

Abstract
Reversible lysine acetylation (RLA) is a widespread regulatory mechanism that modulates the function of proteins involved in diverse cellular processes. A strong case has been made for RLA control exerted by homologues of the Salmonella enterica protein acetyltransferase (SePat) enzyme on the broadly distributed AMP-forming CoA ligase (a.k.a. acyl-CoA synthetases) family of metabolic enzymes, with acetyl-CoA synthetase (Acs) being the paradigm in the field. Here we investigate why the Acs homologue in Streptomyces lividans (SlAcs) is poorly acetylated in vitro by the S. lividans protein acetyltransferase (SlPat) enzyme. Chimeras of S. enterica Acs (SeAcs) and S. lividans Acs (SlAcs) constructed during the course of this work were acetylated by SlPatA in vitro, retained most of their activity, and were under RLA control in a heterologous host. We identified SeAcs residues N- and C-terminal to the target lysine that when introduced into SlAcs, rendered the latter under RLA control. These results lend further support to the idea that Pat enzymes interact with extensive surfaces of their substrates. Finally, we suggest that acetylation of SlAcs depends on factors or conditions other than those present in our in vitro system. We also discuss possible explanations why SlAcs is not controlled by RLA as defined in other bacterial species.
Introduction
Reversible lysine acetylation (RLA) is a post-translational modification that occurs in all domains of life [1] and affects diverse cellular processes and functions. Acetyltransferases transfer the acetyl moiety from acetyl-CoA to the ε-amino group of the target lysine. Lysine acetylation can affect enzyme activity [2], protein stability [3], protein-protein interactions, or DNA binding [4]. Yeast Gcn5 protein (yGcn5p)-related N-acetyltransferases (a.k.a., GNATs), classified by amino acid sequence and structure [5], are the only class of acetyltransferases found in all domains of life [6]. GNATs were first identified for their role in modification of histones [7]. Crystal structures and biochemical analyses of the yGcn5p, the founding member of the GNAT family, with representative peptides from histones has provided valuable information about the substrate specificity and substrate recognition by GNATs [8], [9].
Members of the GNAT family also acetylate metabolic enzymes. For example, in Salmonella enterica, the enzyme acetyl-CoA synthetase (SeAcs) is acetylated by the protein acetyltransferase (SePat), a two-domain acetyltransferase that contains a large domain of unknown function and a C-terminal GNAT domain [10]. SeAcs is a member of the AMP-forming CoA ligase family of enzymes that converts carboxylic acids to their CoA thioesters via an acyl-AMP intermediate [11]. Acetylation of the active site lysine of AMP-forming CoA ligases prevents the adenylylation of the carboxylic acid. In addition to Pat from S. enterica, GNATs are known to acetylate members of the of AMP-forming CoA ligase family (including Acs) in Rhodopseudomonas palustris [12], [13], Bacillus subtilis [14], and Mycobacterium smegmatis [15]. The Acs homologue from Streptomyces coelicolor is acetylated in vivo [16], but the GNAT responsible for acetylation of S. coelicolor Acs is unknown.
Knowledge of the interactions of GNAT with their proteins substrates is limited. R. palustris encodes a single-domain GNAT (RpKatA) and a homologue of the SePat GNAT (RpPat). RpKatA and RpPat discriminate among members of the AMP-forming CoA ligase family produced by R. palustris [13]. In addition to the target lysine, RpPat recognizes a loop greater than 20 Å from the target lysine, suggesting that Pat enzymes interact with a large surface of the acceptor substrate [17]. As a proof of principle, the introduction of this recognition loop into R. palustris methylmalonyl-CoA mutase (RpMatB), an AMP-forming CoA ligase that is not a substrate of RpPat, rendered RpMatB a target of acetylation by RpPat. Thus, synthetic chimeras of AMP-forming CoA ligases have yielded valuable information about how GNATs recognize protein substrates and have produced AMP-forming CoA ligases that are placed under the regulation of lysine acetylation.
RpPat and SePat enzymes acetylate their cognate Acs proteins. Although the GNAT responsible for the acetylation of Acs in S. coeolicolor is unknown, the closely related actinomycete Streptomyces lividans encodes SlPatA, a two-domain homologue of SePat and RpPat enzymes. Significantly, SlPatA does not efficiently acetylate the S. lividans Acs (SlAcs) in vitro [18], making this the first Acs enzyme that is not efficiently acetylated by a Pat acetyltransferase. In contrast, SlPatA efficiently acetylates SeAcs. Here we probe the amino acid sequences in SeAcs that rendered it a better substrate for SlPatA than SlAcs is. By replacing amino acids from SeAcs into the C-terminus of SlAcs, we constructed SlAcs-SeAcs chimeras that were efficiently acetylated by SlPatA. One SlAcs-SeAcs chimera contained 41 amino acid differences from SlAcs. As a result of these changes, the SlAcs-SeAcs chimera was subject to regulation by SlPatA. We used a heterologous model system to demonstrate that the SlAcs-SeAcs chimera was subject to RLA regulation in vivo by SlPatA. In sum, we identified regions in SeAcs that were critical for recognition by SlPatA, and transferring of these residues into the poor substrate SlAcs resulted in a SlAcs variant that was efficiently regulated by SlPatA.
Materials and Methods
Bacterial Strains and Growth Conditions
All strains and plasmids used in this study are listed in Tables 1 and 2, respectively. Escherichia coli and Salmonella enterica strains were grown at 37°C in lysogeny broth (LB, Difco) [19] or no-carbon essential (NCE) minimal medium [20] supplemented with sodium acetate (10 mM), MgSO4 (1 mM), and ampicillin (100 µg ml−1). When necessary, antibiotics were used at the following concentrations: ampicillin, 100 µg ml−1; tetracycline, 10 µg ml−1; chloramphenicol, 12.5 µg ml−1, kanamycin, 50 µg ml−1. L-(+)-arabinose was added at varying concentrations (5 or 200 µM) to induce the expression of S. enterica acs, S. lividans acs, and acs chimeras cloned into the expression vector pBAD30 [21]. Isopropyl β-D-1-thiogalactopyranoside (IPTG) was added to a final concentration of IPTG (0–500 µM) to induce expression of S. lividans patA (EFD66247) clones into the expression vector pSRK-Km [22]. Growth experiments were performed at 37°C using a microtiter plate and a microtiter plate reader (Bio-Tek Instruments). All growth data are plotted as the mean of three data points.
Table 1. Strains used in this study.
| Strain | Relevant Genotype and description | Source |
| S. enterica strains | ||
| TR6583 | metE205 ara-9 | K. Sanderson via J. Roth |
| Derivatives of TR6583 | ||
| JE9152 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+) | Laboratory Collection |
| JE9894 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+) | Laboratory Collection |
| JE13238 | metE205 ara-9 Δacs2 Δpta127 | Laboratory Collection |
| Derivatives of JE9152 | ||
| JE18793 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pBAD30 pSRK-Km | This work |
| JE18794 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pBAD30 pSlPatA9 | This work |
| JE18795 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pSlAcs47 pSRK-Km | This work |
| JE18796 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pSlAcs47 pSlPatA9 | This work |
| JE18797 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pSlAcs48 pSRK-Km | This work |
| JE18798 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pSlAcs48 pSlPatA9 | This work |
| JE18799 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pACS59 pSRK-Km | This work |
| JE18800 | metE205 ara-9 Δacs2 ΔcobB1330 pat1:: Tn10d(tet+)/pACS59 pSlPatA9 | This work |
| Derivatives of JE9894 | ||
| JE18801 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pBAD30 pSRK-Km | This work |
| JE18802 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pBAD30 pSlPatA9 | This work |
| JE18803 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pSlAcs47 pSRK-Km | This work |
| JE18804 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pSlAcs47 pSlPatA9 | This work |
| JE18805 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pSlAcs48 pSRK-Km | This work |
| JE18806 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pSlAcs48 pSlPatA9 | This work |
| JE18807 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pACS59 pSRK-Km | This work |
| JE18808 | metE205 ara-9 Δacs2 pat1:: Tn10d(tet+)/pACS59 pSlPatA9 | This work |
| Derivatives of JE13238 | ||
| JE13787 | metE205 ara-9 Δacs2 Δpta127/pBAD30 bla+ | This work |
| JE14947 | metE205 ara-9 Δacs2 Δpta127/pSlAcs6 bla+ | This work |
| E. coli strains | ||
| JE9314 | C41(λDE3) pka12:: kan+ | Laboratory Collection |
Table 2. Plasmids used in this study.
| Plasmid | Genotype | Source or method |
| pBAD30 | ParaBAD expression vector, bla+ | [21] |
| pSlAcs6 | S. lividans acs+ allele (EFD66247) in pBAD30, bla + | Standard cloning |
| pSlAcs47 | S. lividans acs+ allele (EFD66247) with N-terminal H6 tag in pBAD30, bla + | Standard cloning |
| pSlAcs48 | S. lividans acs –S. enterica acs chimera allele with N-terminal H6 tag in pBAD30, bla + | Standard cloning |
| pACS59 | S. enterica acs+ allele with N-terminal H6 tag in pBAD30, bla + | Standard cloning |
| pSRK-Km | lacIq-lac promoter-operator expression vector, kan+ | [22] |
| pSlPatA9 | S. lividans patA + allele (EFD66247) in pSRK-Km, bla+ | [18] |
| pKLD66 | N-terminal, rTEV-cleavable MBP-His6-tag overexpression vector, bla + | [25] |
| pSlAcs7 | S. lividans acs + (EFD66247) C-terminal domain (D519-D649) in pKLD66, bla + | Standard cloning |
| pTEV5 | N-terminal, rTEV-cleavable His6-tag overexpression vector, bla+ | [25] |
| pSlAcs1 | S. lividans acs+ allele (EFD68454) in pTEV5, bla+ | [18] |
| pSlPatA1 | S. lividans patA + allele (EFD66247) in pTEV5, bla+ | [18] |
| pACS38 | S. enterica acs+ C-terminal domain (D518-S652) in pTEV5, bla+ | Standard cloning |
| pSlAcs8 | A1 chimera: SlAcs 520 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs9 | A2 chimera: SlAcs 550 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs12 | A3 chimera: SlAcs 560 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs22 | A4 chimera: SlAcs 566 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs10 | A5 chimera: SlAcs 582 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs11 | A6 chimera: SlAcs 617 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs14 | B1 chimera: SlAcs 550–582 SeAcs in pTEV5, bla + in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs15 | B2 chimera: SlAcs 550–603 SeAcs in pTEV5, bla + in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs23 | B3 chimera: SlAcs 550–618 SeAcs in pTEV5, bla + in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs17 | B4 chimera: SlAcs 550–627 SeAcs in pTEV5, bla + in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs18 | B5 chimera: SlAcs 550–638 SeAcs in pTEV5, bla + in pTEV5, bla + | Standard cloning |
| pSlAcs19 | B6 chimera: SlAcs 550–643 SeAcs in pTEV5, bla + in pTEV5, bla + | Standard cloning |
| pSlAcs26 | C1 chimera: SlAcs 550–581 SeAcs, 591–627 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs27 | C2 chimera: SlAcs 550–590 SeAcs, 598–627 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs28 | C3 chimera: SlAcs 550–597 SeAcs, 603–627 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs29 | C4 chimera: SlAcs 550–581 SeAcs, 603–627 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs44 | C5 chimera: SlAcs 615–626 SeAcs in pTEV5, bla + | Overlap-extension PCR |
| pSlAcs49 | K610A variant of C3 chimera in pTEV5, bla + | Site-directed mutagenesis |
Molecular Techniques
DNA manipulations were performed using standard techniques [23]. Restriction endonucleases were purchased from Fermentas. DNA was amplified using Pfu Ultra II Fusion DNA polymerase (Agilent) or Herculase II Fusion DNA polymerase (Agilent). Site-directed mutagenesis was performed using the Quikchange™ Site Directed Mutagenesis kit (Agilent). Plasmids were isolated using the Wizard Plus SV Miniprep kit (Promega) and PCR products were purified using the Wizard SV Gel and PCR Clean-Up System (Promega). DNA sequencing was performed using BigDye® (ABI PRISM) protocols, and sequencing reactions were resolved at the University of Georgia Genomics Facility.
Plasmids Used for Protein Overproduction
Chimeric proteins encoded by fusing different regions of S. lividans acs (EFD68454) and S. enterica acs genes were generated by amplifying genomic DNA from S. lividans TK24 genomic DNA from S. enterica strain TR6583, respectively. Fusion plasmids encoding proteins in which the N-terminal domain of SlAcs was fused to the C-terminal domain of SeAcs at residues 520, 550, 560, 566, 582, 617 were generated by overlap-extension PCR [24], followed by standard cloning into plasmid pTEV5 [25]. Fusion plasmids encoding a protein in which an internal sequence of SlAcs was replaced by the corresponding sequence SeAcs were constructed as described below and in Table 2.
Plasmid pSlAcs14 (SlAcs 550–582 SeAcs) – the nucleotides encoding the first 582 residues of SlAcs fused to SeAcs were amplified from pSlAcs9, fused to the C-terminus of SlAcs, and cloned into pTEV5.
Plasmid pSlAcs15 (SlAcs 550–603 SeAcs) – the nucleotides encoding the first 603 residues of SlAcs fused to SeAcs were amplified from pSlAcs9, fused to the C-terminus of SlAcs, and cloned into pTEV5.
Plasmid pSlAcs23 (SlAcs 550–618 SeAcs) – the nucleotides encoding the first 618 residues of SlAcs fused to SeAcs were amplified from pSlAcs9, fused to the C-terminus of SlAcs, and cloned into pTEV5.
Plasmid pSlAcs17 (SlAcs 550–627 SeAcs) – the nucleotides encoding the first 627 residues of SlAcs fused to SeAcs were amplified from pSlAcs9, fused to the C-terminus of SlAcs, and cloned into pTEV5.
Plasmid pSlAcs18 (SlAcs 550–638 SeAcs) – the nucleotides encoding the first 638 residues of SlAcs fused to SeAcs were amplified from pSlAcs9 and cloned into pTEV5.
Plasmid pSlAcs19 (SlAcs 550–643 SeAcs) – the nucleotides encoding the first 643 residues of SlAcs fused to SeAcs were amplified from pSlAcs9 and cloned into pTEV5.
Plasmid pSlAcs26 (SlAcs 550–581 SeAcs, 591–627 SeAcs) – the nucleotides encoding the first 581 residues of SlAcs fused to SeAcs were amplified from pSlAcs9 with primers incorporating residues 582–590 from SlAcs, fused to the nucleotides encoding the 64 residues of SeAcs fused to SlAcs amplified from pSlAcs17, and cloned into pTEV5.
Plasmid pSlAcs27 (SlAcs 550–590 SeAcs, 598–627 SeAcs) – the nucleotides encoding the first 590 residues of SlAcs fused to SeAcs were amplified from pSlAcs9 with primers incorporating residues 591–597 from SlAcs, fused to the nucleotides encoding the 57 residues of SeAcs fused to SlAcs amplified from pSlAcs17, and cloned into pTEV5.
Plasmid pSlAcs28 (SlAcs 550–597 SeAcs, 603–627 SeAcs) – the nucleotides encoding the first 597 residues of SlAcs fused to SeAcs were amplified from pSlAcs9 with primers incorporating residues 598–602 from SlAcs, fused to the nucleotides encoding the 52 residues of SeAcs fused to SlAcs amplified from pSlAcs17, and cloned into pTEV5.
Plasmid pSlAcs29 (SlAcs 550–581 SeAcs, 603–627 SeAcs) – the nucleotides encoding the first 581 residues of SlAcs fused to SeAcs were amplified from pSlAcs9 with primers incorporating residues 582–602 from SlAcs, fused to the nucleotides encoding the 52 residues of SeAcs fused to SlAcs amplified from pSlAcs17, and cloned into pTEV5.
Plasmid pSlAcs44 (SlAcs 615–626 SeAcs) – the nucleotides encoding the first 614 residues of SlAcs were amplified from pSlAcs1, the nucleotides encoding the final 40 residues of SeAcs fused to SlAcs amplified from pSlAcs28, and cloned into pTEV5.
The C-terminal domain of SeAcs was amplified from strain TR6583. DNA fragments were cut with NheI and EcoRI and ligated into pTEV5 [25] cut with the same enzymes. The resulting plasmids directed the synthesis of SlAcs-SeAcs chimeras or SeAcs C-terminal domain (pACS38) each with an N-terminal H6 tag cleavable by recombinant tobacco etch virus (rTEV) protease prepared as described [26].
The C-terminal domain of SlAcs was amplified from S. lividans TK24 genomic DNA. The DNA fragments were cut with KpnI and HinDIII and ligated into pKLD66 [25] cut with the same enzymes. The resulting plasmid pSlAcs7 directed synthesis of the SlAcs C-terminal domain with an N-terminal maltose-binding protein-His6 tag cleavable by rTEV protease as described above.
Construction of Untagged SlAcs Complementation Plasmid
The S. lividans acs was amplified from pSlAcs1 with the primers that included an optimized ribosome-binding site. The DNA fragment was cut with EcoRI and HindIII and ligated into pBAD30 [21], cut with the same enzymes. The resulting plasmid pSlAcs6 expresses SlAcs under the control of the ParaBAD promoter.
Construction of SeAcs, SlAcs, and SlAcs
Complementation vectors encoding H6-tagged SeAcs chimera C3
Genes encoding S. lividans Acs and the S. lividans/S. enterica Acs chimeras were amplified from pSlAcs1 and pSlAcs28, respectively, using primers that included an optimized ribosome-binding site and an N-terminal His6-tag. S. enterica acs was amplified from genomic DNA isolated from JE6583 using primers that included an optimized ribosome-binding site and an N-terminal His6-tag. The DNA fragments were cut with EcoRI and HindIII and ligated into pBAD30, cut with the same enzymes. The resulting plasmids pSlAcs47, pSlAcs48, and pACS59 produce SlAcs, S. lividans/S. enterica Acs chimera C3, and SeAcsWT, respectively, with His6-tags fused N-terminal with a Gly-Ser-Gly linker under the control of at the ParaBAD promoter.
Purification of SlAcs-SeAcs chimeras, SlAcs C-terminal domain, and SeAcs C-terminal domain
Plasmids encoding tagged proteins were transformed with pRARE2 (EMD Millipore) into a Δpka derivative of E. coli strain C41λ(DE3) [27] (JE9314) to prevent acetylation prior to overproduction. The resulting strains were grown overnight and sub-cultured 1∶100 (v/v) into two liters of LB containing ampicillin (100 µg ml−1) and chloramphenicol (12.5 µg ml−1). The cultures were grown shaking at 25°C to A600∼0.7 and protein synthesis was induced with IPTG (0.25 mM). Upon induction, the cultures were grown overnight at 25°C. Cells were harvested at 6000×g for 10 min at 4°C in a Avanti J-2 XPI centrifuge fitted with rotor JLA-8.1000 (Beckman Coulter). Cell pellets were re-suspended in 30 ml of cold His-bind buffer (buffer A) [tris(hydroxymethyl)aminomethane-HCl (Tris-HCl) buffer (50 mM, pH 8), NaCl (500 mM)], and imidazole (5 mM) containing phenylmethanesulfonylfluoride (PMSF, 1 mM). Cells were placed on ice and lysed by sonication for 2 min (2-s pulse followed by 4 s of cooling) at level 7 in a model 550 sonic dismembrator (Fisher). The extract was cleared by centrifugation at 4°C for 30 min at 43,367×g. H6-SlAcs-SeAcs chimera was purified from clarified cell extract using a 1 ml settled bed volume of HisPur™ Ni-NTA Resin (Pierce). Unbound proteins were eluted off the column by washing with buffer A. The resin was washed with 10 column volumes of buffer B [Tris-HCl buffer (50 mM, pH 8), NaCl (500 mM), and imidazole (15 mM)]. H6-SlAcs-SeAcs chimera was eluted with 5 column volumes of buffer C [Tris-HCl buffer (50 mM, pH 8), NaCl (500 mM), and imidazole (250 mM)]. All fractions containing H6-SlAcs-SeAcs chimera were combined. rTEV protease was added to H6-SlAcs-SeAcs chimera and the SlAcs-SeAcs chimera/rTEV mixture was incubated at room temperature for 3 h. PMSF was added to the protein mixture and incubated 15 min at room temperature. The SlAcs-SeAcs chimera/rTEV mixture was dialyzed at 4°C against buffer D (Tris-HCl (50 mM, pH 8), NaCl (500 mM)) twice for 3 h and again against buffer D containing imidazole (5 mM) for 12 h. After cleavage and dialysis, protein mixtures were passed over 1 ml HisPur™ Ni-NTA Resin (Pierce) using the buffers described above. Cleaved SlAcs-SeAcs chimera passed through the resin and eluted in the flow-through fractions. Purified SlAcs-SeAcs chimera was analyzed by SDS-PAGE. Fractions containing SlAcs-SeAcs chimera were pooled together. SlAcs-SeAcs chimera was stored in Tris-Cl buffer (50 mM, pH 8.0) containing NaCl (100 mM) and glycerol (20%, v/v). SlAcs concentration was determined by measuring absorbance at 280 nm. The molecular weights and molar extinction coefficients used to calculate H6-SlAcs-SeAcs chimera concentrations are listed in Table 3. All enzymes were purified to >95% homogeneity.
Table 3. Molecular mass and molar extinction coefficients of proteins used in this study.
| Protein | MM (Da) | ε (M−1 cm−1) |
| SlAcs | 71045 | 135455 |
| SeAcs | 72153 | 138770 |
| A1 chimera | 71527 | 150925 |
| A2 chimera | 71541 | 150925 |
| A3 chimera | 71500 | 150800 |
| A4 chimera | 71432 | 147945 |
| A5 chimera | 71352 | 146455 |
| A6 chimera | 71115 | 135455 |
| B1 chimera | 71234 | 139925 |
| B2 chimera | 71471 | 150800 |
| B3 chimera | 71471 | 150925 |
| B4 chimera | 71530 | 150925 |
| B5 chimera | 71466 | 150925 |
| B6 chimera | 71751 | 150925 |
| C1 chimera | 71297 | 145425 |
| C2 chimera | 71627 | 150925 |
| C3 chimera | 71429 | 145425 |
| C4 chimera | 71293 | 139800 |
| C5 chimera | 71104 | 135330 |
| C3 chimera K609A variant | 71429 | 145425 |
| SlPatA | 108369 | 57760 |
SeAcs Protein Purification
Plasmid pACS10 was transformed into a Δpka derivative of E. coli strain C41λ(DE3) (JE9314). The resulting strain was grown overnight and sub-cultured 1∶100 (v/v) into two liters of LB containing ampicillin (100 µg ml−1). The culture was grown shaking at 37°C to A600∼0.7 and protein synthesis was induced with IPTG (0.25 mM). Upon induction, the cultures were grown overnight at 30°C. SeAcs was purified and stored as described [2]. SlAcsWT and SlPatAWT were purified as described [18].
In vitro CoA Ligase Assays
Activity of SlAcsWT, SeAcsWT, and SlAcs-SeAcs chimera activities were measured using an NADH-consuming assay [12], [28] with modifications. Reactions (100 µl total volume) contained 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 50 mM, pH 7.5), tris(2-carboxyethyl)phosphine (TCEP, 1 mM), ATP (2.5 mM) CoA (0.5 mM), MgCl2 (5 mM), KCl (1 mM), phosphoenolpyruvate (3 mM), NADH (0.1 mM), pyruvate kinase (1 U), myokinase (5 U), lactate dehydrogenase (1.5 U) and acetate (0.2 mM). Reactions were started by the addition of Acs (5–100 pmol). The absorbance at 340 nm was monitored in a 96-well plate using the Spectramax Plus UV-visible spectrophotometer (Molecular Devices). Enzyme activities were determined to be in the linear range of the assay and were calculated as described [28].
In vitro Protein Acetylation Assay
Protein acetylation was observed using radiolabeled Ac-CoA as described [10], [12], [29]. Acetylation reactions contained 2-(bis(2-hydroxyethyl)imino)-2-(hydroxymethyl)-1,3-propanediol (Bis-Tris-HCl) buffer (50 mM, pH 6.0), [1-14C]-Ac-CoA (20 mM), acyl-CoA synthetase or acyl-CoA synthetase C-terminal domain (3 µM), glycerol (10%, v/v), and SlPatAWT (1 µM). Reactions (20 µl total volume) were incubated for 60 min at 30°C. Samples (5 µl) were resolved using SDS-PAGE [30] and proteins were visualize by Coomassie Blue R staining [31]. Gels were dried and exposed 16 h to a multipurpose phosphor screen (Packard). Labeled proteins were visualized using a Typhoon Trio+ Imager (GE Healthcare) equipped with ImageQuant TL software (GE Healthcare). Acetylation was quantified as digital light units and is reported relative to SeAcsWT acetylation.
The effect of acetylation on activity of SlAcsWT, SeAcsWT, and SlAcs-SeAcs chimera activity was determined as described [12] with modifications. SlAcsWT, SeAcsWT, or SlAcs-SeAcs (3 µM) was incubated with SlPatAWT (1 µM) and 50 µM Ac-CoA for 90 min at 30°C using the buffer system described above. After 90 min, reactions were diluted into HEPES buffer (50 mM, pH 7.5 at 4°C). SlAcsWT, SeAcsWT, and SlAcs-SeAcs chimera activities were measured as described above.
In vitro Deacetylation Assays
Acetylated SlAcs-SeAcs chimera C3 was deacetylated with S. enterica CobBS (SeCobBS) sirtuin deacetylase as described [29]. In vitro acetylated SlAcs-SeAcs chimera C3 (3 µM, radiolabeled) was incubated with SeCobBS (3 µM) in deacetylation buffer containing HEPES buffer (50 mM, pH 7.0), NAD+ (1 mM) for 60 min at 37°C (10 µl reaction volume). Reaction mixture samples (5 µl) were resolved by SDS-PAGE, and subjected to phosphor imaging analysis to assess the acetylation state of SlAcs-SeAcs chimera C3 after incubation with SeCobBS.
H6-SlAcs, H6-SeAcs, or H6-Chimera C3 enzymes isolated from S. enterica were deacetylated with SeCobBS as described above with modifications. H6-SlAcs, H6-SeAcs, or H6-Chimera C3 enzymes (1 mM) were incubated with SeCobBS (1 µM) in deacetylation buffer containing HEPES (50 mM, pH 7.0), NAD+ (1 mM) for 60 min at 37°C (25 µl reaction volume). Acs activity was measured using the CoA synthetase assay described above.
Results
S. lividans Acetyl-CoA Synthetase (SlAcs) is Functional in vivo in a Heterologous System
The SeAcs homologue from S. lividans converts acetate to acetyl-CoA in vitro [18]. Alignment of the SeAcs and SlAcs amino acid sequences using BLAST revealed 52% sequence identity and 62% sequence similarity in amino acid sequence. To determine whether or not SlAcs functioned in vivo, we expressed S. lividans acs+ ectopically in a Δacs Δpta S. enterica strain (JE13238) demanding growth on low concentrations of acetate (10 mM). S. enterica uses two pathways for the conversion of acetate to acetyl-CoA (Fig. 1A) [11], [32]. One pathway is comprised of SeAcs, which catalyzes a two-step conversion of acetate to acetyl-CoA via an acetyl-AMP intermediate. RLA controls SeAcs activity [2]. The protein acetyltransferase SePat acetylates and inactivates of SeAcs (discussed further below) [10], and SeAcs is deacetylated and reactivated by the sirtuin type deacetylase SeCobB [2], [29]. In the second pathway, acetate kinase (Ack) and phosphotransacetylase (Pta) catalyze the conversion of acetate to acetyl-CoA via an acetyl-phosphate intermediate. Acs activity is used by the cell when the concentration of acetate in the environment is <10 mM, whilst Pta/Ack is the preferred pathway when S. enterica is growing on concentrations of acetate ≥25 mM. A S. enterica strain lacking the Acs and Ack/Pta pathways failed to grow on acetate (10 mM, Fig. 1B, squares). When SlAcs was produced ectopically, growth of an S. enterica Δacs Δpta strain was restored (Fig. 1B, circles), demonstrating that SlAcs was active and could substitute for SeAcs function in vivo.
Figure 1. SlAcsWT can substitute for SeAcsWT in S. enterica during growth on acetate.

A. S. enterica encodes a one-enzyme and a two-enzyme pathway for acetate activation. The one-enzyme pathway is composed of acetyl-CoA synthetase (Acs), whose activity is modulated post-translationally by the protein acetyltransferase (Pat) and sirtuin deacetylase (CobB) enzymes. The two-enzyme pathway is comprised of acetate kinase (Ack) and phosphotransacetylase (Pta). B. Growth behavior of Δacs Δpta S. enterica strain JE13238 as a function of SlAcsWT. Experiments were performed on NCE minimal medium supplemented with acetate (10 mM), at 37°C using a microtiter plate and a plate reader (Bio-Tek Instruments). Synthesis of SlAcsWT was ectopically encoded (plasmid pSlAcs6) and induced using L-(+)-arabinose (5 mM). Cloning vector (pBAD30) lacking S. lividans acs+ was used as negative control. All S.D. <0.01 absorbance units.
SlPatA Acetylates the C-terminal Domain of SeAcs, but not SlAcs
AMP-forming CoA synthetases are two-domain enzymes that activate carboxylic acids to CoA thioesters in a two-step reaction. In the first half-reaction, an invariant lysine in the C-terminal domain (K609 of SeAcs) is buried in the active site cleft located between the N- and C-terminal domains [33]. Upon adenylylation of the carboxylic acid substrate, the C-terminal domain undergoes a ∼140° domain rotation to allow for the thioesterification of the fatty acyl-AMP intermediate [34]. The catalytic lysine of AMP-forming CoA ligases is surface exposed when the enzyme is in the thioester-forming conformation [33], and this likely represents the conformation that is subject to acetylation by Pat.
Previously, we demonstrated that SlAcs was a poor substrate for the SlPatA enzyme in vitro [18]. That work identified SlAcs as the first example of an acetyl-CoA synthetase that was not recognized by the cognate Pat protein acetyltransferase in vitro [10], [12]. However, SlPatA efficiently acetylated and inactivated the acetoacetyl-CoA synthetase SlAacS from S. lividans, and the orthologous SeAcs enzyme [18], indicating that SlPatA was catalytically active, but somehow unable to acetylate SlAcs in vitro.
We considered the possibility that SlAcs favored the adenylyation conformation in vitro, which would likely render the target K610 inaccessible to SlPatA due to its location in the SlAcs active site. To differentiate the inaccessibility of SlAcs K610 from the inability of SlPatA to recognize SlAcs, we isolated the C-terminal domains of SeAcs (a good substrate of SlPatA) and SlAcs. In the absence of the N-terminal domain, the target lysine is no longer protected, thus it is accessible to the acetyltransferase.
Homogeneous C-terminal domains of SlAcs (residues D519–D649, 131 aa) and SeAcs (residues D518-S652, 135 aa) were incubated in the presence of SlPatA and radiolabeled [1-14C] acetyl-CoA. Differential migration of the C-terminal domains is likely due to differences in hydropathy of (grand average of hydropathy [GRAVY] scores [35] for SlAcs and SeAcs C-terminal domains are +0.023 and −0.160, respectively), which has been shown to affect gel mobility of protein in SDS-PAGE [36]. As shown in figure 2A, the C-terminal domain of SeAcs was acetylated, but the SlAcs C-terminal domain was not. These data showed that the N-terminal domain of SeAcs was not required for acetylation by SlPatA. Additionally, these results strongly suggested that inaccessibility of residue K610 was likely not the reason why SlAcs was poorly acetylated in vitro. We hypothesized that regions within the C-terminal domain of SlAcs enzyme prevented acetylation of SlPatA. As shown in figure 2B, the C-terminal domains of SlAcs and SeAcs share ∼50% sequence identity.
Figure 2. SlPatA efficiently acetylates the C-terminal domain of SeAcs.

A. The C-terminal domain of SlAcsWT or SeAcsWT was incubated with [1-14C]-acetyl-CoA in the presence or absence of SlPatAWT. Proteins were separated by SDS-PAGE and stained with Coomassie Blue R to visualize proteins. Acetylation was visualized by phosphor imaging. B. Alignment of the C-terminal domain of SlAcs and SeAcs. “ * ” denotes conserved residues; “.” denotes similar residues; light gray boxes denote conserved loops of the AMP-forming CoA ligase family [39]; dark gray box denotes catalytic lysine.
Chimeras of SlAcs and SeAcs Reveal Regions in the SeAcs C-terminal Domain that are Critical for Acetylation by SlPatA
Based on regions of sequence conservation (Fig. 2B), we generated a set of precise fusions between the SlAcs and SeAcs that contained varying amounts of the SeAcs protein. A SlAcs chimera containing the SlAcs N-terminal domain fused to the SeAcs C-terminal (chimera A1) was strongly acetylated by SlPatA, confirming that the C-terminal domain of SlAcs was responsible for the poor acetylation of SlAcsWT (Figs. 3A, B).
Figure 3. Construction and acetylation of SlAcs-SeAcs chimeras.

A. A scheme of SlAcsWT (white) and SeAcsWT (grey) drawn to scale. Target lysine K610 for SlAcsWT and K609 aligned and depicted by “K”. The N-terminal domains (520 residues) are shortened with a hatch in all remaining panels. B. Schematic representation of SlAcs-SeAcs chimeras A1–A6 in which the C-terminal portion of SlAcsWT (white) was replaced with the corresponding amino acid sequence from SeAcsWT (gray). All chimeras are drawn to scale for reference to the target lysine (denoted by “K”). Numbers all denote the fusion points with respect to the SlAcs protein sequence (i.e. either the first residue of SlAcsWT replaced by SeAcsWT sequence or the first residue of SlAcsWT after the SeAcsWT amino acid sequence). C. Acetylation of SlAcs-SeAcs chimeras A1–A6 and B1–B6 using SlPatAWT and [1-14C]acetyl-CoA. D. Schematic or SlAcs-SeAcs chimers B1–B6 in which internal portion of the C-terminal SlAcs domain are replaced with the corresponding sequence from SeAcsWT. E. Schematic of chimeras C1–C5. F. Acetylation of SlAcs-SeAcs chimeras C1–C6.
We identified regions of the SeAcs C-terminal domain important for acetylation by SlPatA by constructing chimeras that contained decreasing amounts of the SeAcs C-terminal domain relative to chimera A1. To measure the efficiency of acetylation, each chimera was incubated with SlPatA and radiolabeled [1-14C] acetyl-CoA. SlPatA strongly acetylated chimeras containing at least the final 86 amino acids of SeAcs (chimeras A1, A2, A3, A4; Fig. 3). These chimeras contained at least 43 amino acids N-terminal to the acetylation site, a region previously reported to be important for acetylation of homologous AMP-forming CoA ligase enzymes by the R. palustris Pat homologue [17].
To narrow down the number of SeAcs residues required for acetylation of the SlAcs-SeAcs chimeras, we focused on SlAcs-SeAcs chimera A2, which had the fewest SeAcs-derived residues (Fig. 3B), and the highest level of acetylation (Fig. 3C).
We generated a second set of chimeras in which various stretches of SlAcs-derived residues were substituted into SlAcs-SeAcs chimera A2 (Fig. 3D). SlAcs-SeAcs chimeras B4, B5, and B6 that contained at least 45 residues of SeAcs (including the SeAcsK609 acetylation site) were strongly acetylated (Fig. 3C). Notably, the A10 loop of Acs, which contains the target lysine, is completely conserved between SeAcs and SlAcs (Fig. 2B). However, 17 amino acids C-terminal to the acetylation site of SeAcs were required for acetylation by SlPatA. This revealed a previously unrecognized region of the protein important for acetylation. Of this set of SlAcs-SeAcs chimeras, chimera B4 was the best substrate of SlPatA and contained the fewest SeAcs-derived amino acids.
To determine whether the 77 contiguous SeAcs-derived residues of chimera B4 were critical for acetylation, we identified regions of SlAcs and SeAcs with low amino acid sequence conservation and introduced those sets of SlAcs residues into chimera B4 (Fig. 3E). Of those tested, only SlAcs-SeAcs chimera C3 was acetylated with similar efficiency as SeAcs (Fig. 3F). Acetylation of each chimera was quantified relative to acetylation of SeAcs (Fig. 4A, gray bars).
Figure 4. SlAcs-SeAcs Chimera C3 is active and efficiently acetylated.

A. Acetyl-CoA synthetase activity of each chimera and SlAcsWT relative to SeAcsWT (gray bars). Amount of acetylation in figure 3C and 3F was quantified and normalized to the total acetylation of SeAcs (black bars). SlAcs-SeAcs chimera C3, the most efficiently acetylated, active chimera with the fewest SeAcsWT-derived residues, is noted with a star. Values are reported as the mean ± S.D. of three experiments. B. Sequence alignment of SlAcsWT, SeAcsWT, chimera C3, Rhodopseudomonas palustris CGA009 Acs (RpAcs), and Mycobacterium smegmatis mc2155 Acs (MsAcs). Residues in chimera C3 that are derived from the SeAcsWT amino acid sequence are highlighted in black. SlAcs residues conserved in the MsAcs homologue are shown in bold typeface in the sequence of the latter. Black box indicates the target lysine.
Importantly, chimeras containing only the 60 SeAcs-derived residues N-terminal to K610 (chimera B2) or 11 SeAcs-derived residues C-terminal to K610 (chimera C5) were <30% acetylated relative to SeAcs. Thus, amino acid sequences N- and C-terminal to the target lysine were important for acetylation by SlPatA, and neither set of amino acids rendered SlAcs a strong acetylation target when introduced independently.
Assessment of the Enzymatic Activity of the Chimeras
Chimeras were tested for their AMP-forming acetyl-CoA ligase forming activity. Although the SlAcsWT and SeAcsWT C-terminal domains share a high degree of sequence conservation, not all chimeras were active (Fig. 4A, black bars). To identify active chimeras that were also targets of acetylation, the acetylation of each chimera was measured relative to SeAcs (Fig. 4A, gray bars). SlAcs-SeAcs chimera C3 (hereafter referred to as chimera C3) was identified as the single chimera with the fewest SeAcs residues that was active and efficiently acetylated by SlPatA. As shown in figure 4B, chimera C3 contained 41 amino acid differences from SlAcs. For comparison, we include the analogous sequence from Acs homologues known to acetylated by protein acetyltransferases in other bacteria. Notably, the wildtype SlAcs amino acid sequence replaced by SeAcs sequences shares some sequence homology with these Acs homologues.
Chimera C3 Activity is Modulated by Acetylation and Deacetylation
As shown in figure 5A, the catalytic residue K610 residue is the only residue of chimera C3 that was acetylated. To test whether the activity of chimera C3 was under the control of acetylation, the protein was incubated with SlPatA acetyltransferase in the presence and absence of the acetyl donor, acetyl-CoA. Upon acetylation, chimera C3 activity decreased >98%, similar to the regulation of SeAcs activity (Fig. 5B, gray bar). The SlAcs enzyme retains >75% activity upon incubation with SlPatA and Ac-CoA (Fig. 5B, gray bar). As mentioned above, acetylation of SeAcsWT is reversed by the NAD+-dependent sirtuin deacetylase CobB in S. enterica, and deacetylation reactivates the SeAcsWT enzyme [2]. We tested whether chimera C3 could be deacetylated by incubating acetylated chimera C3 with SeCobB, the co-substrate NAD+, or both. When SeCobB and NAD+ were present in the reaction mixture, chimera C3Ac was completely deacetylated (Fig. 5C), demonstrating that the reversibility of the acetylation process was not affected by the substitutions in chimera C3.
Figure 5. Chimera C3 is regulated by reversible lysine acetylation.
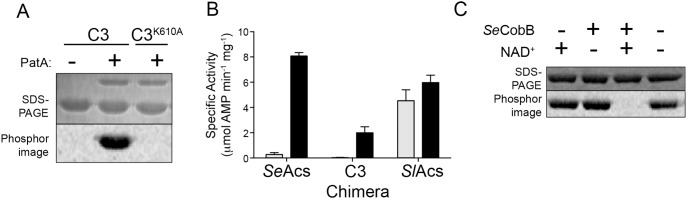
A. Chimera C3 or chimera C3K610A was incubated with [1-14C]-acetyl-CoA in the presence or absence of SlPatAWT. Proteins were separated by SDS-PAGE and stained with Coomassie Blue R to visualize proteins. Acetylation was visualized by phosphor imaging. B. Chimera C3, SeAcsWT, or SlAcsWT was incubated in the presence (grey bars) or absence (black bars) of SlPatAWT. Samples were removed, diluted, and assayed to measure acetyl-CoA synthetase activity after 90 min incubation with SlPatAWT. Acs activity was measured in an NADH-consumption assay. Values are reported as the mean ± S.D. of three experiments. C. Chimera C3 previously acetylated by SlPatAWT with [1-14C]-acetyl-CoA was incubated with the addition of SeCobBWT and/or NAD+. Proteins were resolved by SDS-PAGE and stained with Coomassie Blue R to visualize proteins. Acetylation was visualized by phosphor image.
SlAcs-SeAcs Chimera C3 is Acetylated in vivo in S. enterica by SlpatA
To determine the efficiency of SlPatA acetylation of chimera C3 in vivo, we used S. enterica acetate utilization (Fig. 1A) as a heterologous model to demonstrate the effects of SlPatA acetylation on activity of the Acs homologues. In this system, His-tagged chimera C3, SlAcs, and SeAcs (H6-chimera C3, H6-SlAcs, H6-SeAcs, respectively) were produced from plasmids in S. enterica acs pat cobB and S. enterica acs pat cobB + strains JE9152 and JE9894, respectively. All the experiments were conducted in S. enterica pat strains to prevent acetylation by SePat. We characterized the effect of SlPatA acetylation on the H6-Acs homologues by measuring growth of each strain harboring a plasmid with an inducible SlPatA allele or an empty cloning vector. Additionally, we isolated H6-SeAcsWT, H6-SlAcsWT, and H6-chimera C3 from cells grown in the presence or absence of SlPatA to quantify the effects of SlPatA acetylation on each Acs protein. As shown in figure 6A, production of H6-chimera C3, H6-SlAcs, or H6-SeAcs supported growth of S. enterica acs pat cobB strain (open symbols). This was the expected result, since the strain lacked Pat activity, thus the cell could not acetylate (i.e., inactivate) any of the Acs enzymes. We attributed the lag in the strain producing H6-chimera C3 to the decreased activity of this chimera (Fig. 5B). When production of SlPatA was induced in each strain (25 µM inducer), growth of S. enterica acs pat cobB strains producing H6-SeAcsWT or H6-chimera C3 was significantly reduced, while growth of the S. enterica acs pat cobB strain producing H6-SlAcsWT was unaffected. Importantly, inhibition of an S. enterica acs cobB strain producing H6-SlAcsWT required high levels of SlPatAWT induction (500 µM inducer, Fig. 6B). No growth inhibition occurred when SlPatAWT was induced at low levels (≤5 µM inducer, Fig. 6C).
Figure 6. Chimera C3 is regulated by SlPatA in vivo in S. enterica.

Growth behavior of S. enterica in NCE minimal medium supplemented with acetate (10 mM). A. Growth of S. enterica Δacs pat ΔcobB producing H6-SeAcsWT (triangles), H6-SlAcsWT (circles), or H6-Chimera C3 (squares) harboring either a plasmid expressing SlPatAWT (filled shapes) or an empty vector (open shapes). All media was supplemented with 25 µM IPTG. B. Growth of S. enterica Δacs pat ΔcobB (JE9152) producing H6-SlAcsWT harboring a plasmid producing SlPatAWT induced with IPTG concentrations of 25 µM (open circles), 50 µM (light gray), 100 µM (medium gray), 250 µM (dark gray), or 500 µM (black). For reference, half-filled circles denote an equivalent strain producing H6-SlAcsWT harboring an empty vector induced with 500 µM IPTG. C. Growth of S. enterica Δacs pat ΔcobB (JE9152) producing H6-SeAcsWT (triangles), H6-SlAcsWT (circles), H6-Chimera C3 (squares), or empty vector (diamonds) harboring either a plasmid expressing SlPatAWT (filled shapes) or an empty vector (open shapes). All media was supplemented with 5 µM IPTG. D. Growth of S. enterica Δacs pat cobB + (JE9894) producing H6-SeAcsWT (triangles), H6-SlAcsWT (circles), or H6-Chimera C3 (squares) harboring either a plasmid expressing SlPatAWT (filled shapes) or an empty vector (open shapes). All media was supplemented with 25 µM IPTG. E. Growth of S. enterica Δacs pat cobB + (JE9894) producing H6-SlAcs-SeAcs chimera C3 harboring a plasmid producing SlPatAWT induced with IPTG concentrations of 10 µM (open triangle), 25 µM (light gray), 500 µM (medium gray), 100 µM (dark gray), or 250 µM (black). For reference, the inverted, filled triangles denote the growth of an equivalent strain producing H6-SlAcs-SeAcs chimera C3 harboring an empty vector (no SlPatAWT) induced with 500 µM IPTG. F. S. enterica Δacs pat cobB + strains (JE9894) producing H6-SeAcsWT (circles) or H6-SlAcsWT (squares) are shown growing with high induction (250 µM IPTG) of empty vector control (open symbols) or a plasmids expressing SlPatA. F. All S.D. <0.015 absorbance units.
As expected, the presence of SeCobBWT in a S. enterica acs pat cobB+ strain resulted in no significant growth defects upon SlPatAWT induction in strains expressing H6-SlAcsWT or H6-SeAcsWT (Fig. 6D). However, we did note a slight inhibition of growth of a S. enterica acs pat cobB+ strain producing H6-chimera C3. We surmised that such an effect was likely due to a decreased ability of SeCobBWT to deacetylate and reactivate H6-chimera C3 and restore growth. This idea was supported by the observation that increased induction of SlPatAWT inhibited a S. enterica acs pat cobB+ strain producing H6-chimera C3 (Fig. 6E), but not those producing H6-SlAcsWT nor H6-SeAcsWT (Fig. 6F).
Since high levels of SlPatA induction were required to inhibit growth of an S. enterica acs cobB strain producing H6-SlAcsWT, we expected that H6-SlAcsWT to be poorly acetylated by SlPatAWT and thus more active in vivo. We also expected higher proportions of acetylated to non-acetylated H6-SeAcsWT and H6-chimera C3 in vivo. To measure the effect of SlPatAWT acetylation on the activity of H6-SlAcs of, H6- SeAcs of, and H6-chimera C3, we grew S. enterica acs cobB strains expressing the corresponding acs alleles while inducing SlPatAWT at low levels (5 µM) to allow for growth and biomass accumulation for all strains (Fig. 6C). H6-SlAcsWT, H6-SeAcsWT and H6-chimera C3 enzymes were isolated from strains harboring plasmid-borne SlPatAWT or empty vector.
As shown in figure 7, activity of the H6-SlAcsWT enzyme isolated from a strain producing SlPatAWT was not significantly reduced compared to H6-SlAcsWT isolated from a strain with no SlPatAWT. However, activities of the H6-SeAcsWT and H6-chimera C3 enzymes were significantly lower when isolated from strains expressing SlPatAWT compared to those with no SlPatAWT. Activities of the SeAcs and H6-chimera C3 were restored upon incubation with SeCobB deacetylase. These data suggested that SlPatAWT more efficiently acetylated H6-SeAcsWT and H6-chimera C3 than it did H6-SlAcs in a heterologous in vivo model.
Figure 7. Activities of Chimera C3 and SeAcsWT are reduced in strains expressing SlPatAWT.
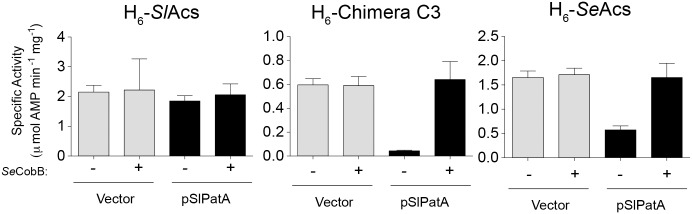
H6-Chimera C3, H6-SlAcs, and H6-SeAcs were produced in S. enterica Δacs pat ΔcobB strain JE9152 harboring either a plasmid producing SlPatAWT or an empty vector. Strains were grown in NCE minimal medium supplemented with acetate (10 mM). Acs proteins were incubated in the presence or absence of SeCobB deacetylase and its co-substrate NAD+. Acs activity was measured in an NADH-consumption assay. Values are reported as the mean ± S.D. of three activity measurements.
Discussion
Herein we report the first Acs enzyme that is not a substrate of Pat homologues in vitro. This finding is important, since Acs is the paradigm for the analysis RLA in all metabolic systems reported thus far. Our results begin to shed some light onto why the SlAcs is not efficiently acetylated by the SlPatAWT enzyme of S. lividans. By constructing chimeras of SlAcs that are acetylated by SlPatWT and retain biological activity we gained insights into structural, physiological and possibly evolutionary questions raised by this work.
Is Acs Activity under RLA Control in Streptomycetes?
At present, the answer to this question is unclear. It is not known whether SlAcsWT is a bona fide substrate of SlPatAWT in vivo in S. lividans. The literature adds to the challenge of determining whether or not in streptomycetes Acs is under RLA control. Work performed by others in Streptomyces coelicolor suggested that the Acs enzyme of this actinomycete may be under RLA control, because results of in vitro experiments showed that acetylated ScAcs was a substrate of a sirtuin deacetylase present in that bacterium [16]. The same authors also reported the isolation of acetylated ScAcs from S. coelicolor. Since the S. coelicolor genome contains a gene encoding a SlPatA homologue, they concluded that ScAcs was under RLA control.
Our initial work with the S. lividans SlPatAWT and SlAcsWT enzymes paints a complex picture for the regulation of SlAcsWT function in this organism, and by extrapolation, maybe in S. coelicolor. Because the specific activity of SlAcsWT is similar to that of SeAcsWT in vitro (Fig. 5B), we hypothesize that SlAcsWT activity is also tightly controlled by S. lividans. To account for the inability of SlPatAWT to acetylate SlAcsWT, we propose that SlPatAWT has evolved unique strategies for substrate recognition, or SlPatAWT is not the primary modifier of SlAcsWT. We discuss each possibility further below.
In vitro, SlPatAWT does not Recognize SlAcsWT
Pat homologues acetylate Acs in R. palustris and S. enterica [10], [12]. Clearly, acetylation of SlAcsWT by SlPatAWT does not occur efficiently in vitro or in a heterologous model system (Figs. 4B, 5A, 6A, 7) [18]. The following possibilities should be taken into consideration when thinking about the potential regulation of SlAcsWT by RLA. First, it is possible that SlAcsWT may have evolved to evade acetylation by SlPatAWT. Secondly, since S. lividans encodes ∼72 predicted GNAT-type acetyltransferases (Pfam00583) it is possible that one of these GNATs, not SlPatAWT, acetylates SlAcsWT. If a GNAT other than SlPatA acetylated SlAcs, it begs the questions of what selective pressure drove the conformational change SlAcs to avoid recognition by SlPatA, and what the physiological benefits of such a change are. And thirdly, the reversed domain organization of SlPatA, relative to RpPat and SePat, may prevent recognition of SlAcsWT by SlPatAWT.
Substantial Changes in the C-terminal Domain of SlAcsWT Lead to its Recognition by SlPatAWT
Forty-one amino acid changes in the C-terminal domain of SlAcsWT were needed to allow SlPatWT to recognize and acetylate SlAcs (Fig. 3). If we assumed that the domain organization of SlPatWT was not a factor in SlAcsWT recognition, such a large number of substitutions would suggest that the protein underwent dramatic evolutionary changes to prevent modification by SlPatWT. Importantly, we note that some SlAcs sequences that were replaced in the C3 chimera exhibit homology to Acs homologues that are acetylated by GNAT enzymes in other bacteria (Fig. 4B). This suggests acetylation of Acs and other AMP-forming acyl-CoA synthetases cannot be predicted by amino acid sequence [17]. Our results indicate, however, that SlAcs recognition by SlPatAWT is reversible by mutation, and that the resulting SlAcs variant can be reversibly acetylated.
How do Changes in the C-terminal Domain of SlAcs Affect its Acetylation and Activity?
Studies of R. palustris Pat (RpPat) substrate specificity indicate that this enzyme recognizes a loop >20 residues N-terminal to the target lysine in the substrate protein, suggesting that the RpPat interacts with a relatively large surface of substrate proteins [17]. Here, we demonstrate that the identities of residues ranging from 8–52 amino acids N-terminal to the target lysine of SeAcsWT, in combination with 5–17 amino acids C-terminal to the target lysine of SeAcsWT are critical for recognition of this substrate by SlPatAWT in isolation. This indicates that SlPatAWT recognizes several regions of the SeAcs C-terminal domain including the target lysine, residues N-terminal to the target lysine, and residues C-terminal to the target lysine. It is possible that these regions of the SeAcs C-terminal domain are necessary for direct interactions with the SlPatA protein. Alternatively, these regions may be necessary to position the target lysine for entry into the SlPatA active site. The crystal structure of SlPatAWT substrate SeAcsWT is known (PDB 1PG3, 1PG4) [33]. Comparison of this structure with the structure of SlAcs (structure not known) may distinguish these possibilities. Efforts to obtain the crystal structure of SlAcs are ongoing.
Is SlAcsWT Regulated by One or More Protein Acetyltransferases?
As mentioned above, SlAcsWT may have evolved to evade acetylation specifically by SlPatAWT. However SlAcsWT may be acetylated in vivo by one of the additional 72 predicted GNAT-type acetyltransferases (Pfam00583) encoded by the genome of this bacterium or by an enzyme independent pathway. The possibility that an alternative GNAT acetylates SlAcsWT more efficiently than SlPatAWT does is not unprecedented. It is known that the genome of R. palustris encodes a Pat homologue and a single-domain GNAT protein acetyltransferase that share overlapping protein acetyltransferase substrates, and that both enzymes acetylate these substrates with different affinities [13]. Alternatively, SlAcsWT may be acetylated directly and non-enzymatic by the reactive metabolite acetyl-phosphate. This phenomenon has been characterized in E. coli and has been shown to affect the activity of the target enzymes [37], [38]. Therefore, the possibility of SlPatAWT not being the sole regulator of SlAcsWT activity in S. lividans needs to be further investigated.
Does the Unique Domain Organization of SlPatAWT Affect Substrate Specificity?
Pat acetyltransferases are two-domain enzymes composed of a GNAT (acetyltransferase) domain and a large domain whose function is likely regulatory. In SlPatAWT, the GNAT domain is located at the N-terminus of the protein [18]. In contrast, in R. palustris and S. enterica, the domain order is reversed (i.e., GNAT domain is at the C-terminus of protein). SlPatAWT also has a collagen-like Gly-Pro-Ser motif in the large domain [18]. S. enterica and R. palustris Pat homologues efficiently acetylate their cognate Acs enzymes in vitro [10], [12]. The alternate domain organization of SlPatAWT may account for the poor acetylation of SlAcsWT compared to SeAcsWT and SlAacSWT in vitro [18]. If SlPatAWT has evolved strategies for recognition of protein substrates differently from SePat and RpPat, our in vitro assay may be missing a factor that promotes efficient SlPatAWT recognition of SlAcsWT such as a small molecule, macromolecule (e.g. protein), or an as-yet-unidentified intracellular condition. If this were the case, the amino acid changes introduced into SlAcsWT to generate the SlAcs-SeAcs chimera C3 obviate the need for additional factors or conditions for SlPatAWT recognition.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All data are included in the manuscript.
Funding Statement
Funding was provided by United States Public Health Service, National Institutes of Health grant R01 GM062203. The funders had no role in the design, data collection and analysis, decision to publish or preparation of the manuscript.
References
- 1.Soppa J (2010) Protein acetylation in archaea, bacteria, and eukaryotes. Archaea 2010: pii: 820681. [DOI] [PMC free article] [PubMed]
- 2. Starai VJ, Celic I, Cole RN, Boeke JD, Escalante-Semerena JC (2002) Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298: 2390–2392. [DOI] [PubMed] [Google Scholar]
- 3. Liang W, Malhotra A, Deutscher MP (2011) Acetylation regulates the stability of a bacterial protein: growth stage-dependent modification of RNase R. Mol Cell. 44: 160–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kouzarides T (2007) Chromatin modifications and their function. Cell 128: 693–705. [DOI] [PubMed] [Google Scholar]
- 5. Vetting MW, Carvalho LPSd, Yu M, Hegde SS, Magnet S, et al. (2005) Structure and functions of the GNAT superfamily of acetyltransferases. Arch Biochem Biophys 433: 212–226. [DOI] [PubMed] [Google Scholar]
- 6. Thao S, Escalante-Semerena JC (2011) Control of protein function by reversible N(epsilon)-lysine acetylation in bacteria. Curr Opin Microbiol 14: 200–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brownell JE, Allis CD (1995) An activity gel assay detects a single, catalytically active histone acetyltransferase subunit in Tetrahymena macronuclei . Proc Natl Acad Sci U S A 92: 6364–6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rojas JR, Trievel RC, Zhou J, Mo Y, Li X, et al. (1999) Structure of Tetrahymena GCN5 bound to coenzyme A and a histone H3 peptide. Nature 401: 93–98. [DOI] [PubMed] [Google Scholar]
- 9. Kuo YM, Andrews AJ (2013) Quantitating the specificity and selectivity of Gcn5-mediated acetylation of histone H3. PLoS One 8: e54896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Starai VJ, Escalante-Semerena JC (2004) Identification of the protein acetyltransferase (Pat) enzyme that acetylates acetyl-CoA synthetase in Salmonella enterica . J Mol Biol 340: 1005–1012. [DOI] [PubMed] [Google Scholar]
- 11. Starai VJ, Escalante-Semerena JC (2004) Acetyl-coenzyme A synthetase (AMP forming). Cell Mol Life Sci 61: 2020–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Crosby HA, Heiniger EK, Harwood CS, Escalante-Semerena JC (2010) Reversible N(epsilon)-lysine acetylation regulates the activity of acyl-CoA synthetases involved in anaerobic benzoate catabolism in Rhodopseudomonas palustris . Mol Microbiol 76: 874–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Crosby HA, Pelletier DA, Hurst GB, Escalante-Semerena JC (2012) System-wide studies of N-lysine acetylation in Rhodopseudomonas palustris reveal substrate specificity of protein acetyltransferases. J Biol Chem 287: 15590–15601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gardner JG, Grundy FJ, Henkin TM, Escalante-Semerena JC (2006) Control of acetyl-coenzyme A synthetase (AcsA) activity by acetylation/deacetylation without NAD(+) involvement in Bacillus subtilis . J Bacteriol 188: 5460–5468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Xu H, Hegde SS, Blanchard JS (2011) Reversible acetylation and inactivation of Mycobacterium tuberculosis acetyl-CoA synthetase is dependent on cAMP. Biochemistry 50: 5883–5892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mikulik K, Felsberg J, Kudrnacova E, Bezouskova S, Setinova D, et al. (2012) CobB1 deacetylase activity in Streptomyces coelicolor . Biochem Cell Biol 90: 179–187. [DOI] [PubMed] [Google Scholar]
- 17. Crosby HA, Rank KC, Rayment I, Escalante-Semerena JC (2012) Structural insights into the substrate specificity of the protein acetyltransferase RpPat: identification of a loop critical for recognition by RpPat. J Biol Chem 287: 41392–41404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Tucker AC, Escalante-Semerena JC (2013) Acetoacetyl-CoA synthetase activity is controlled by a protein acetyltransferase with unique domain organization in Streptomyces lividans . Mol Microbiol 87: 152–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bertani G (1951) Studies on lysogenesis. I. The mode of phage liberation by lysogenic Escherichia coli . J Bacteriol 62: 293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berkowitz D, Hushon JM, Whitfield HJ Jr, Roth J, Ames BN (1968) Procedure for identifying nonsense mutations. J Bacteriol 96: 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Guzman LM, Belin D, Carson MJ, Beckwith J (1995) Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177: 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan SR, Gaines J, Roop RM 2nd, Farrand SK (2008) Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol 74: 5053–5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Elion EA, Marina P, Yu L (2007) Constructing recombinant DNA molecules by PCR. In: F. M Ausubel, R. E. R Brent, D. D Kingston, J. G Moore, J. A Seidman, a Smith and K Struhl, editors. Current protocols in molecular biology. New York, N.Y.: Greene Publishing Associates & Wiley Interscience. Unit 3.17.11–13.17.12. [DOI] [PubMed]
- 24. Horton RM, Ho SN, Pullen JK, Hunt HD, Cai Z, et al. (1993) Gene splicing by overlap extension. Methods Enzymol 217: 270–279. [DOI] [PubMed] [Google Scholar]
- 25. Rocco CJ, Dennison KL, Klenchin VA, Rayment I, Escalante-Semerena JC (2008) Construction and use of new cloning vectors for the rapid isolation of recombinant proteins from Escherichia coli . Plasmid 59: 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Blommel PG, Fox BG (2007) A combined approach to improving large-scale production of tobacco etch virus protease. Protein Expr Purif 55: 53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Miroux B, Walker JE (1996) Over-production of proteins in Escherichia coli: mutant hosts that allow synthesis of some membrane proteins and globular proteins at high levels. J Mol Biol 260: 289–298. [DOI] [PubMed] [Google Scholar]
- 28. Garrity J, Gardner JG, Hawse W, Wolberger C, Escalante-Semerena JC (2007) N-lysine propionylation controls the activity of propionyl-CoA synthetase. J Biol Chem 282: 30239–30245. [DOI] [PubMed] [Google Scholar]
- 29. Tucker AC, Escalante-Semerena JC (2010) Biologically active isoforms of CobB sirtuin deacetylase in Salmonella enterica and Erwinia amylovora . J Bacteriol 192: 6200–6208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685. [DOI] [PubMed] [Google Scholar]
- 31.Sasse J (1991) Detection of proteins. In: F. A Ausubel, R Brent, R. E Kingston, D. D Moore, J. G Seidman, J. A Smith and K Struhl, editors. Current Protocols in Molecular Biology. New York: Wiley Interscience. 10.16.11–10.16.18.
- 32. Starai VJ, Takahashi H, Boeke JD, Escalante-Semerena JC (2003) Short-chain fatty acid activation by acyl-coenzyme A synthetases requires SIR2 protein function in Salmonella enterica and Saccharomyces cerevisiae . Genetics 163: 545–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Gulick AM, Starai VJ, Horswill AR, Homick KM, Escalante-Semerena JC (2003) The 1.75Å crystal structure of acetyl-CoA synthetase bound to adenosine-5′-propylphosphate and coenzyme A. Biochemistry. 42: 2866–2873. [DOI] [PubMed] [Google Scholar]
- 34. Reger AS, Wu R, Dunaway-Mariano D, Gulick AM (2008) Structural characterization of a 140° domain movement in the two-step reaction catalyzed by 4-chlorobenzoate: CoA ligase. Biochemistry 47: 8016–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kyte J, Doolittle RF (1982) A simple method for displaying the hydropathic character of a protein. J Mol Biol 157: 105–132. [DOI] [PubMed] [Google Scholar]
- 36. Shirai A, Matsuyama A, Yashiroda Y, Hashimoto A, Kawamura Y, et al. (2008) Global analysis of gel mobility of proteins and its use in target identification. J Biol Chem 283: 10745–10752. [DOI] [PubMed] [Google Scholar]
- 37. Weinert BT, Iesmantavicius V, Wagner SA, Scholz C, Gummesson B, et al. (2013) Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli . Mol Cell 51: 265–272. [DOI] [PubMed] [Google Scholar]
- 38. Kuhn ML, Zemaitaitis B, Hu LI, Sahu A, Sorensen D, et al. (2014) Structural, kinetic and proteomic characterization of acetyl phosphate-dependent bacterial protein acetylation. PLoS One 9: e94816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gulick AM (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem Biol 4: 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All data are included in the manuscript.