

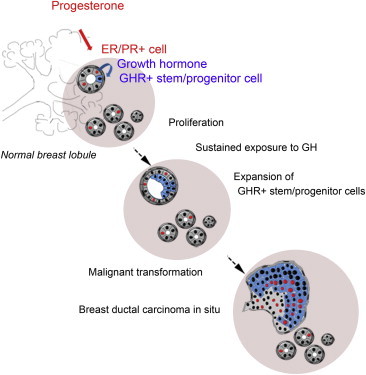
Summary
Using in vitro and in vivo experimental systems and in situ analysis, we show that growth hormone (GH) is secreted locally by normal human mammary epithelial cells upon progesterone stimulation. GH increases proliferation of a subset of cells that express growth hormone receptor (GHR) and have functional properties of stem and early progenitor cells. In 72% of ductal carcinoma in situ lesions, an expansion of the cell population that expresses GHR was observed, suggesting that GH signaling may contribute to breast cancer development.
Graphical Abstract
Highlights
-
•
Growth hormone is produced by normal breast epithelium upon progesterone stimulation
-
•
Stem/progenitor cells express GHR and proliferate in response to GH stimulation
-
•
Expansion of GHR-positive cells is seen in precursor lesions of breast malignancies
Growth hormone (GH) is secreted locally by normal human mammary epithelial cells upon progesterone stimulation. GH signaling increases proliferation of mammary stem and progenitor cells. Analysis of ductal carcinoma in situ lesions shows an expansion of the cell population that expresses GH receptor, suggesting that GH signaling may contribute to breast cancer development.
Introduction
Mammary gland development is controlled by the endocrine system, in particular by the ovarian steroid hormones, estrogen and progesterone, and by the pituitary hormones, growth hormone (GH) and prolactin. Studies in animal models showed that GH deficiency impairs mammary gland development. Spontaneous dwarf rats, which bear a loss-of-function mutation in GH, have deficient alveolar development that can be rescued by GH reinfusion (Swanson and Unterman, 2002). Ghr knockout (KO) mice have retarded duct development and limited side branching (Bocchinfuso and Korach, 1997; Zhou et al., 1997). In humans, mutations affecting the expression and function of the GH receptor (GHR) are collectively known as Laron syndrome (LS). Similar to Ghr KO mice, these patients have short stature and reduced body weight (Laron and Klinger, 1994). Mammary gland development is affected but can support normal lactation.
Sustained exposure to steroid hormones constitutes one of the best established factors of risk for breast cancer (Russo and Russo, 2006). There is compelling evidence, from both animal work and epidemiological studies, that elevated levels of GH also increase the risk of breast cancer (De Stavola et al., 2004; Gunnell et al., 2001). The incidence of cancers is higher in patients with acromegaly, a condition associated with hypersecretion of GH (Jenkins, 2004; Perry et al., 2008; van Garderen and Schalken, 2002; Waters and Barclay, 2007), and in individuals with taller height (Ahlgren et al., 2004; Green et al., 2011; De Stavola et al., 2004; Gunnell et al., 2001). Conversely, no cancers have been diagnosed so far in patients with LS (two cohorts studied, of 169 and 230 patients), although they have a higher longevity than the general population (Laron, 2008). Their blood relatives had an incidence of cancers of 24%.
There is evidence that GH can be secreted by breast cancer cells (Chiesa et al., 2011; Raccurt et al., 2002). Studies from Lobie’s group have reported that autocrine GH signaling in MCF7 cells confers a mesenchymal, invasive phenotype in vitro and generates more aggressive tumors in vivo (Mukhina et al., 2004). Although the molecular mechanisms underlying steroid hormones and GH signaling have been elucidated in studies spanning decades of research, it is still poorly understood how exposure to these hormones increases risk of breast cancer.
In this study, we utilized a combination of in vitro and in vivo functional assays and in situ analysis of normal breast epithelium to show that GH selectively exerts its effects on normal mammary stem/progenitor cells. We demonstrated that GHR is expressed in a distinct subpopulation of cells with phenotypic and functional properties of stem and early progenitor cells. We also showed that a subpopulation of breast epithelial cells produces GH upon progestin stimulation. GH/GHR signaling increases proliferation of mammary stem and progenitor cells. We speculate that sustained GH stimulation, linked to sustained progesterone stimulation, can increase the risk of malignant transformation by expanding the stem/progenitor cell population and increasing their proliferation rate. Consistent with this concept, we found that 90% of ductal carcinoma in situ (DCIS) lesions have a GHR+ cell population detectable by immunohistochemistry (IHC). In 72% of DCIS, the GHR+ cell population is expanded compared to normal tissue. We also showed that inhibition of GH signaling halts the growth of a patient-derived breast cancer xenografted in immunodeficient mice.
Results
GHR Is Present in a Subset of Normal Human Breast Epithelium Cells that Express Stem Cell Markers and Lack Lineage Differentiation Markers
GHR Is Expressed in the Normal Human Mammary Epithelium
We performed immunofluorescent (IF) staining for GHR on normal human breast sections (aesthetic mammoplasty samples). GHR was detected in all samples analyzed, originating from eight patients. The vast majority of GHR+ cells in the epithelium were present in cell clusters, and a small minority were present as scattered, isolated cells (Figures 1A–1C). GHR+ cells were present in 1.2%–5% of mammary epithelial cells (four patients, three paraffin blocks/ sample, 4,359 ± 2,555 average number cells analyzed/sample). We utilized flow cytometry analysis for a more sensitive and quantitative assessment and found that GHR was expressed in 3.5%–19% of normal breast epithelial cells (mean = 9.7 ± 6.27 SD, n = 6) (staining controls are shown in Figures S1A–S1F available online).
Figure 1.
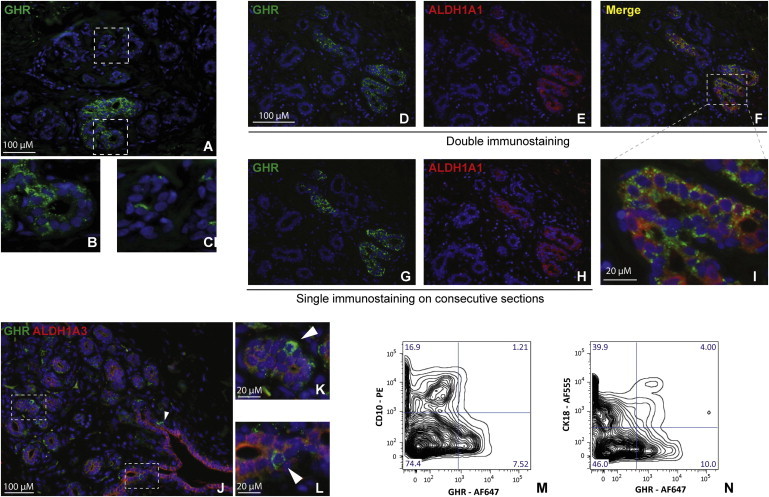
GHR Is Expressed in the Human Mammary Gland Epithelium
(A–C) Immunostaining of normal breast epithelium shows that GHR is expressed mostly in clusters and rarely as scattered cells. Representative images of eight samples.
(D–F) Double IF of normal breast epithelium shows that GHR is expressed mostly in ALDH1A1+ cells or in their immediate proximity.
(G and H) Single IF for GHR and ALDH1A1 in consecutive normal breast sections.
(I) Detail of (E), showing ALDH1A1+/GHR+ cells.
(J–L) Double IF for ALDH1A3 and GHR shows isolated GHR+ cells adjacent to ALDH1A3+ cells (arrowheads). Representative images of five samples.
(M) Flow cytometry for GHR and CD10 showing no overlap between cells expressing these markers. Representative data from two different samples.
(N) Flow cytometry for GHR and CK18 showing a small overlap (4%) between these markers. Representative data from two different samples.
GHR Colocalizes with Stem/Progenitor Cell Markers
In previous global profiling studies, we found that GHR transcript was 6-fold upregulated in progenitor versus more differentiated cells in normal breast epithelium (Dontu et al., 2003). We enquired now if GHR protein was present in breast stem/progenitor cells. Two distinct sets of markers were found to be associated with the highest proliferation potential and the broadest differentiation potential of breast cells in vitro and in vivo: aldehyde dehydrogenase (ALDH) activity, as determined by the ALDEFLUOR assay, and CD49high/EpCAM−/lineage− cells, as determined by flow cytometry (Ginestier et al., 2007; Lim et al., 2009). We identified the equivalent of these cell populations in situ, by IF staining for ALDH1A1 and ALDH1A3 and double IF staining for CD49f and EpCAM. The combination of ALDH1A1 and ALDH1A3 isoforms identifies normal breast epithelial cells with ALDH activity (Honeth et al., 2014). ALDH1A1+ and ALDH1A3+ cells were present primarily as rare clusters of cells and infrequently as scattered, isolated cells (Figures 1D–1L; Figures S2A–S2H). Double IF staining showed that GHR was expressed mainly in ALDH1A1+ cells or in their immediate proximity (75% of GHR+ cell clusters overlapped with ALDH1A1+ areas) (Figures 1D–1I; Figures S2A–S2F). Single staining of consecutive sections confirmed these conclusions (Figures 1G and 1H; Figures S2E and S2F). GHR+ cells were also found adjacent to ALDH1A3+ areas (Figures 1J–1L; Figures S2G and S2H). More than half of GHR+ cells (56%) present as isolated cells or small cell clusters were adjacent to ALDH1A3+ cells. Given the small size of these cell populations (all less than 10% average), the probability of this overlap/juxtaposition to be random is low (p < 0.0001, Fisher’s exact test). For quantitative assessment, we used ALDEFLUOR and fluorescence-activated cell sorting (FACS) to separate ALDH+ and ALDH− cells from normal breast epithelium. Subsequent GHR staining and analysis of sorted cells showed that approximately two-thirds of the ALDH+ cells were GHR+ (mean 59.3 ± 7.0 SD, n = 3) (Figure S2I). GHR+ cells were also found adjacent and/or overlapping with CD49f+/EpCAM− cells (stem/progenitor cells), as well as with CD49f+/EpCAM+ cells (luminal progenitor cells) (Figures S2J–S2Q) (Lim et al., 2009). Flow cytometry using CD49f, EpCAM, and GHR showed the presence of CD49f+EpCAM-GHR+ and EpCAM+GHR+ cells, consistent with the results of in situ analysis (Figures S2R–S2T).
GHR+ Cells Do Not Express Markers of Lineage Differentiation
In order to determine if GHR+ cells associate with a specific cell lineage, we performed double IF staining for GHR and CD10 or cytokeratin 18 (CK18) on single cell suspension from dissociated mammoplasty tissue. CD10 and CK18 are present in the vast majority of myoepithelial and luminal cells, respectively, that form the two main layers of the mammary tree duct. Flow cytometry analysis did not detect CD10+ cells within the GHR+ cell population (Figure 1M). A subpopulation of CK18+ cells was detected in the GHR+ cell population (approximately 0.4% of the total epithelial population) (Figure 1N). Analysis of sections immunostained for GHR and lineage markers showed similar results. The majority of the GHR-expressing cells did not express EpCAM, CK18, CD10, or smooth muscle actin (SMA). This latter marker is associated with myoepithelial cells. We found only very rare, isolated GHR+ cells expressing EpCAM, CK18, or CD10. We did not find any SMA+ cells that expressed GHR (Figures S3A–S3F). We detected GHR colocalizing with CK14 and CK5 in cells of the intralobular epithelium, but not those of larger ducts (Figures S3G–S3J). These cytokeratins are associated with progenitor cells (Villadsen et al., 2007).
GHR+ Cells Have Functional Properties of Stem/Progenitor Cells
GHR+ Cells from the Normal Breast Epithelium Can Form Mammospheres
In order to assess the functional potential of GHR+ cells, we separated GHR+ and GHR− normal human mammary epithelial cells (HMECs) from mammoplasty tissue by FACS and assessed their ability to form mammospheres (Figures 2A and 2B). Several groups have demonstrated that mammosphere-initiating cells from both mouse and human mammary gland have the highest ability to repopulate the cleared mammary fat pad in vivo (as low as ten cells generate outgrowths [Cicalese et al., 2009; Pece et al., 2010]). We found that only GHR+ cells generated mammospheres, whereas GHR− cells failed to do so (Figure 2B). The efficiency of mammosphere formation was constant over three passages in vitro (data not shown).
Figure 2.
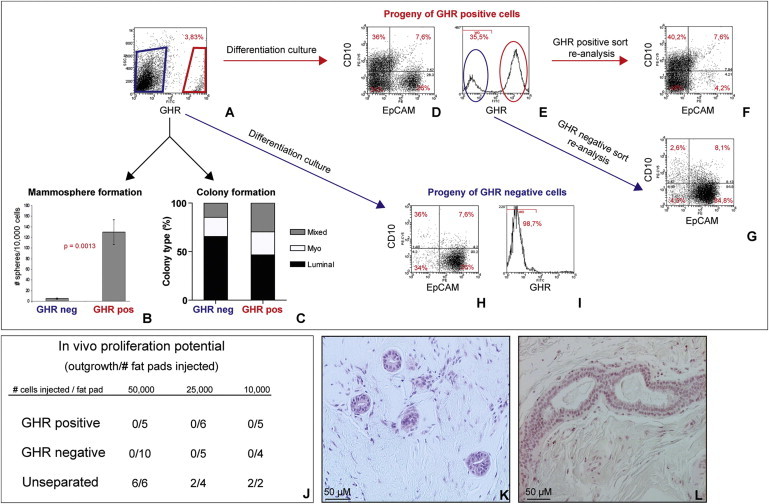
Functional Potential of GHR+ Cells In Vitro and In Vivo
(A) Primary HMECs were stained for GHR and then separated by FACS into GHR+ and GHR− cells.
(B) GHR+ cells generated mammospheres in suspension culture, whereas GHR− cells did not (five independent experiments on different patient samples, each performed in triplicate, n = 5).
(C) Colony-forming ability of sorted GHR+ and GHR− populations. The GHR+ population gave rise to more colonies with mixed phenotype and less pure luminal colonies compared to the GHR− population (representative example from three independent experiments, on two different patient samples, each performed in triplicate, n = 3).
(D–I) Lineage differentiation potential of the progeny derived from GHR+ (D–E) and GHR− cells (H–I), analyzed by flow cytometry. Progeny of GHR+ cells comprised of myoepithelial cells (CD10+) (D), luminal cells (EpCAM+), and EpCAM−/CD10− cells (E). GHR+ cells generated both GHR+ (red circle) and GHR− (blue circle) cells. Progeny of GHR− cells were predominantly EpCAM+ and CD10− (H), and they were comprised of only GHR− cells (I). (F–G) After 10 days in culture the progeny of GHR+ cells was sorted in GHR+ and GHR− cells and analyzed by flow cytometry. The GHR+ population comprised myoepithelial cells (CD10+) and EpCAM−/CD10− cells (F). A majority of GHR− cells expressed the luminal marker EpCAM (G) (three independent experiments on different patients samples, n = 3).
(J) Ability of GHR+, GHR−, or unseparated cells (sorted on viability alone) to generate outgrowths in vivo in humanized fat pads of NOD/SCID mice.
(K–L) Representative images of outgrowths generated in vivo by unseparated breast epithelial cells.
GHR+ Mammary Epithelial Cells Can Differentiate along Luminal and Myoepithelial Lineages
In order to assess the differentiation potential of GHR+ and GHR− cells, FACS-separated cells were plated at clonogenic densities, in differentiating conditions. Colonies were assessed for expression of lineage differentiation markers, SMA and CK18. There was a higher representation of progenitor cells with bipotent and myoepithelial differentiation potential in the GHR+ cell population compared to the GHR− cell population (Figure 2C). The sorted CD49f+EpCAM+GHR+ cells generated predominantly luminal colonies (90%) and CD49f-EpCAM+GHR+ cells generated mixed, luminal, and myoepithelial colonies (data not shown). Additionally, we plated GHR+ and GHR− sorted cells onto a collagen substratum and analyzed the expression of lineage markers in the entire population of progeny cells, after 10 days in culture. As shown in Figure 2D, GHR+ cells generated single positive CD10+ and EpCAM+ cells, as well as double-positive and double-negative cells, for these markers. This cell progeny included both GHR+ and GHR− cells (Figure 2E). Analysis of resorted cell populations showed that the GHR+ progeny was EpCAM-CD10+ or EpCAM-CD10− (Figure 2F), whereas the majority of GHR− progeny were EpCAM+CD10− (Figure 2G). GHR− cells generated predominantly luminal EpCAM+/CD10− cells and only GHR− progeny (Figures 2H and 2I). Collectively, these data show that GHR+ cells have a broader differentiation potential compared to GHR− cells.
Potential of GHR+ Cells to Generate Outgrowths In Vivo
To test the functional potential of GHR+ cells in vivo, we separated GHR+ and GHR− cells by FACS. Cells sorted on viability alone (unseparated cells) were used as controls. We implanted cells from these three subpopulations into the humanized cleared mammary fat pads of NOD/scid mice, as previously described (Ginestier et al., 2007; Kuperwasser et al., 2004). Outgrowths were only observed in the fat pads implanted with unseparated cells (Figures 2J–2L). These results, consistent in five mammoplasty samples, suggest that paracrine or juxtacrine signaling between GHR+ and GHR− cell populations may be important for proliferation of xenotransplanted cells. This conclusion was supported by experiments in which GHR+ and GHR− separated cells were mixed and implanted together in the fat pad (total of 50,000 cells/fat pad), which resulted in outgrowths in all implantations. Paracrine communication between GHR+ and GHR− cells are likely to involve additional signals beside GH, because treatment with GH antagonist or knockdown (kd) of GHR did not affect proliferation of mammary cells in suspension culture (data shown below). Moreover, although Ghr KO in vivo impairs mammary gland development, it does not abolish it (Laron and Klinger, 1994; Zhou et al., 1997). Alternatively, these results may reflect conditions specific to xenotransplantation that do not entirely recapitulate the physiological human breast environment.
GH/GHR Activation Increases Proliferation of Mammary/Stem Progenitor Cells
GH Treatment Increases Mammosphere Formation
To determine the functional significance of GHR expression in normal mammary stem/progenitor cells, we assessed the effect of GH treatment on mammosphere formation. Human recombinant GH was used at doses of 3 ng, 10 ng, 30 ng, 100 ng, and 1 μg/ml, corresponding to physiological serum levels of GH in adulthood, in childhood, and in pathologic conditions, such as acromegaly and pituitary tumors, respectively (Corneli et al., 2007). GH dose was optimized in MCF10A cells. Increased levels of phosphorylated Stat5, a known downstream target of GHR, were observed upon treatment with doses of GH that increased mammosphere formation (Figure S4A). MCF10A cells are normal-like mammary epithelial cells that express both GHR and GH (Figures S4B and S4C). The effect of GH and GHA treatment on proliferation of MCF10A cells as spheres was similar to that seen in normal mammary epithelial cells (Figure S4D).
In HMEC cultures, we observed a dose-dependent increase in mammosphere formation upon treatment with GH. Concomitant treatment with GH antagonist (GHA) abolished this effect (Figure 3A). Increased sphere formation was maintained in secondary and tertiary passages, when GH treatment was applied in primary culture only (10 ng/ml) (Figure 3B). The cell composition of these spheres was similar in all passages, indicating that GH treatment did not favor differentiation along a particular lineage (Figure 3C). Because GH can also activate the prolactin receptor, we investigated if the effect on mammosphere formation was dependent on GHR. To this end, we knocked down GHR in HMECs. GHR kd cells formed spheres with similar frequency as control cells but failed to respond to GH stimulation. GH treatment increased sphere formation in cells infected with nonsilencing small hairpin RNA (shRNA) (Figures 3D and S4E). Analysis of PKH26 dye retention, performed on mammosphere-derived cells, indicated that treatment with GH did not change the ratios of the PKH26 high, medium, and low cells, which correspond to stem, early progenitor, and late progenitor cells (Pece et al., 2010) (Figure S4F).
Figure 3.
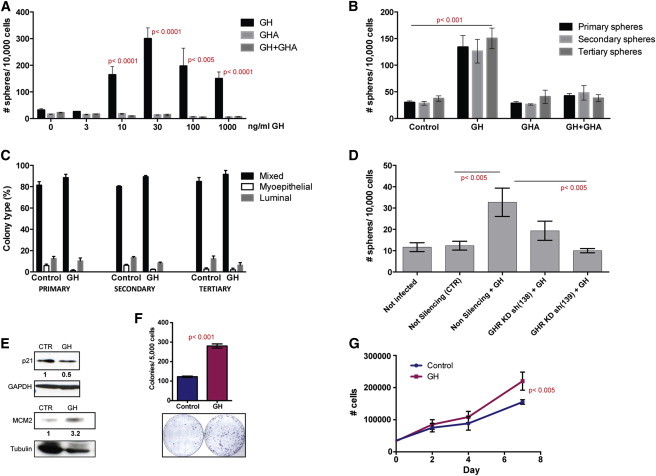
GH/GHR Signaling Is Active in Mammary Progenitor Cells
(A) A dose-dependent increase in mammosphere formation was observed upon treatment of primary HMECs with human recombinant GH. GHA abolished this effect, but did not have an effect when used alone (five independent experiments in different patient samples, each performed in triplicate, n = 5).
(B) Increased sphere formation was maintained in subsequent passages, although treatment (GH 10 ng/ml, GHA 100 ng/ml, GH+GHA) was applied only in primary culture (representative example from three independent experiments on different patient samples, each performed in triplicate, n = 3).
(C) GH treatment did not change the differentiation potential of cells composing the mammospheres. Primary, secondary, and tertiary mammospheres were dissociated, and single cells suspensions were plated at clonogenic densities on a collagen substratum in the presence of serum. After 12 days, colonies were immunostained with lineage markers (EpCAM and CD10 or CK18 and CK14). Colonies were scored based on cell composition, and percentages are shown for GH-treated and control samples (p > 0.05 for all three passages and types of colonies) (three independent experiments on two patient samples, each performed in triplicate, n = 3).
(D) Knockdown (kd) of GHR in primary HMECs did not affect sphere formation. In GHR kd cell cultures, mammosphere formation was not changed by GH treatment (representative example from four independent experiments on three patient samples, each performed in triplicate, n = 3).
(E) WB analysis showed increased level of proliferation marker MCM2 and decreased level of quiescence marker p21 after GH treatment (10 ng/ml).
(F) GH treatment (10 ng/ml) increased more than 2-fold clonogenicity of primary HMECs in adherent culture (representative example from four independent experiments on different patient samples, each performed in triplicate, n = 3).
(G) Proliferation of primary HMECs grown in adherent conditions was increased in GH-treated cultures compared to control (representative example from three independent experiments, on different patient samples, each performed in triplicate, n = 3).
Data are presented as means ± SD.
The effect on sphere formation suggested that GH may stimulate stem/progenitors to enter the cell cycle. Indeed, western blot (WB) analysis showed a decreased level of p21 (a marker associated with quiescence) and an increased level of MCM2 (a marker associated with dividing cells) in mammosphere-derived cells treated with GH compared to control (Figure 3E).
GH Treatment Increases Clonogenicity of Normal Mammary Epithelial Cells in Adherent Culture
We assessed the effect of GH treatment (10 ng/ml) on proliferation of HMECs in adherent conditions, at high- or low- (clonogenic) density plating. GH treatment increased clonogenicity 2-fold, an effect similar to that seen in suspension culture (Figure 3F). The increase in proliferation in high-density culture was more modest but statistically significant (Figures 3G and S4G). These findings support the conclusion that GH treatment increases the number of cycling stem/progenitor cells, but not that of more differentiated progenitors, that represent the majority of proliferating cells in adherent culture. This is consistent with the higher representation of GHR+ progenitor cells in mammospheres, compared to adherent culture (Figures S4D–S4G).
Progestins Induce GH Secretion by Normal Human Mammary Epithelium
Experiments in female dogs showed that GH is secreted in the normal mammary epithelium in vivo, upon treatment with progestins (Selman et al., 1994). We found that GH was also present in a subset of human mammary epithelial cells (Figures 4A and 4B). Double staining for GH and ALDH1A1 showed that GH is expressed in ALDH1A1+ areas or in their immediate proximity (Figures 4C, S5A, and S5B). The majority of cells secreting GH were not ALDH1A1+ however. A subpopulation of HMECs stained positive for GH in primary culture as well (Figure 4D).
Figure 4.
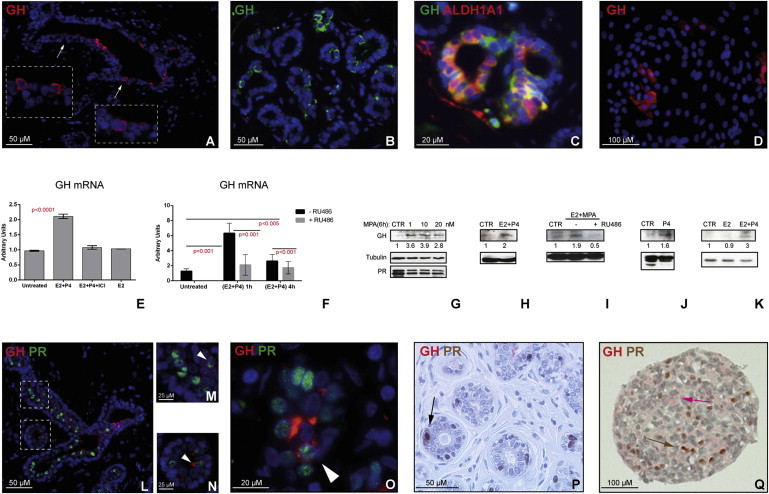
GH Is Secreted by Human Normal Mammary Epithelium, upon P4 or MPA Treatment Alone or in Combination with E2
(A–D) GH is secreted by primary HMECs in vivo and in vitro, as shown by immunostaining for GH in situ, in normal breast sections (A and B). Double staining for GH and ALDH1A1 on normal breast sections shows that GH is present in cells adjacent to ALDH1A1+ cells (C). GH is detected in vitro in HMECs grown in primary culture on a collagen substratum (D).
(E and F) Treatment with E2+P4 increases levels of GH mRNA in mammary epithelial cells grown as mammospheres (E) or on collagen substratum (F). Cells were treated with a combination of E2+P4, a combination of E2+P4+ICI182.780 (ER antagonist) or with E2 alone for 24 hr (representative example from four independent experiments on different patient samples, each performed in triplicate, n = 3) (E). Cells were treated with a combination of E2+P4 for 1 and 4 hr, in presence or absence of PR antagonist RU486 (representative example from three independent experiments on different patient samples, each performed in triplicate, n = 3) (F).
(G) WB analysis of GH protein levels in T47D cells treated with MPA (0, 1, 10, and 20 nM) for 6 hr was performed to assess dose effect. Treatment did not change PR protein levels.
(H–K) WB analysis of GH protein level in primary mammospheres treated with E2+P4 (H), E2+ MPA (I), or P4 alone (J). PR antagonist RU486 abolished the increase in GH level induced by P4 (I). Treatment with E2 alone did not increase GH level (K) (representative WB from experiments using four different mammoplasty samples are shown). Densitometry using ImageJ was performed for quantification. Intensity of bands was normalized against loading controls. Ratios between treated versus control samples are shown. Difference between GH levels in control cells and cells treated with P4 was statistically significant (p < 0.05), as well as cells treated with E2 and P4 (p < 0.005, two-way ANOVA).
(L–P) Double immunostaining for GH and PR on normal mammary epithelium shows rare PR+ cells secreting GH (M–P, arrowheads).
(Q) IHC for PR and GH on mammosphere sections.
Data are presented as means ± SD.
To test if progestins stimulate GH production, as is the case in dogs, we treated human mammosphere cultures with progesterone (P4) and medroxyprogesterone acetate (MPA), alone or in combination, and performed qRT-PCR and WB analysis for GH. (Henceforth “progestins” refers to synthetic compounds P4 and MPA, and progesterone refers to endogenous hormone.) We found a significant increase in GH mRNA and protein level in mammospheres treated with P4 or P4+estrogen (E2) (Figures 4E–4K). Doses of progestin treatment were optimized in T47D cells, which express high levels of PR (Figures 4G and S5C). Treatment with PR antagonist RU468 abolished these effects, even in combined treatment E2+P4 (Figures 4F and 4I). E2 alone did not have a significant effect (Figures 4E and 4K). Treatment with estrogen receptor alpha (ER) antagonist ICI 182.780 (ICI) decreased the effect of E2+P4 treatment, probably by decreasing the number of PR+ cells, capable of responding to P4 (Figure 4E). Similar results were obtained for treatment with MPA or P4+MPA (data not shown). In the mammary tissue of dogs, GH was detected in PR+ cells (Lantinga-van Leeuwen et al., 2000). Double IF for PR and GH in sections of normal breast epithelium showed rare GH+PR+ cells; the majority of GH+ cells were adjacent to PR+ cells (Figures 4L–4P). Similar results were observed in primary HMECs in adherent culture (Figures S5D and S5E) and in suspension culture (Figures 4Q, S5F, and S5G).
GH Is an Effector of Paracrine Signaling Initiated by Progesterone and Acting on Mammary Stem/Progenitor Cells
There is evidence that human normal mammary stem cells do not express ER and PR (Honeth et al., 2014; Lim et al., 2009). Double IF staining of normal breast sections showed that GHR+ cells were ER− and PR− (Figures 5A–5C). As GH can be secreted in response to progestins and acts on GHR+ cells, we hypothesized that GH/GHR activation is part of the paracrine communication that conveys proliferation signals initiated by steroid hormones to ER−/PR− mammary stem/progenitor cells.
Figure 5.
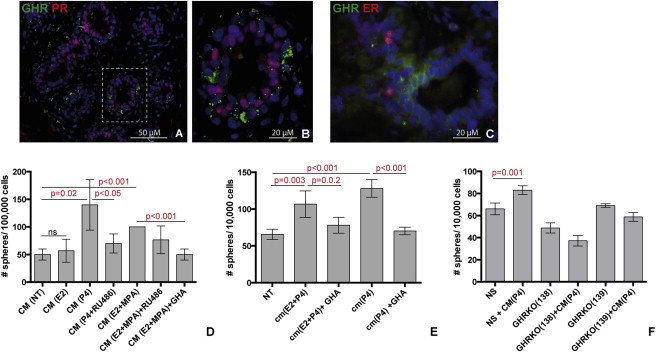
GH Is One of the Paracrine Signals that Increases Mammosphere Formation following Treatment with P4
(A–C) Double immunostaining for GHR with PR (A and B) or ER (C) on normal mammary epithelium shows no colocalization of GHR with either of these markers.
(D) Mammosphere formation was increased by treatment with conditioned medium (CM) from previous culture treated with MPA, P4, or E2+MPA, compared to control (CM from nontreated culture, NT). The increased mammosphere formation was inhibited by PR antagonist RU486 or by GHA. No effect was observed when conditioned medium from E2 alone was used (three independent experiments on different patient samples, each performed in triplicate, n = 3).
(E) Mammosphere formation was increased by treatment with conditioned medium (CM) from previous culture treated with P4 or E2+P4, compared to control (CM(NT)). The increase in mammosphere formation was inhibited by GHA (representative example form three independent experiments on different patient samples, each performed in triplicate, n = 3)
(F) Mammosphere formation was increased upon treatment with conditioned medium from P4-treated cultures CM (P4) in cultures of primary mammary epithelial cells infected with nonsilencing shRNA, but not in cultures of GHR kd cells (representative example from three independent experiments in different mammoplasty samples, each performed in triplicates, n = 3).
Data are presented as means ± SD.
To test this hypothesis, we utilized mammosphere cultures, which enrich in GHR-expressing stem/progenitor cells. Conditioned medium (CM) from mammosphere culture treated with progestins or progestins+E2 significantly increased mammosphere formation in new culture. The effect was specific to progestins because it was abolished by the PR antagonist RU468 (Figure 5D). GHA treatment significantly decreased sphere formation in these cultures (Figures 5D and 5E). The effect was dependent on GHR, because kd of GHR rendered mammary epithelial cells nonresponsive to stimulation with CM from P4-treated cultures (Figure 5F). Collectively, these data indicate that in the human mammary gland, GH secretion and GH/GHR activation are linked to steroid hormone stimulation. A role of GH in mediating paracrine effects initiated by progesterone and conveyed to an ER/PR− stem/progenitor cell population is also consistent with the results of in vivo experiments shown above, in which HMECs separated based on GHR expression loose ability to generate outgrowths upon xenotransplantation in NOD/scid mice.
GHR Defines a Cell Population that Is Expanded in DCIS Lesions
Our findings suggested that a higher rate of proliferation in the stem/progenitor cell compartment, induced by GH/GHR signaling, may account for the association between GH exposure and increased cancer risk. We hypothesized that an expanded GHR+ cell population may be associated with early stages of malignant transformation. DCIS is a recognized, although nonobligate, precursor of invasive breast cancer. We analyzed tissue microarrays (TMAs) from a series of 175 pure DCIS (no associated invasive carcinoma) samples from Guy’s and St. Thomas’s Breast Tissue and Data Bank. These patients presented between 1976 and 2004, with either symptomatic or screen-detected DCIS. In this cohort, 58% of the DCIS were of high cytonuclear grade; 34% were of intermediate grade; and 7% were of low grade. Twenty-eight percent of women were 50 years of age or less, and 74% of DCIS cases were ER+ (Allred score of three or more). Tumors were considered positive for GHR, where more than 10% cancer epithelial cells stained positive. We found that 90% of DCIS lesions had GHR+ cells ranging from 10% to 100% of the cell population, with 72% of cases having more than 20% GHR+ cells (the maximum seen in normal breast tissue by flow cytometry or IHC). Almost half of the lesions (47%) had ≥50% GHR+ cells (Figure 6). The intensity of immunostaining varied. A granular pattern, previously described for GHR staining and observed by us in normal tissue, was associated with both cytoplasmic and membrane localizations (Mertani et al., 1998). In addition, a more uniform cytoplasmic staining was observed in DCIS lesions (Figure 6). These data support the concept that an expansion of the GHR+ progenitor cell population is an early event associated with malignant transformation.
Figure 6.
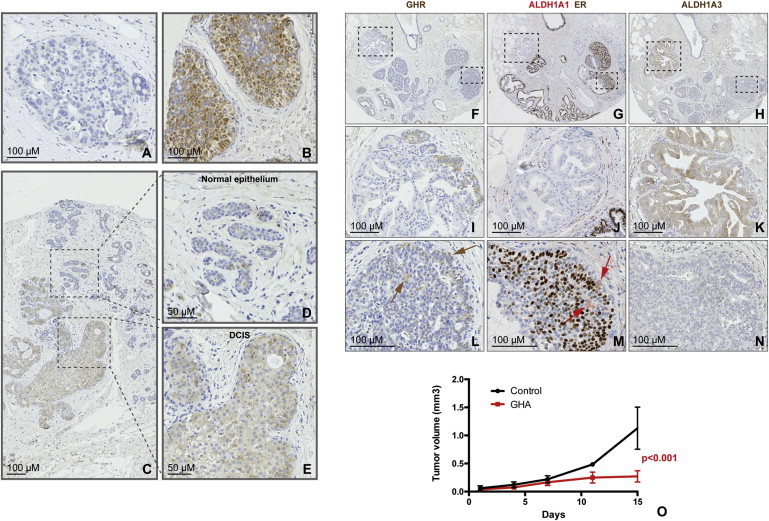
GHR Defines a Cell Population that Is Expanded in DCIS
(A and B) Representative images of GHR immunostaining of DCIS cores on TMAs. Examples of DCIS areas showing no expanded GHR+ population (A) or of positive areas with homogeneous high cytonuclear GHR expression (B).
(C–E) Expanded GHR+ cells population were found in DCIS areas (E) adjacent to areas of normal epithelium, with isolated GHR+ cells (D).
(F–N) Immunostaining for GHR, ALDH1A1/ER, and ALDH1A3 on consecutive sections of DCIS samples on TMAs. The same TMA core contains DCIS areas that are GHR−/ALDH1A1−/ER−/ALDH1A3+ (I–K), as well as areas that are GHR+/ALDH1A1+/ER+/ALDH1A3− (GHR and ALDH1A1 are present in a minority of cells, brown and red arrows, respectively) (L–N).
(O) Effects of GHR antagonist Pegvisomant on growth of established tumors. Patient-derived breast cancer xenografts, injected in the mammary fat pad of NOD/Scid mice, were used. Tumor growth was considerably reduced in treated animals compared to control (animals injected with vehicle alone, n = 5 animals per group).
In order to assess if a correlation between GHR expression and ALDH1A1 and/or ALDH1A3 is present in DCIS, as was the case in the normal tissue, we immunostained consecutive sections of the same TMAs for these markers. Samples were compared with respect to GHR and ALDH1A1 expression (75 patients) and GHR and ALDH1A3 expression (67 patients). ALDH1A1 and ER double IHC also allowed us to investigate correlation with ER. Among the analyzed samples, 24 (32%) were positive for ALDH1A1 (threshold 0) and 16 (24%) for ALDH1A3 (moderate or strong staining present). These criteria were established by the pathologist (SP). GHR-positive samples showed positive correlation with ER expression (more than 10% cells positive), by Fisher’s two-sided analysis (p = 0.02). Of the 24 samples that had detectable ALDH1A1+ cells, 19 were also positive for GHR, and of the 16 DCIS samples with moderate or strong ALDH1A3 staining, 11 were positive for GHR. There was no statistically significant correlation between GHR expression and either ALDH1A1 or ALDH1A3, possibly due to the high percentage of GHR-positive DCIS lesions and the relatively small number of samples available for comparison. ALDH1A1+ cells appeared to be overlapping or juxtaposed with GHR in both normal areas adjacent to the DCIS lesions and within the DCIS lesions (Figures 6F–6N; Figure S5). GHR+ cells appeared to be ER+ in the majority of cases, but not always. This latter observation, and the positive correlation of GHR and ER across patient samples, supports the hypothesis that autocrine and paracrine signaling involving GH and steroid hormones may be involved in breast cancer initiation and/or progression.
Inhibition of GH Signaling Significantly Reduces Breast Tumor Growth
It has been shown that growth of tumors generated by subcutaneously injecting the ER-positive breast cancer cell line MCF7 in immunodeficient mice is considerably reduced by treatment with the GHA Pegvisomant, a drug used in patients with acromegaly (Divisova et al., 2006). We investigated the effect of GHA treatment in an orthotopic xenograft model established from a patient-derived sample. Tumors were allowed to reach 4 mm in the largest diameter before treatment was initiated. Control animals were treated with vehicle alone. As shown in Figure 6O, tumor growth was considerably diminished in treated animals. This xenograft was established from a triple-negative breast cancer; therefore, inhibition of GH may be effective in this subtype of breast cancers as well.
Discussion
Initially, the role of GH in both normal breast development and tumorigenesis was linked to that of prolactin, because human GH activates both GHR and prolactin receptor. Subsequently, its role in insulin growth factor-1 (IGF-1) synthesis by the liver and the importance of the GH/IGF-1 axis in cancer initiation and progression were brought to the foreground (Kleinberg et al., 2009; Laban et al., 2003). More recently, a novel hypothesis was proposed, that autocrine/paracrine mechanisms employing locally secreted GH may promote growth in certain tumors, independent of IGF-1 signaling (Mukhina et al., 2004; Zhu et al., 2005). It has been also speculated that the association between GH and cancer risk is due to an effect on the stem/progenitor cell population (Green et al., 2011).
In this study we explored this latter hypothesis and investigated the effect of GH/GHR signaling on a subpopulation of mammary cells with functional properties of stem/progenitor cells. We detected GHR in normal breast epithelium in distinct clusters of cells, either overlapping with or in proximity of stem/progenitor cells (ALDH1A1+ or CD49f+/EpCAM− cells) or luminal progenitor cells (ALDH1A3+ or CD49f+/EpCAM+ cells). GHR+ cells did not express markers of lineage differentiation, ER, or PR. In functional assays, GHR+ cells displayed properties of stem and progenitor cells, being capable of initiating mammosphere formation and generating both luminal and myoepithelial lineage. GHR+ and GHR− cells did not proliferate in vivo when separated but were capable of generating outgrowths when recombined. These results suggest the existence of paracrine or juxtacrine communication between GHR+ and GHR− cells, necessary for proliferation in the mouse environment. Treatment of normal breast epithelial cells with recombinant GH in adherent and nonadherent primary cultures increased clonogenicity, an effect abolished by GHA inhibitor or by GHR kd. Upregulation of the proliferation marker MCM2 and downregulation of the cell-cycle inhibitor p21 accompanied these effects.
We also showed that GH can be secreted locally in the mammary gland, by a population of cells situated in proximity of PR+ cells. Treatment with progestins increased GH mRNA and protein levels in normal breast epithelial cells. Indirect evidence for a link between GH secretion and progesterone also comes from studies in human patients in which serum peaks of GH correlate with those of progesterone in 24 hr cycles or during the lutheal phase of the menstrual cycle (Caufriez et al., 2009, 2011).
Signaling through ER and PR is critical for mammary morphogenesis (Bocchinfuso and Korach, 1997; Sternlicht, 2006). Mouse mammary stem cells do not express ER or PR but respond to progesterone stimulation through indirect mechanisms, involving RANK-L, amphiregulin, and Wnt (Asselin-Labat et al., 2010; Beleut et al., 2010; Brisken et al., 2000; Joshi et al., 2010). There is evidence that human mammary stem/progenitor cells are ER−/PR− as well (Honeth et al., 2014; Lim et al., 2009). ALDH1A1+ cells and mammosphere-initiating cells of human origin, are also ER−, but can generate ER+/PR+ cells (Honeth et al., 2014). We showed here that treatment with progestins increased mammosphere formation through paracrine mechanisms mediated by GH. GHA treatment and GHR kd abolished this effect. We propose that part of the communication between PR+ mammary epithelial cells and GHR+ progenitor cells is mediated by GH.
We speculate that in vivo, serum GH of pituitary and placental origin may be responsible for the expansion of mammary stem/progenitor cells during developmental windows when the mammary gland grows isometrically (intrauterine life, childhood). In adulthood, when GH secretion from the pituitary gland decreases, this hormone is secreted locally in the mammary gland, in response to progesterone stimulation. This process may contribute to additional expansion of the stem/progenitor cell populations during the lutheal phase of the menstrual cycle and during pregnancy, when the mammary gland does not grow isometrically. In a process reminiscent of “niche formation,” ER− stem/progenitor cells generate sensory ER+ cells (Honeth et al., 2014). A paracrine signaling loop is initiated when estrogen peaks at the end of follicular phase and upregulates PR. Subsequently, progesterone peaks in the lutheal phase and activates PR molecular targets. We propose that GH is secreted as long as progesterone levels are high. Mitogenic signals are conveyed by GH to neighboring GHR+ stem/progenitor cells (Figures 7A and 7B). If these cycling progenitor cells divide further and generate differentiated cells, the ratio between stem/progenitor and differentiated cells will be maintained relatively constant. Unlike other tissues, however, proliferation of breast cells follows the cyclic fluctuation of ovarian hormones (Russo and Russo, 2006). The cumulative effect of prolonged exposure to steroid hormones and/or GH during many menstrual cycles would lead eventually to changed ratios between progenitor and differentiated cells, in favor of the former. An expanded undifferentiated cell population with a higher rate of proliferation would be at higher risk for transformation through oncogenic hits (Figure 7C).
Figure 7.
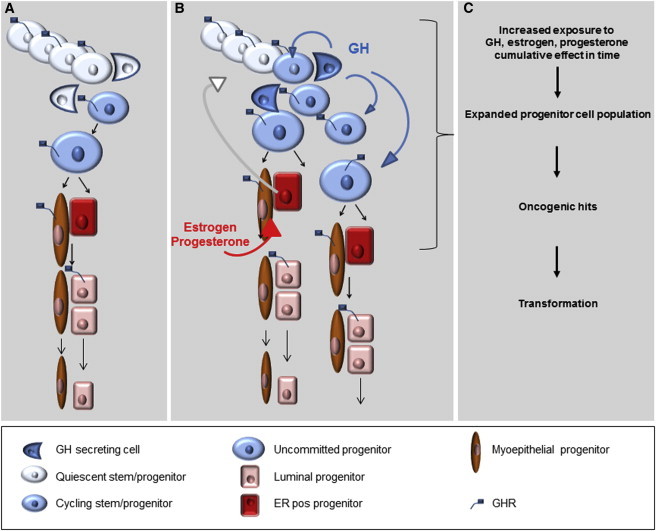
Effect of GH Stimulation on Normal Mammary Stem/Progenitor Cells and Implications for Oncogenic Transformation: A Model
(A) Normal mammary stem cells undergo asymmetric divisions, which generate a stem cell that goes into quiescence and a proliferating progenitor that gives rise to differentiated progenies. In the presence of GH, the daughter cell destined to become quiescent divides again, doubling the final total number of progenies.
(B) GH is part of an intercellular signaling loop triggered by E2, which drives expression of PR, followed by P4/PR signaling and local production of GH. This signaling loop is active as long as steroid hormones levels are elevated (puberty, middle of the menstrual cycle, pregnancy) and results in an increased number of stem/progenitor cells entering the cell cycle, through GH/GHR activation.
(C) Increased levels or prolonged exposure steroid hormones and/or GH would lead eventually to an expanded undifferentiated population at a higher risk for transformation through oncogenic hits.
Consistent with this model, our analysis of DCIS series showed the presence of an expanded GHR+ cell population in a majority of these lesions and a positive correlation between GHR and ER. Cellular colocalization of these two receptors in DCIS suggests that, upon malignant transformation, GHR+ cells may secrete GH and proliferate through autocrine stimulation.
Animals treated with GH develop hyperproliferative lesions in the mammary gland and have increased incidence of spontaneous mammary tumors (Törnell et al., 1992; van Garderen and Schalken, 2002; Waters and Barclay, 2007). Conversely, animals deficient in GH signaling, such as spontaneous dwarf rats or Ghr KO mice, are resistant to carcinogen-induced cancers, including mammary cancer (Swanson and Unterman, 2002). Blocking GH signaling in mice xenografted with the ER-positive breast cancer cell line MCF7 considerably reduced tumor size and prolonged latency of tumor formation (Divisova et al., 2006). In our study, we utilized an xenograft model based on orthotopic implantation of a triple-negative breast cancer propagated in vivo only. The tumorigenic cell population of this tumor is represented by cells with ALDH activity (Ginestier et al., 2007). In this model, a significant reduction in tumor growth was observed in animals treated with GHA, compared to controls.
Taken together, these findings indicate that exposure to elevated levels of GH may represent a cumulative factor of risk for breast cancer as a consequence of effects of this hormone on the breast stem/progenitor cell population. This may confer a risk similar to that represented by exposure to steroid hormones and may have clinical implications for breast cancer risk assessment, prevention, and treatment.
Experimental Procedures
Dissociation of Normal Breast Epithelium
Normal breast tissue was obtained from patients undergoing mammoplasty for aesthetic reasons, under protocols approved by the Institutional Review Board and by Guy’s Research Ethics Committee, in compliance with the Human Tissue Act. The tissue was processed as previously described (Ginestier et al., 2007).
Mammosphere Culture
Mammosphere culture and dissociation were performed as previously described (Dontu et al., 2003), at a density of 20,000 viable cells/ml in primary culture and 5,000 cells/ml in subsequent passages. Assessing mammosphere formation in cell fractions sorted by FACS was performed at 1,000 cells/ml. The medium did not contain B27 or phenol red.
Adherent Culture
HMECs were placed on collagen-coated plates in Dulbecco’s modified Eagle’s medium-F12 with 5% serum, insulin (5 μg/ml), hydrocortisone (1 μg/ml), EGF (10 ng/ml), cholera toxin (10 ng/ml), and 1× Pen/Strep/Fungizone Mix. All media were phenol red free. Charcoal-stripped serum was used. All reagents were from GIBCO.
Immunostaining and Flow Cytometry
Cells were stained fresh or after fixation in methanol. Antibodies’ source and working dilutions are shown in Table S1. Cells were stained 20 min on ice in Hank’s balanced salt solution (HBSS; GIBCO) 2% FBS, then washed in HBSS 2% FBS, and resuspended in HBSS 5% FBS. Viability stain was performed using 1 μg/ml propidium iodide (PI) (Sigma) for 5 min. The cells were gated on viability and forward scattered in all experiments.
ALDEFLUOR Assay
The ALDEFLUOR Kit (StemCell Technologies) was used in accordance with the manufacturer’s protocol.
TMAs were constructed from 2 mm cores of 175 DCIS cases, in compliance with the Guy’s and St. Thomas Research Tissue and Data Bank procedures and with Guy’s Research Ethics Committee approval.
IHC
Source and working dilutions of antibodies and antigen retrieval procedures are shown in Table S1. All primary antibodies were incubated for 1 hr, except GH, which was incubated for 15 hr. Peroxidase Histostain-Plus Kit (Zymed) and EnVision G2 Doublestain System (Dako) were used in accordance with the manufacturer’s protocols.
Immunostaining on Coverslips
Cells were fixed 10 min in 4% paraformaldehyde, washed twice in PBS, treated with 0.1% Triton X- 100 for 5 min, and incubated in blocking buffer (PBS 2% BSA) for 1 hr. Primary and secondary antibodies were incubated for 1 hr in blocking buffer.
WB Analysis
Total cell extracts were electrophoresed on 10% SDS-PAGE gels. Antibodies were used at dilutions shown in Table S1, in 5% BSA or 5% milk in TBS buffer with 0.1% Tween. Incubation was 15 hr at 4°C for primary antibodies and 1 hr at room temperature for secondary antibodies. A Super Signal West Pico Chemiluminescent Substrate Kit System (Thermo Scientific) was used for signal detection.
GH, E2 P4, and GHA Treatment
Recombinant human GH and GH blocking antibody (GHA) (R&D Systems) were used in concentration of 3, 10, 30, 100, and 1,000 ng/ml GH and 30, 100, 300, 1,000, and 10,000 ng/ml GHA alone or in combination. E2, P4, and MPA (Sigma) were used in concentration of 10−7 M; ICI 182.780 and RU486 (SLS) were used at 10−8 M. To test GH levels by WB, HMECs were treated with E2 (10−7 M) for 6 hr and then P4 or MPA (10−7 M) for 24 hr. T47D cells were treated with MPA (1, 10, and 20 nM) for 6 hr. Cells were serum starved for 24 hr before treatment. For assessing GH mRNA level, cells were treated for 1 or 4 hr with P4, E2, E2+P4 with or without ICI 182.780, RU486. CM medium was used in a 1:3 ratio with fresh medium.
GHR Knockdown
Lentiviral vectors (pGIPZ) expressing two selected shRNAs against GHR (Applied Biosystems) were used in accordance with the manufacturer’s protocol (see also the Supplemental Experimental Procedures).
In Vivo Treatment of Xenografts
Animal studies were performed under protocols approved by the University Committee for Use and Care of Animals, University of Michigan and the Institutional Committees on Animal Welfare of the United Kingdom Home Office. A patient-derived breast tumor xenograft was implanted in the fourth mammary fat pat of NOD/scid mice. Tumors were allowed to reach 4 mm in the largest diameter before treatment with GHA Pegvisomant (Pfizer) was initiated. Treatment was done by subcutaneous injection 40 mg/kg, 1/day for 2 weeks. Control animals were treated with vehicle alone. Each group had five animals. Tumors were measured with calipers every 2–3 days, and the weight of tumors was calculated using the following formula: weight = l × h2/2. Animals were humanely sacrificed when tumors in the control group grew bigger than 1.2 cm in the largest diameter.
Statistical Analysis
The effect on mammosphere formation was tested for statistical significance by one-way Anova one tail, unequal variance. The effect of treatment on tumor growth was assessed by two-way Anova with Sidak’s multiple comparison tests. The correlation between markers in normal samples was analyzed using Fisher’s exact test and the correlation between markers in DCIS by two-way Anova with Sidak’s multiple comparison tests.
Acknowledgments
This work was supported by a Komen for the Cure grant (BCTR0707974) and in part by the Avon Foundation, Breakthrough Breast Cancer, and the Marion Burn Foundation. M.W. has financial holdings and is a scientific advisor for OncoMed Pharmaceuticals.
Footnotes
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/3.0/).
Supplemental Information
References
- Ahlgren M., Melbye M., Wohlfahrt J., Sørensen T.I. Growth patterns and the risk of breast cancer in women. N. Engl. J. Med. 2004;351:1619–1626. doi: 10.1056/NEJMoa040576. [DOI] [PubMed] [Google Scholar]
- Asselin-Labat M.L., Vaillant F., Sheridan J.M., Pal B., Wu D., Simpson E.R., Yasuda H., Smyth G.K., Martin T.J., Lindeman G.J., Visvader J.E. Control of mammary stem cell function by steroid hormone signalling. Nature. 2010;465:798–802. doi: 10.1038/nature09027. [DOI] [PubMed] [Google Scholar]
- Beleut M., Rajaram R.D., Caikovski M., Ayyanan A., Germano D., Choi Y., Schneider P., Brisken C. Two distinct mechanisms underlie progesterone-induced proliferation in the mammary gland. Proc. Natl. Acad. Sci. USA. 2010;107:2989–2994. doi: 10.1073/pnas.0915148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocchinfuso W.P., Korach K.S. Mammary gland development and tumorigenesis in estrogen receptor knockout mice. J. Mammary Gland Biol. Neoplasia. 1997;2:323–334. doi: 10.1023/a:1026339111278. [DOI] [PubMed] [Google Scholar]
- Brisken C., Heineman A., Chavarria T., Elenbaas B., Tan J., Dey S.K., McMahon J.A., McMahon A.P., Weinberg R.A. Essential function of Wnt-4 in mammary gland development downstream of progesterone signaling. Genes Dev. 2000;14:650–654. [PMC free article] [PubMed] [Google Scholar]
- Caufriez A., Leproult R., L’Hermite-Balériaux M., Moreno-Reyes R., Copinschi G. A potential role of endogenous progesterone in modulation of GH, prolactin and thyrotrophin secretion during normal menstrual cycle. Clin. Endocrinol. (Oxf.) 2009;71:535–542. doi: 10.1111/j.1365-2265.2009.03539.x. [DOI] [PubMed] [Google Scholar]
- Caufriez A., Leproult R., L’Hermite-Balériaux M., Kerkhofs M., Copinschi G. Progesterone prevents sleep disturbances and modulates GH, TSH, and melatonin secretion in postmenopausal women. J. Clin. Endocrinol. Metab. 2011;96:E614–E623. doi: 10.1210/jc.2010-2558. [DOI] [PubMed] [Google Scholar]
- Chiesa J., Ferrer C., Arnould C., Vouyovitch C.M., Diaz J.J., Gonzalez S., Mares P., Morel G., Wu Z.S., Zhu T. Autocrine proliferative effects of hGH are maintained in primary cultures of human mammary carcinoma cells. J. Clin. Endocrinol. Metab. 2011;96:E1418–E1426. doi: 10.1210/jc.2011-0473. [DOI] [PubMed] [Google Scholar]
- Cicalese A., Bonizzi G., Pasi C.E., Faretta M., Ronzoni S., Giulini B., Brisken C., Minucci S., Di Fiore P.P., Pelicci P.G. The tumor suppressor p53 regulates polarity of self-renewing divisions in mammary stem cells. Cell. 2009;138:1083–1095. doi: 10.1016/j.cell.2009.06.048. [DOI] [PubMed] [Google Scholar]
- Corneli G., Gasco V., Prodam F., Grottoli S., Aimaretti G., Ghigo E. Growth hormone levels in the diagnosis of growth hormone deficiency in adulthood. Pituitary. 2007;10:141–149. doi: 10.1007/s11102-007-0031-0. [DOI] [PubMed] [Google Scholar]
- De Stavola B.L., dos Santos Silva I., McCormack V., Hardy R.J., Kuh D.J., Wadsworth M.E. Childhood growth and breast cancer. Am. J. Epidemiol. 2004;159:671–682. doi: 10.1093/aje/kwh097. [DOI] [PubMed] [Google Scholar]
- Divisova J., Kuiatse I., Lazard Z., Weiss H., Vreeland F., Hadsell D.L., Schiff R., Osborne C.K., Lee A.V. The growth hormone receptor antagonist pegvisomant blocks both mammary gland development and MCF-7 breast cancer xenograft growth. Breast Cancer Res. Treat. 2006;98:315–327. doi: 10.1007/s10549-006-9168-1. [DOI] [PubMed] [Google Scholar]
- Dontu G., Abdallah W.M., Foley J.M., Jackson K.W., Clarke M.F., Kawamura M.J., Wicha M.S. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev. 2003;17:1253–1270. doi: 10.1101/gad.1061803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginestier C., Hur M.H., Charafe-Jauffret E., Monville F., Dutcher J., Brown M., Jacquemier J., Viens P., Kleer C.G., Liu S. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell. 2007;1:555–567. doi: 10.1016/j.stem.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J., Cairns B.J., Casabonne D., Wright F.L., Reeves G., Beral V., Million Women Study collaborators Height and cancer incidence in the Million Women Study: prospective cohort, and meta-analysis of prospective studies of height and total cancer risk. Lancet Oncol. 2011;12:785–794. doi: 10.1016/S1470-2045(11)70154-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunnell D., Okasha M., Smith G.D., Oliver S.E., Sandhu J., Holly J.M. Height, leg length, and cancer risk: a systematic review. Epidemiol. Rev. 2001;23:313–342. doi: 10.1093/oxfordjournals.epirev.a000809. [DOI] [PubMed] [Google Scholar]
- Honeth G., Lombardi S., Ginestier C., Hur M., Marlow R., Buchupalli B., Shinomiya I., Bombelli S., Ramalingham V., Purushotham A.D., Pinder S.E., Dontu G. Aldehyde dehydrogenase and estrogen receptor define a hierarchy of cellular differentiation in the normal human mammary epithelium. Breast Cancer Res. 2014;16:R52. doi: 10.1186/bcr3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins P.J. Acromegaly and cancer. Horm. Res. 2004;62(Suppl 1):108–115. doi: 10.1159/000080768. [DOI] [PubMed] [Google Scholar]
- Joshi P.A., Jackson H.W., Beristain A.G., Di Grappa M.A., Mote P.A., Clarke C.L., Stingl J., Waterhouse P.D., Khokha R. Progesterone induces adult mammary stem cell expansion. Nature. 2010;465:803–807. doi: 10.1038/nature09091. [DOI] [PubMed] [Google Scholar]
- Kleinberg D.L., Wood T.L., Furth P.A., Lee A.V. Growth hormone and insulin-like growth factor-I in the transition from normal mammary development to preneoplastic mammary lesions. Endocr. Rev. 2009;30:51–74. doi: 10.1210/er.2008-0022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuperwasser C., Chavarria T., Wu M., Magrane G., Gray J.W., Carey L., Richardson A., Weinberg R.A. Reconstruction of functionally normal and malignant human breast tissues in mice. Proc. Natl. Acad. Sci. USA. 2004;101:4966–4971. doi: 10.1073/pnas.0401064101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laban C., Bustin S.A., Jenkins P.J. The GH-IGF-I axis and breast cancer. Trends Endocrinol. Metab. 2003;14:28–34. doi: 10.1016/s1043-2760(02)00003-6. [DOI] [PubMed] [Google Scholar]
- Lantinga-van Leeuwen I.S., van Garderen E., Rutteman G.R., Mol J.A. Cloning and cellular localization of the canine progesterone receptor: co-localization with growth hormone in the mammary gland. J. Steroid Biochem. Mol. Biol. 2000;75:219–228. doi: 10.1016/s0960-0760(00)00173-4. [DOI] [PubMed] [Google Scholar]
- Laron Z. The GH-IGF1 axis and longevity. The paradigm of IGF1 deficiency. Hormones (Athens) 2008;7:24–27. doi: 10.14310/horm.2002.1111034. [DOI] [PubMed] [Google Scholar]
- Laron Z., Klinger B. Laron syndrome: clinical features, molecular pathology and treatment. Horm. Res. 1994;42:198–202. doi: 10.1159/000184193. [DOI] [PubMed] [Google Scholar]
- Lim E., Vaillant F., Wu D., Forrest N.C., Pal B., Hart A.H., Asselin-Labat M.L., Gyorki D.E., Ward T., Partanen A., kConFab Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat. Med. 2009;15:907–913. doi: 10.1038/nm.2000. [DOI] [PubMed] [Google Scholar]
- Mertani H.C., Garcia-Caballero T., Lambert A., Gerard F., Palayer C., Boutin J.M., Vonderhaar B.K., Waters M.J., Lobie P.E., Morel G. Cellular expression of growth hormone and prolactin receptors in human breast disorders. Int. J. Cancer. 1998;79:202–211. doi: 10.1002/(sici)1097-0215(19980417)79:2<202::aid-ijc17>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Mukhina S., Mertani H.C., Guo K., Lee K.O., Gluckman P.D., Lobie P.E. Phenotypic conversion of human mammary carcinoma cells by autocrine human growth hormone. Proc. Natl. Acad. Sci. USA. 2004;101:15166–15171. doi: 10.1073/pnas.0405881101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pece S., Tosoni D., Confalonieri S., Mazzarol G., Vecchi M., Ronzoni S., Bernard L., Viale G., Pelicci P.G., Di Fiore P.P. Biological and molecular heterogeneity of breast cancers correlates with their cancer stem cell content. Cell. 2010;140:62–73. doi: 10.1016/j.cell.2009.12.007. [DOI] [PubMed] [Google Scholar]
- Perry J.K., Mohankumar K.M., Emerald B.S., Mertani H.C., Lobie P.E. The contribution of growth hormone to mammary neoplasia. J. Mammary Gland Biol. Neoplasia. 2008;13:131–145. doi: 10.1007/s10911-008-9070-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raccurt M., Lobie P.E., Moudilou E., Garcia-Caballero T., Frappart L., Morel G., Mertani H.C. High stromal and epithelial human gh gene expression is associated with proliferative disorders of the mammary gland. J. Endocrinol. 2002;175:307–318. doi: 10.1677/joe.0.1750307. [DOI] [PubMed] [Google Scholar]
- Russo J., Russo I.H. The role of estrogen in the initiation of breast cancer. J. Steroid Biochem. Mol. Biol. 2006;102:89–96. doi: 10.1016/j.jsbmb.2006.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selman P.J., Mol J.A., Rutteman G.R., van Garderen E., Rijnberk A. Progestin-induced growth hormone excess in the dog originates in the mammary gland. Endocrinology. 1994;134:287–292. doi: 10.1210/endo.134.1.7506206. [DOI] [PubMed] [Google Scholar]
- Sternlicht M.D. Key stages in mammary gland development: the cues that regulate ductal branching morphogenesis. Breast Cancer Res. 2006;8:201. doi: 10.1186/bcr1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanson S.M., Unterman T.G. The growth hormone-deficient Spontaneous Dwarf rat is resistant to chemically induced mammary carcinogenesis. Carcinogenesis. 2002;23:977–982. doi: 10.1093/carcin/23.6.977. [DOI] [PubMed] [Google Scholar]
- Törnell J., Carlsson B., Pohjanen P., Wennbo H., Rymo L., Isaksson O. High frequency of mammary adenocarcinomas in metallothionein promoter-human growth hormone transgenic mice created from two different strains of mice. J. Steroid Biochem. Mol. Biol. 1992;43:237–242. doi: 10.1016/0960-0760(92)90213-3. [DOI] [PubMed] [Google Scholar]
- van Garderen E., Schalken J.A. Morphogenic and tumorigenic potentials of the mammary growth hormone/growth hormone receptor system. Mol. Cell. Endocrinol. 2002;197:153–165. doi: 10.1016/s0303-7207(02)00259-9. [DOI] [PubMed] [Google Scholar]
- Villadsen R., Fridriksdottir A.J., Rønnov-Jessen L., Gudjonsson T., Rank F., LaBarge M.A., Bissell M.J., Petersen O.W. Evidence for a stem cell hierarchy in the adult human breast. J. Cell Biol. 2007;177:87–101. doi: 10.1083/jcb.200611114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters M.J., Barclay J.L. Does growth hormone drive breast and other cancers? Endocrinology. 2007;148:4533–4535. doi: 10.1210/en.2007-0855. [DOI] [PubMed] [Google Scholar]
- Zhou Y., Xu B.C., Maheshwari H.G., He L., Reed M., Lozykowski M., Okada S., Cataldo L., Coschigamo K., Wagner T.E. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse) Proc. Natl. Acad. Sci. USA. 1997;94:13215–13220. doi: 10.1073/pnas.94.24.13215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu T., Starling-Emerald B., Zhang X., Lee K.O., Gluckman P.D., Mertani H.C., Lobie P.E. Oncogenic transformation of human mammary epithelial cells by autocrine human growth hormone. Cancer Res. 2005;65:317–324. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.