger” sign in pure akinesia with gait freezing. Neurol Sci; 2011;32:703–705. doi: 10.1007/s10072-011-0589-1. [DOI] [PubMed] [Google Scholar]
- Keogh MJ, et al. Neuroferritinopathy: a new inborn error of iron metabolism. Neurogenetics; 2012;13:93–96. doi: 10.1007/s10048-011-0310-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota A, et al. A novel ferritin light chain gene mutation in a Japanese family with neuroferritinopathy: description of clinical features and implications for genotype-phenotype correlations. Mov Disord; 2009;24:441–445. doi: 10.1002/mds.22435. [DOI] [PubMed] [Google Scholar]
- Lehn A, et al. Neuroferritinopathy. Parkinsonism Relat Disord; 2012;18:909–915. doi: 10.1016/j.parkreldis.2012.06.021. [DOI] [PubMed] [Google Scholar]
- McNeill A, et al. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology; 2008;70:1614–1619. doi: 10.1212/01.wnl.0000310985.40011.d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mir P, et al. Adult-onset generalized dystonia due to a mutation in the neuroferritinopathy gene. Mov Disord. 2005;20:243–245. doi: 10.1002/mds.20280. [DOI] [PubMed] [Google Scholar]
- Mizuno S, et al. CSF iron, ferritin and transferrin levels in restless legs syndrome. J Sleep Res; 2005;14:43–47. doi: 10.1111/j.1365-2869.2004.00403.x. [DOI] [PubMed] [Google Scholar]
- Mancuso M, et al. Hereditary ferritinopathy: a novel mutation, its cellular pathology, and pathogenetic insights. J Neuropathol Exp Neurol; 2005;64:280–294. doi: 10.1093/jnen/64.4.280. [DOI] [PubMed] [Google Scholar]
- Muhoberac BB, Vidal R. Abnormal iron homeostasis and neurodegeneration. Front Aging Neurosci. 2013;5:32. doi: 10.3389/fnagi.2013.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta E, et al. Neuroferritinopathy in a Japanese family with a duplication in the ferritin light chain gene. Neurology; 2008;70:1493–1494. doi: 10.1212/01.wnl.0000310428.74624.95. [DOI] [PubMed] [Google Scholar]
- Ohta E, Takiyama Y. MRI findings in neuroferritinopathy. Neurol Res Int. 2012:197438. doi: 10.1155/2012/197438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ondo WG, Adam OR, Jankovic J, Chinnery PF. Dramatic response of facial stereotype/tic to tetrabenazine in the first reported cases of neuroferritinopathy in the United States. Mov Disord; 2010;25:2470–2472. doi: 10.1002/mds.23299. [DOI] [PubMed] [Google Scholar]
- Ory-Magne F, et al. Clinical phenotype and neuroimaging findings in a French family with hereditary ferritinopathy (FTL498-499InsTC) Mov Disord; 2009;24:1676–1683. doi: 10.1002/mds.22669. [DOI] [PubMed] [Google Scholar]
- Storti E, et al. De novo FTL mutation: A clinical, neuroimaging, and molecular study. Mov Disord; 2013;28:252–253. doi: 10.1002/mds.25275. [DOI] [PubMed] [Google Scholar]
- Vidal R, et al. Intracellular ferritin accumulation in neural and extraneural tissue characterizes a neurodegenerative disease associated with a mutation in the ferritin light polypeptide gene. J Neuropathol Exp Neurol; 2004;63:363–380. doi: 10.1093/jnen/63.4.363. [DOI] [PubMed] [Google Scholar]
- Vidal R, et al. Hereditary Ferritinopathies. In: Dickson Dennis W, Weller Roy O., editors. Neurodegeneration: The Molecular Pathology of Dementia and Movement Disorders. 2. Blackwell Publishing Ltd; 2011. pp. 461–466. [Google Scholar]
- Wills AJ, et al. Palatal tremor and cognitive decline in neuroferritinopathy. J Neurol Neurosurg Psychiatry; 2002;73:91–92. doi: 10.1136/jnnp.73.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]