
Fig. 1.
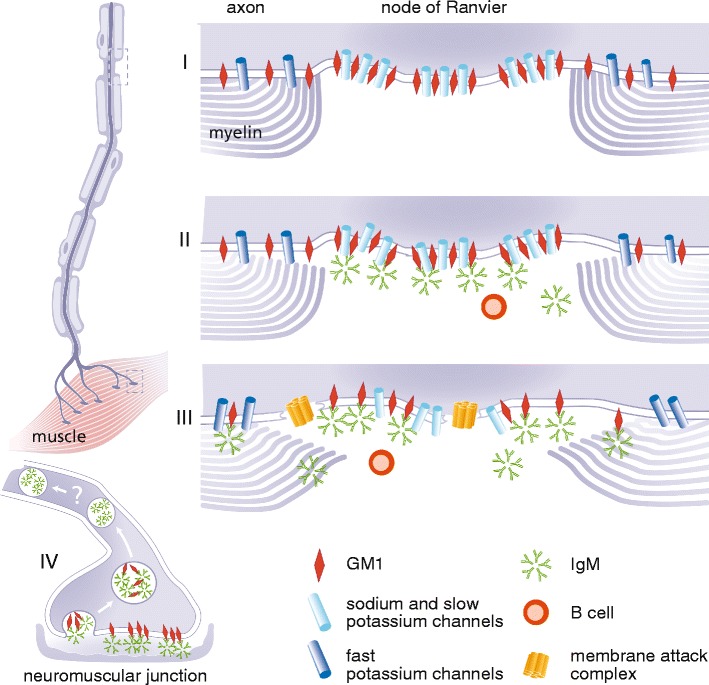
Schematic model of putative disease mechanisms in MMN. Anti-GM1 IgM antibodies may trigger direct and complement dependent damage to axons. In the normal physiological situation the node of Ranvier is characterized by clusters of ion channels, held together by GM1 and other lipids in so called lipid rafts (I). These voltage gated sodium and slow voltage gated potassium channels, together with fast voltage gated potassium channels in the paranodal region, maintain normal saltatory conduction. Paranodal myelin is attached to the axon by GM1. Activated B cells (plasma cells) produce the pentameric IgM antibodies that bind to GM1, possibly to heteromeric complexes containing GM1, cholesterol and galactocerebroside (not depicted in this figure) (II). The binding of these anti-GM1 antibodies can lead to the first signs of demyelination and possible dysfunction of the voltage gated sodium and slow voltage gated potassium channels. Once there is binding of anti-GM1 antibodies to GM1 the classical complement pathway is activated, and deposition of complement factors such as membrane attack complex (MAC) can take place (III). While focal demyelination continues, deposition of MAC may lead to further disruption of the Schwann-cell-axolemma junctions, displacement of ion-channel clustering and disturb membrane integrity at the (para)nodal region. Loss of fast voltage gated potassium channels through severe demyelination in the paranodal region can lead to leakage of potassium and subsequent hyperpolarization. At the site of the neuromuscular junction (NMJ) (IV), anti-ganglioside antibodies are rapidly internalised after binding, thus preventing the activation and deposition of complement factors. It is as of yet unknown whether retrograde transportation into the proximal part of the axon plays a role in the pathogenesis of MMN