
Figure 1.
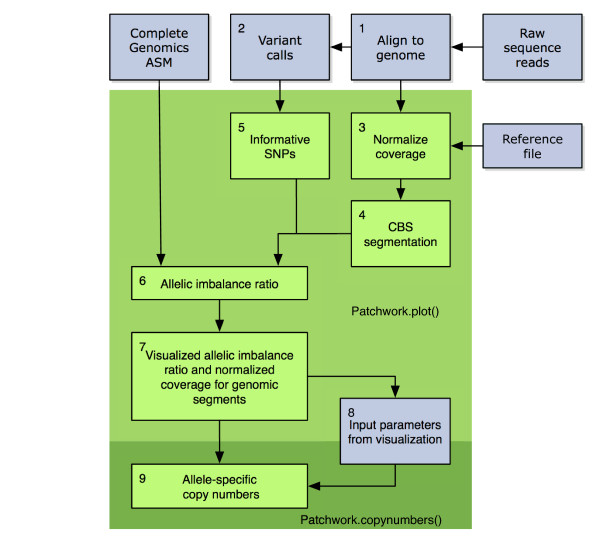
Patchwork flowchart. 1) Sequenced reads are aligned to the genome. 2) Single-nucleotide (and optionally short indel) variants that do not match the reference genome are extracted. 3) Systematic bias is removed by normalizing for GC content and other position-related effects. Coverage information from one or more diploid samples sequenced with the same method is used in this process. 4) The genome is segmented, based on the normalized coverage. 5) Informative heterozygous variants are identified. 6) Allelic imbalance ratio is calculated for each segment. 7) Visualization of allelic imbalance ratio and normalized coverage for genomic segments. 8) Manual interpretation of visualization to obtain input parameters for next step. 9) Allele-specific copy number is calculated for all genomic segments. Steps 3 to 7 and 9 are handled by the Patchwork.plot() and Patchwork.copynumbers() modules respectively.