

Abstract
Microbial metalloenzymes constitute a large library of biocatalysts, a number of which have already been shown to catalyze the breakdown of toxic chemicals or industrially relevant chemical transformations. However, while there is considerable interest in harnessing these catalysts for biotechnology, for many of the enzymes, their large-scale production in active, soluble form in recombinant systems is a significant barrier to their use. In this work, we demonstrate that as few as three mutations can result in a 300-fold increase in the expression of soluble TrzN, an enzyme from Arthrobacter aurescens with environmental applications that catalyzes the hydrolysis of triazine herbicides, in Escherichia coli. Using a combination of X-ray crystallography, kinetic analysis, and computational simulation, we show that the majority of the improvement in expression is due to stabilization of the apoenzyme rather than the metal ion-bound holoenzyme. This provides a structural and mechanistic explanation for the observation that many compensatory mutations can increase levels of soluble-protein production without increasing the stability of the final, active form of the enzyme. This study provides a molecular understanding of the importance of the stability of metal ion free states to the accumulation of soluble protein and shows that differences between apoenzyme and holoenzyme structures can result in mutations affecting the stability of either state differently.
INTRODUCTION
Approximately half of all known proteins contain a metal ion cofactor in their mature form (1). These metal ions can play structural or catalytic roles, and their incorporation into apoenzyme folding intermediates is an important step in the overall folding pathway (2). One of the most diverse superfamilies of enzymes, which includes a large number of microbial enzymes that are capable of catalyzing the hydrolysis of toxic synthetic compounds (3–5), is the metal ion-dependent amidohydrolase superfamily (6, 7). Unfortunately, many of these proteins form insoluble aggregates or are produced only at very low levels when overexpressed in recombinant systems.
There is a need to improve heterologous protein expression in order to enable us to characterize proteins in the laboratory or to produce proteins on a large scale for industrial or medical purposes. Heterologous overexpression of proteins is often less than ideal because of protein aggregation or degradation (8). This can arise due to differences between the native folding situation and that which occurs when expression levels are much higher and in a different host, such as a lack of suitable folding chaperones, different pH, high local concentration of the protein, or the absence of a cofactor, which is particularly important for metalloproteins. The technique of laboratory evolution has great potential for improving soluble-protein production and involves subjecting the protein to random mutagenesis and recombination, followed by screening of the many variants that are produced for enhanced expression (9, 10). Although there have been a few successful examples of rationally engineering proteins to enhance expression of the soluble form (11), our limited understanding of protein folding and stability currently imposes a limit on this approach. Thus, a simple means by which mutations that increase expression can be identified would be very useful.
Stabilizing mutations are also of interest in the context of molecular evolution and protein engineering; the “trade-off” between protein stability and function as a result of the accumulation of mutations during evolution is relatively well understood at a conceptual level. Because most mutations that change protein function are destabilizing, each step toward a new function increases the probability that the protein will become less stable (12–14). To overcome this, proteins must rely on the actions of molecular chaperones to sustain them in their unstable states or accumulate compensatory, stabilizing mutations (15, 16). Indeed, there is a clear association between the operational stability of a protein (the point at which it irreversibly unfolds) and its tolerance for amino acid substitutions, or evolvability (17–19). However, recent work has shown that some compensatory mutations that increase or maintain protein solubility do not affect the stability of the functional protein (20), leading to the suggestion that they might instead specifically stabilize folding intermediates along the pathway to the mature protein. Although this is well characterized at the phenotypic level, our structural understanding of the phenomenon is lagging, and it is unclear how these compensatory mutations can improve or rescue poorly expressed proteins.
The triazine hydrolase (TrzN) from Arthrobacter aurescens TC1 (21, 22) is one example of a potentially useful enzyme for which low levels of soluble-protein production in recombinant systems is a problem. This Zn2+-dependent metalloenzyme has evolved since the introduction of the s-triazine herbicides atrazine and ametryn (23) to efficiently hydrolyze and detoxify these compounds (21, 24). TrzN is potentially of great environmental value; atrazine and ametryn are widely used (25) and environmentally persistent (26), which has resulted in toxicologically relevant concentrations of atrazine being detected in both surface and ground water in several countries (27–29). Although not acutely toxic to vertebrates (23), both short- and long-term exposure to atrazine concentrations above the maximum contaminant level in drinking water can potentially cause a number of chronic health problems (30), while off-target effects on aquatic organisms and vegetation present further environmental concerns (31, 32). The breakdown of triazine herbicides by TrzN provides a possible solution to the environmental and public health problems associated with their use without sacrificing the significant economic benefits they provide in crop production (33). Indeed, the addition of a free-enzyme formulation of TrzN to contaminated waterways has been shown to significantly reduce atrazine concentrations (34). However, native TrzN cannot be produced at even moderate yields in Esherichia coli, thereby limiting its usefulness.
In this work, we subjected TrzN to a directed-evolution experiment in order to increase its soluble-protein production. Variants that were produced at significantly higher levels than the native protein were then analyzed structurally and kinetically to determine the molecular basis of these improvements.
MATERIALS AND METHODS
Mutagenesis and library screening.
Codon-optimized trzN was kindly provided by S. Kotsonis (Orica Australia, Ltd., Australia) and was used as the template in all subsequent work. Random mutagenesis was performed using GeneMorph II (Stratagene, USA) and the primers TrzNF and TrzNR (see Table S2 in the supplemental material), with a mutagenesis rate of 1 to 2 per kbp. The amplicons produced by random mutagenesis were cloned into pET14b (Novagen) via the NdeI and BamHI restriction sites (Fermentas). Site-directed mutagenesis of trzN and variants was performed by the overlapping-PCR method using the primers TrzNF and TrzNR and appropriate mutagenic primers (see Table S2 in the supplemental material). The amplicons produced by site-directed mutagenesis were cloned into pET14b via the NdeI and BamHI restriction sites.
Libraries of TrzN variants were screened by plate-clearing assay. LB agar was supplemented with 1 g liter−1 Gesaprim 900 WG (90% [wt/wt] atrazine; Syngenta), producing a homogeneous suspension of atrazine. E. coli BL21 λDE3 (Novagen) cells were transformed with libraries of random trzN mutants, plated to a density of approximately 500 CFU per petri dish (9-cm diameter), and grown at 37°C for up to 2 weeks. At least 40,000 colonies were screened in each round of mutagenesis. Colonies that developed clearing zones more rapidly than colonies containing the mutagenesis template were picked, their plasmids were isolated (Nucleospin; Macherey-Nagel), and their trzN genes were sequenced (Micromon; Monash University, Melbourne, Australia).
Protein expression, purification, and quantification.
Derivatives of pET14b containing trzN and its variants were used to transform BL21 λDE3 cells. Protein expression was induced by incubation at 37°C for 48 h in LB medium containing 1 μM isopropyl-β-d-1-thiogalactopyranoside (IPTG) (Sigma). The expression cultures were harvested by centrifugation (5,800 × g), resuspended in binding buffer (50 mM HEPES [Sigma], 50 mM NaCl [Ajax Finechem], pH 7.5), and then lysed using either Bugbuster (Novagen) or a French pressure cell. The cell lysates were clarified by centrifugation (20,000 × g), and soluble TrzN and variants were purified from the clarified cell extracts by immobilized metal affinity chromatography (Talon Co2+ resin; Clontech) and size exclusion chromatography (Superdex 200; GE Healthcare Life Sciences) on an AKTA fast protein liquid chromatography (FPLC) system (Amersham Biosciences). Fractions (0.5 ml) were collected at a flow rate of 0.1 ml/min, and absorbance was measured at 280 nm. Protein purity was monitored using precast reducing 4 to 20% SDS-PAGE gels (NuSep) stained with Coomassie brilliant blue (Sigma). The protein was stored in 50 mM HEPES, pH 7.5, 100 mM NaCl. The enzyme concentration was determined by measuring absorbance at 280 nm in 6 M urea, using the extinction coefficient 61,670 M−1 cm−1 for a monomer. The level of expression of variants, relative to the total soluble protein was estimated using the public domain software Image J (35), which can calculate area and pixel value statistics of user-defined selections. Briefly, scanned SDS-PAGE images were converted to 8-bit grayscale JPEG files, two-dimensional (2D) plots of gel lanes were produced, and the area of the peak corresponding to TrzN was calculated and compared to the area corresponding to the total soluble protein.
Proteins were coexpressed with a variety of chaperone systems to test whether expression could be enhanced. The chaperone coexpression systems (TaKaRa, Japan) were (i) the GroES/GroEL complex (pGro7) and (ii) GroES/GroEL in combination with DnaK, DnaJ, and GrpE (pG-KJE8) (36). Cultures containing these plasmids were grown in the presence of the antibiotic markers ampicillin (50 μg/ml) and chloramphenicol (20 μg/ml). Coexpression of the chaperones was carried out using the inducing agent arabinose at a concentration of 0.15 mg/liter, consistent with previous work (37). Coexpression of chaperones was confirmed using SDS-PAGE.
Enzyme assays.
Hydrolysis of atrazine was quantified spectrophotometrically (SpectraMax 190) by the decrease in absorbance at 264 nm that accompanies atrazine dechlorination, as described previously (38). Assays and standard curves used an analytical standard of >99.5% pure atrazine (Sigma), and all measurements were made in triplicate. The values for Vmax and Km were determined from a fit of the data to the following equation (equation 1):
| (1) |
where υ is the initial velocity, Vmax is the maximum velocity, Km is the Michaelis constant, and A is the substrate concentration. The kcat was calculated by dividing Vmax by the concentration of enzyme used in the reaction (Vmax = kcat[E]).
To measure the metal ion binding affinity, apo-TrzN was incubated with increasing concentrations of Zn2+ for 30 min before measurement of the rate of atrazine dechlorination (Vmax). The data were fitted to the following equation (equation 2):
| (2) |
where Kd is the metal ion concentration at which 50% maximum activity is regained, assuming one metal ion per subunit.
Protein stability measurements.
Enzyme stability was measured by incubating the enzymes at six temperatures between 40 and 60°C for 15 min prior to kinetic analysis at 25°C. The residual activity (Vmax) was then plotted as a function of the incubation temperature; this generated a sigmoidal curve in which the inflection point, or Tm50(app) (melting point), of the transition between folded and unfolded protein is calculated using a Boltzmann function (equation 3):
| (3) |
where ymin and ymax are the values of the minimum and maximum activities, T is the temperature of preincubation, Tm [Tm50(app) for residual-activity measurements] is the melting temperature, and λ denotes the slope of the curve within Tm.
For the measurement of apoenzyme stability and comparison to the holoenzyme, thermal unfolding of the proteins was studied by measuring the molar ellipticity of protein samples using a Chirascan circular-dichroism (CD) spectropolarimeter (Applied Photophysics) with a quartz cell at 220 nm. A linear ramp of temperature was performed from 20°C to 80°C, with a band width of 1 nm and a ramping rate of 1°C/minute. The signal from a buffer sample was subtracted from each run. The transition between folded and unfolded protein was calculated using equation 3.
Crystallization, data collection, and structure determination.
Both proteins were concentrated to 20 mg/ml in crystallization buffer (50 mM HEPES, pH 7.5, 100 mM NaCl). The crystallization used a protocol slightly modified from that previously used to obtain crystals of native TrzN (38). For holo-TrzN-G3, the crystallization buffer also included 100 μM ZnCl2. Crystals grew several days after mixing the protein solution with precipitant solution (100 mM Bis-Tris [2,2-bis(hydroxymethyl)-2,2′,2″-nitrilotriethanol], pH 5.5, 200 mM NH4SO4, 30% [wt/vol] polyethylene glycol [PEG] 3350) in the hanging droplets (2 μl reservoir:1 μl protein) over a reservoir consisting of the precipitant solution. The concentration of PEG 3350 was increased to 35% for use as a cryoprotectant during flash cooling of the crystals to 100 K under a stream of nitrogen gas. Diffraction data for apo-TrzN-G3 were collected at the MX2 beamline of the Australian Synchrotron at a wavelength of 1.2398 Å. Diffraction data for holo-TrzN-G3 were collected at ID23-2 of the European Synchrotron Radiation Facility at a wavelength of 0.8726 Å. The diffraction data were indexed and integrated with the XDS package (39) and scaled using AIMLESS (40). Molecular replacement solutions were found using MOLREP (41), and the structure was refined with REFMAC (42) and phenix.refine (43). Iterative model building was performed using COOT (44). Anomalous difference density maps were produced for the apo-TrzN-G3 structure as described previously (45). All protein structure diagrams were produced using PyMol (46).
Computational simulations.
Starting structures for apo-TrzN-G3 were generated from the 1.8-Å-resolution crystal structure (see Table 2). The positions of residues in the native TrzN structure were taken from native holo-TrzN (Protein Data Bank [PDB] accession no. 3LS9) (38). Hydrogens were constructed with ideal stereochemistry. Protonation states of histidines were assigned by calculating pKa values with the PROPKA program (47). The orientations of His, Asn, and Gln side chains in the active site were taken from the crystal structure and verified by visual inspection. We employed a strategy that has been calibrated and shown to give reliable results when calculating free energies for other protein systems. Specifically, we deleted residues that did not have at least one nonhydrogen atom within a sphere with a radius of 24 Å centered on the active site. This gave a protein system ∼50 Å in size that contained all the important catalytic residues. The resulting system was then solvated in a cubic 70-Å box of water that was replicated periodically in all directions to allow infinite bulk simulations and an accurate calculation of the long-range electrostatic interactions. Significant distortions of the protein were prevented by harmonically restraining all protein atoms between 20 and 24 Å from the sphere's center to their experimentally determined positions. All long-range electrostatic interactions were computed with the particle mesh Ewald (PME) method. Fourteen potassium counterions were included in the complex to render the simulation systems neutral. Simulations were performed at constant temperature and pressure with Berendsen's algorithm. The complexes were simulated for 5 ns each following 200 ps of thermalization. The CHARMM27 force field (48) was used for the protein and a slightly modified TIP3P model for the water, in which the difference is in the Lennard-Jones parameters, as in the CHARMM version of the model the hydrogens, as well as the oxygen, have Lennard-Jones interactions (49). We used the NAMD program (50), version 2.7, for all molecular dynamics (MD) simulations.
TABLE 2.
Data collection and refinement statistics for structures reported in this work
| Statistic | Valuea |
|
|---|---|---|
| Holo-TrzN-G3 | Apo-TrzN-G3 | |
| Space group | P1 21 1 | P1 21 1 |
| Unit cell parameters | ||
| a (Å) | 57.09 | 56.11 |
| b (Å) | 101.80 | 101.27 |
| c (Å) | 79.89 | 77.23 |
| β (°) | 103.2 | 100.98 |
| Data collection | ||
| Wavelength (Å) | 0.87260 | 1.2398 |
| Resolution range (Å) | 48.78–1.80 (1.90–1.80) | 48.34–1.85 (1.95–1.85) |
| No. of unique reflections | 82,249 | 72,046 |
| Redundancy | 3.9 (3.8) | 3.7 (3.7) |
| Completeness (%) | 100 (100) | 100 (100) |
| Rmerge(I)b | 0.071 (0.610) | 0.025 (0.521) |
| Mean <I/σ(I)> | 11.0 (2.1) | 14.6 (2.4) |
| Refinement | ||
| No. of reflections (total) | 82,036 | 68,057 |
| Resolution range | 48.8–1.8 (1.82–1.80) | 19.8–1.85 (1.90–1.85) |
| Rwork/Rfreec | 0.1767/0.2181 (0.2804/0.3067) | 0.1884/0.1914 (0.292/0.289) |
| RMSd deviations | ||
| Bond lengths (Å) | 0.006 | 0.008 |
| Bond angles (°) | 0.966 | 1.108 |
| PDB ID | 4LH8 | 4L9X |
Values in parentheses are for the highest-resolution shell.
Rmerge(I) = (Σhkl Σj |Ihkl,j − 〈Ihkl〉|)/(Σhkl Σj Ihkl,j), where 〈Ihkl〉 is the average intensity of j symmetry-related observations of reflections with Miller indices hkl.
Rwork = Σhkl|F(obs) − F(calc)|/Σhkl|F(obs)|; 5% of the data that were excluded from the refinement were used to calculate Rfree.
RMS, root mean square.
To compare the stabilities of variant and native TrzN, we employed a standard thermodynamic cycle (51, 52). The molecular dynamics free energy (MDFE) method reversibly transforms alanine into valine during a series of MD simulations. For the folded state, we simulated a portion of the TrzN complex in the center of a 70-Å water box, as explained above. For the unfolded state, we simulated a tripeptide consisting of a central alanine or valine with flanking alanines solvated by the same 70-Å water box. Previous work had shown that this model of the unfolded state gives good agreement between experimental and computed stability free energies for other protein systems (53). In the simulations, we gradually changed alanine into valine by increasing the coupling parameter, λ, from zero to one with the values 0.001, 0.01, 0.05, 0.1, 0.2, 0.4, 0.6, 0.8, 0.9, 0.95, 0.99, and 0.999 (54). The free-energy derivatives were computed at each λ value from a 100-ps MD simulation, or window. The last 80 ps of each window were used for averaging. A complete mutation run consisted of 12 windows and 1.2 ns of simulation. Ten runs were performed in each direction, starting from equilibrated structures separated by 0.2 ns of dynamics. This gave 24 ns of simulation time per protein, or 72 ns of simulation in total. Stability free-energy differences were calculated as the differences in the computed free energies of the unfolded, reference, and folded protein simulations. To estimate the uncertainty in the MDFE simulation results, we performed forward-backward pairs of runs, one in each direction, and took the uncertainty to be the corresponding standard deviation.
To test whether we could computationally predict the thermodynamic stability changes of mutations in a high-throughput manner, we used the protein design tool FoldX (version 2.52), following a four-step procedure that has previously been described in detail (14, 55, 56). The structure of apophosphotriesterase (apo-PTE) (PDB accession no. 1PTA) was obtained from the PDB and optimized using the repair function of FoldX before structures of the A80V variant generated in holo-PTE and apo-PTE were constructed using the buildmodel command, as well as self-mutations (e.g., A80A). The energy calculation function of FoldX was used to calculate the energies of the variants, which were compared with the energies of the native structures to obtain ΔΔG values. These were then normalized based on prior work (ΔΔGEXP = [ΔΔGFOLDX + 0.78]/1.14) (56).
Protein structure accession numbers.
The coordinates of holo-TrzN-G3 and apo-TrzN-G3 have been deposited in the PDB under accession numbers 4LH8 and 4L9X, respectively.
RESULTS AND DISCUSSION
Laboratory evolution to increase whole-cell dechlorinase activity.
When heterologously produced in E. coli, only very small amounts (<0.2 mg protein per liter Terrific Broth [TB] medium) of correctly folded, active native TrzN (TrzN-N) were produced (Fig. 1), consistent with previous work (37). To improve the yield of its soluble form, we performed laboratory evolution, using a method that we have previously used with related metallohydrolases from Pseudomonas sp. strain ADP (AtzA [atrazine chlorohydrolase]) and Agrobacterium radiobacter (phosphotriesterase) (5, 57). This involved screening libraries of random variants using an agar plate-based activity screen in which substrates were included in the agar gel at concentrations in excess of the solubility limit, resulting in opaque plates. Because of the greater solubility of the breakdown products, enzyme-catalyzed substrate hydrolysis conferred a “clearing” phenotype, in which an area of translucent agar is visible around colonies expressing active enzyme.
FIG 1.
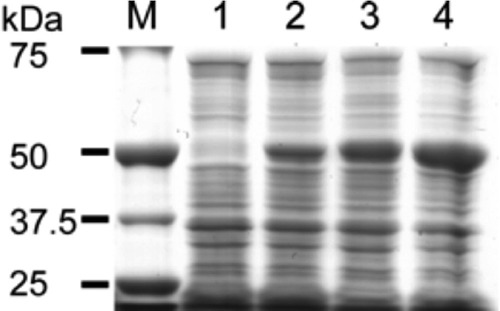
SDS-PAGE showing the increase in expression of soluble TrzN by laboratory evolution. The molecular mass marker is shown in lane M. Lanes 1 to 4 show the increasing cellular concentrations of expression of soluble TrzN in rounds 1 (lane 2; A159V), 2 (lane 3; A159V/L131P), and 3 (lane 4; A159V/L131P/D38N) compared with the native TrzN (lane 1).
Three rounds of random mutagenesis with recombination were performed, screening mutant libraries of ∼50,000 colonies per round. As expected from the very low levels of soluble expression, colonies producing TrzN-N took approximately 350 h of incubation at room temperature before exhibiting a “clearing” phenotype (see Fig. S1 in the supplemental material). However, after three generations of random mutagenesis and recombination, the whole-cell activity of the most active variant (TrzN-G3; D38N/L131P/A159V) had increased so that clearing occurred in less than 12 h. SDS-PAGE and kinetic analysis indicated that the vast majority of this improvement in whole-cell activity was a result of at least 336-fold-increased expression (<0.2 mg/liter versus 67.2 mg/liter), with the kinetic parameters changing little from previously published values (Fig. 1 and Table 1) (37). Similar results were obtained from gel densitometry, where the percentage of total protein increased from less than 0.1% to approximately 7%.
TABLE 1.
Effects of mutations in TrzN on protein yield, stability, and kinetic parametersa
| Amino acidsb | Yield (mg/liter) | Tm50(app) (oC) (holo-TrzN) | kcat (s−1) | Km (μM) | kcat/Km (105 M−1 s−1) |
|---|---|---|---|---|---|
| D, L, A | <0.2 | 52.1 | 2.1 | 19.7 | 1.1 |
| N, L, A | <0.1 | ND | 2.6 | 21.3 | 1.2 |
| D, P, A | 0.6 | 51.3 | 3.1 | 48.6 | 0.6 |
| D, L, V | 8.4 | 51.8 | 2.8 | 18.9 | 1.5 |
| N, P, A | 1.2 | 52.3 | 3.2 | 52.2 | 0.6 |
| N, L, V | 14.4 | 54.4 | 2.6 | 21.7 | 1.2 |
| D, P, V | 28.2 | 51.0 | 3.2 | 54.2 | 0.6 |
| N, P, V | 67.2 | 53.6 | 3.7 | 53.0 | 0.7 |
Yield was measured as purified protein, stability was measured as the temperature at which 50% activity was lost (68), and kinetic parameters were measured with atrazine as the substrate using a previously published protocol (38). The Tm50(app) values could not be determined for the majority of the apoenzyme samples because the protein immediately unfolded upon removal of the metal ion.
Amino acids at positions 38, 131, and 159, respectively.
A combinatorial library of the three mutations from the highest-expressing variant was created to analyze their effects on the expression and stability of the enzyme. The effects of the A159V and L131P mutations in the wild-type background on soluble-protein yield appear to be additive: the A159V mutation has the largest effect in isolation (42-fold), followed by L131P (3-fold). At the double-mutant stage, addition of the L131P mutation to the A159V background again led to a 3-fold improvement in soluble-protein production. In contrast, the D38N mutation had no measurable effect in the wild-type background but resulted in ∼2-fold improvement in soluble-protein production once combined with the A159V mutation in rounds 2 and 3.
We observed that the majority of protein production for TrzN-N was in the insoluble fraction and that the amino acid substitutions did not significantly affect the ratio of soluble to insoluble protein, i.e., TrzN-G3 exhibited an increase in both soluble- and insoluble-protein production (see Fig. S2 in the supplemental material). Additionally, soluble-TrzN-N production decreased over time, from a maximum of 0.4% total protein between 2 and 5 h to 0.2% protein from 6 to 20 h, as measured by gel densitometry, unlike TrzN-G3 (see Fig. S3 in the supplemental material), which comprised approximately 2% of the total soluble protein throughout the 20-hour induction period. This suggests that TrzN-N is significantly less stable and that a significant proportion of the protein that successfully folds to a soluble state eventually unfolds, leading to degradation/proteolysis. The large amount of insoluble protein indicates that protein aggregation is a more common fate than proteolysis/degradation. It is unlikely that the improved levels of soluble-protein production are a result of improved protein translation, since the native protein is expressed at high levels but in an insoluble form. Additionally, the mRNA transcripts from these variants differ by only 1 bp out of 1,359.
There are three points at which synthesized metalloprotein can be degraded or aggregate, leading to a reduction in soluble-protein accumulation: (i) folding of the nascent polypeptide, (ii) unfolding/aggregation of the apoenzyme, or (iii) unfolding/aggregation of the holoenzyme. To understand how these mutations have resulted in such a large increase in soluble-TrzN-G3 accumulation, we investigated all three possibilities.
Folding chaperones do not significantly improve production of soluble TrzN.
Previous work has shown that overexpression of native folding chaperones does not significantly improve the expression and stability of TrzN-N (37). To confirm this and to investigate in more detail, we coexpressed TrzN-N with two sets of chaperonins/chaperones known to aid the folding of nascent polypeptides at different stages: (i) the GroES/GroEL complex, which binds to polypeptides and encapsulates them in an environment that strongly favors compact protein folding and (ii) GroES/GroEL in combination with DnaK, DnaJ, and GrpE, which prevent aggregation/degradation of nascent polypeptides via binding to hydrophobic patches as they are released from the ribosome (36).
The use of folding chaperones/chaperonins did not significantly improve the yield of soluble protein (see Fig. S4 in the supplemental material). Although this is not uncommon for recombinant proteins, it does suggest that the initial folding steps of TrzN-N are less likely to be the bottleneck to its soluble expression than instability of one of the folded states, either with or without bound metal ions.
Holoenzyme stabilization has minimal effect on the level of soluble-protein accumulation.
To test the effects of the three mutations on the stability of the fully folded, active, Zn2+-bound holoenzyme (holo-TrzN-N), we measured residual activity after heating at a range of temperatures (Table 1). Of the three substitutions, the only one that appeared to have a stabilizing effect on holo-TrzN-N is D38N: in the L131P background it resulted in a 1°C increase in Tm50(app), in the A159V background a 2.6°C increase, and in the L131P/A159V background a 2.6°C increase. In contrast, the A159V mutation had virtually no effect on holoenzyme stability, and the L131P mutation was marginally destabilizing in most combinations. Given the large change in soluble expression, it is notable that the Tm50(app) of the final 3rd-round variant holo-TrzN-G3 (D38N/L131P/A159V) is only 1.5°C higher than that of holo-TrzN-N.
To investigate the effects of these mutations on the metal-bound form of the enzyme in more detail, we solved the crystal structure of holo-TrzN-G3 by molecular replacement, using the published native holo-TrzN coordinates (Table 2) (38). N38 is located in a solvent-exposed loop between strands 12 and 13 in the peripheral β-sandwich domain and extends into the solvent in the same orientation as D38 in TrzN-N (Fig. 2A). Although there is no obvious structural explanation for its stabilizing effect, it is possible that this mutation results in a subtle change to the protein hydration shell. L131 is located on a loop of the upper face of the (β/α)8 barrel domain between strands 2 and 3 and partly constitutes the substrate-binding pocket (Fig. 2C). The only significant difference in the kinetic parameters between TrzN-N and the variants in Table 1 is a slight increase in the kcat and Km in variants containing L131P. This is consistent with a minor increase in the volume of the active site as a result of the L131P mutation. Finally, A159 is located on strand 3 of the (β/α)8 barrel domain, beneath the metal ion binding site, and fills a small hydrophobic cavity (Fig. 2E). The A159V mutation does not significantly change the structure in this region. Thus, apart from a slight increase in the size of the active-site binding pocket, the tertiary structures of TrzN-G3 and TrzN-N are almost identical.
FIG 2.
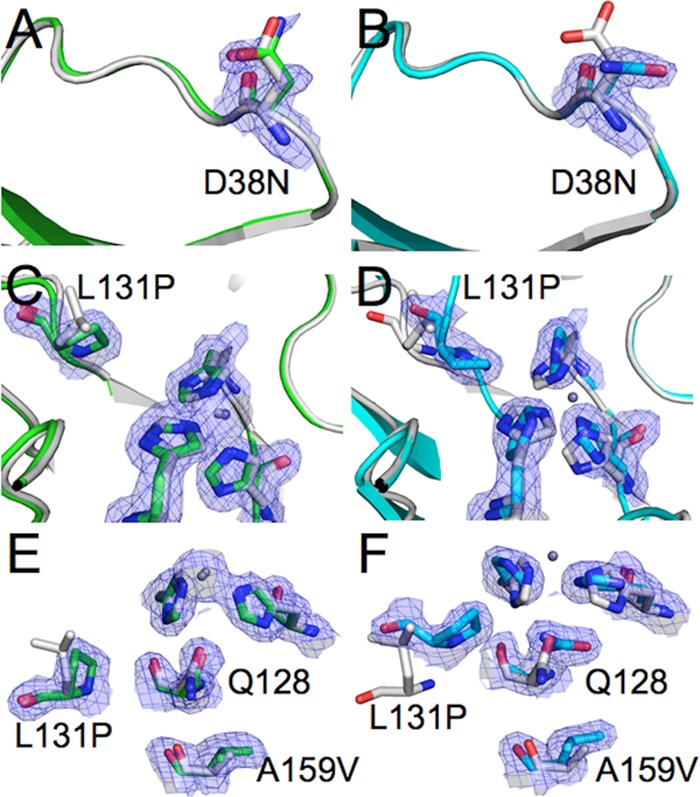
Effects of the mutations in holo-TrzN-G3 (green) (A, B, and C) and apo-TrzN-G3 (cyan) (D, E, and F). (A and D) The structure of native holo-TrzN is shown in gray for comparison. The D38N mutation results in little conformational change in the holo-TrzN-G3 structure, while an alternative rotamer is adopted in apo-TrzN-G3. (B and E) The L131P mutation results in an increase in the volume of the active-site binding pocket but no other backbone changes in holo-TrzN-G3, whereas in apo-TrzN-G3, the proline residue stabilizes a tight turn in the alternative backbone conformation. (C and F) The A159V mutation results in a slight increase in an already very small cavity in native holo-TrzN, whereas in the rearranged structure of apo-TrzN-G3, a much larger cavity is formed, which is filled by the A159V mutation.
The insignificant structural changes shown here are consistent with the insignificant changes to the stability of the holoenzyme. To test the possibility that the mutations affected another state of the protein, we turned our attention to the metal-ion-free apoenzyme, apo-TrzN.
Apo-TrzN is less stable than holo-TrzN.
The publication of the structure of native TrzN confirmed that it is a Zn2+-dependent metalloenzyme (38). Closer inspection of the active site reveals a unique metal ion coordination sphere in the amidohydrolase superfamily, in which a conserved aspartic acid ligand has been lost. Eight subtypes of metal ion coordination sites have been reported previously in the amidohydrolase family (4, 7), making TrzN the first characterized member of the 9th subtype. It is known that the loss of an aspartic acid metal ion ligand can lower the metal ion binding affinity of enzymes (58). Indeed, the metal ion affinity of TrzN-G3 is low (49 μM) (see Fig. S5 in the supplemental material) in comparison to intracellular levels of free Zn2+ in E. coli (femtomolar) (59), which is potentially a major problem when metalloproteins, such as TrzN, are overexpressed recombinantly from strong promoters at levels several orders of magnitude higher than in their native host. Indeed, prior work has shown that even when excess Zn2+ is added to the expression medium, overexpressed metalloenzymes often accumulate primarily as the apoenzyme and form the holoenzyme only once they bind metal ions postlysis (60). By knocking out the genes involved in Zn2+ homeostasis in E. coli, it was established that tight control of the free Zn2+ concentration is essential to prevent Zn2+-induced cell toxicity (61). Thus, although excess Zn2+ was provided during cell growth, the ability of E. coli to maintain low intracellular free-Zn2+ concentrations means that, when overproduced, TrzN is likely to spend a significant amount of time as an apoenzyme, making the stability of this state crucial to the accumulation of the protein.
It was impossible to analyze the stability of apo-TrzN-N quantitatively because the purified protein immediately denatured upon chelation/extraction of the metal ion. Although full quantification of its stability was not possible, the half-life of apo-TrzN-N at 4°C is less than 1 h, unlike holo-TrzN-N, which was stable at this temperature for several weeks. Therefore, it can be concluded that apo-TrzN-N is significantly less stable than holo-TrzN-N. In contrast, apo-TrzN-G3 was substantially more stable, so much so that when purified without excess metal ions in the lysis or purification buffers, the apoenzyme form was the dominant species isolated from E. coli cells and was stable in purified form for several weeks. We were thus able to measure its unfolding using circular-dichroism spectroscopy, determining that it is 3.5°C less stable than the holo-TrzN-G3 (see Fig. S6 in the supplemental material).
Stabilizing apo-TrzN results in increased soluble-protein accumulation.
Having established that the apoenzyme forms of both TrzN-N and TrzN-G3 are less stable than the holoenzyme forms and that the stability of apo-TrzN-G3 is significantly increased through incorporation of the mutations, we investigated the effects of the three mutations on the apoenzyme in detail by solving the crystal structure of apo-TrzN-G3 (Table 2). To confirm that no metal ions were present in the crystal, we used anomalous X-ray scattering by collecting diffraction data slightly above the Zn-K absorption edge using tuneable synchrotron radiation (45). Neither subunit within the asymmetric unit was found to contain significant levels of Zn (see Fig. S7 in the supplemental material).
The structure of apo-TrzN-G3 is significantly different from that of holo-TrzN-G3 in two regions, with the C-α main chain adopting a different position in the adjacent loops 2 (residues 131 to 139) and 3 (residues 161 to 179) (Fig. 3a), which constitute part of the metal ion binding site and substrate binding cavity. In the absence of the metal ion, loop 2 collapses inward to fill the active-site cavity, mainly via the side chain of F132. The ability of TrzN-G3 to fluctuate between these two quite different conformations in the presence and absence of metal ions is shown in Fig. 3b, in which the B subunit of the holo-TrzN-G3 crystal is shown. This subunit has only 50% metal ion occupancy, and the two conformations can clearly be seen simultaneously in the same crystal. There are also a number of changes in the metal-ligating residues: in apo-TrzN-G3, H65 and H67 share a hydrogen bond and Q128 shares hydrogen bonds with H238 and H236 (Fig. 2F). Large conformational changes to metal ion coordination residues and loops in the absence of metal ions are not uncommon in metallohydrolases of this superfamily (62, 63).
FIG 3.
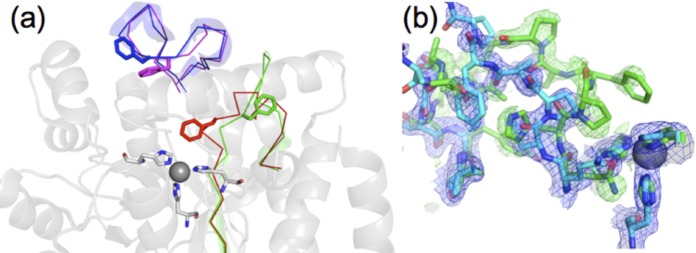
Comparison between apo-TrzN-G3 and holo-TrzN-G3. (a) Two loops undergo conformational change as a result of metal binding, with loop 2 (H130-Y141) in apo-TrzN-G3 (red) moving out of the active site in holo-TrzN-G3 (green). Loop 3 (S161-P180) undergoes a smaller change from apo-TrzN-G3 (magenta) to holo-TrzN-G3 (blue), which results in opening of the gorge to the active site. (b) Density from a single protein crystal structure in which the Zn2+ site is occupied at only 50%. The 2mfo-Dfc, αcalc map (blue) is contoured at 1σ, with the holo-TrzN-like chain model colored cyan (50% occupancy); the mfo-Dfc, αcalc map (green) is contoured at 2.5σ, demonstrating that the apo-TrzN-like chain (green; 50% occupancy) is also present in this structure. The second subunit of the dimer (subunit A) had 100% metal ion occupancy and no detectable density corresponding to apo-TrzN.
Unlike the holo-TrzN-G3 structure, the stabilizing effects of the L131P and A159V mutations can be easily rationalized in the apo-TrzN-G3 structure. The L131P mutation precedes F132 on loop 2, which is inserted into the active-site cavity. This area of the loop undergoes a tight bend. The cyclic backbone formed by proline is known to stabilize tight turns in polypeptide chains (Fig. 2D). Notably, in the holo-TrzN-N and holo-TrzN-G3 structures, where L131P is slightly destabilizing, the amino acid does not exist on a turn. The A159V mutation is the central residue in a large cavity immediately below the active site, which significantly increases in volume in the apoenzyme relative to the metal-bound form (Fig. 2F). The stabilizing effect of this residue, therefore, relates to its ability to fill and stabilize this cavity. In contrast, in the holo-TrzN-N and holo-TrzN-G3 structures, the size of the cavity does not significantly change. The N38D mutation has no clear effect on the apo-TrzN-G3 structure; it extends into the solvent, albeit in a slightly different conformation than in the holo-TrzNG3 or native structure (Fig. 2B). This analysis provides some explanation for the often-cited view that stability effects are too subtle to rationalize, i.e., the effects are apparent only when we investigate the state that they specifically affect.
To better understand the reasons for the stabilizing influence of the A159V substitution in apo-TrzN-G3, we carried out molecular-dynamics free-energy calculations. In brief, A159 was reversibly transformed to V159 during a series of MD simulations of both apo- and holo-TrzN, and also of a model of TrzN in an unfolded state. The stability free energies were then calculated as the differences between the mutation free energies computed for the unfolded reference state and those of the folded forms. The effect on the computed stability free energy of the A159V mutation on apo-TrzN was very large: a ΔΔG value of −4.29 kcal/mol compared to a ΔΔG value of −0.43 kcal/mol for holo-TrzN (see Table S1 in the supplemental material). Thus, our simulations are in agreement with the experimental data, which show that the A159V mutation strongly stabilizes apo-TrzN but that it has essentially no effect on the stability of holo-TrzN.
Compensatory mutations increase expression by increasing the stability of folding intermediates and can be predicted through calculation.
While most attention is understandably focused on engineering or manipulating the active metal-ion-bound form of potentially useful proteins, the work presented here suggests that it is the apoenzyme that is sometimes more important in the context of protein stability, production, and evolvability. Interestingly, over 1,000 apoenzyme crystal structures have been solved and deposited in the PDB, many of which have been unintentionally solved, since the active holoenzyme form is generally of much more interest. We suggest that structural reassessment of apoenzyme structures in combination with in silico screening procedures could be a facile route to rational apoenzyme stabilization for increased heterologous protein production, as opposed to directed-evolution experiments, for which the screening step can often be very difficult and/or costly in terms of time and resources. Because of the high demand for increased protein expression for laboratory, industrial, and medical applications, these findings could be used as a simple and effective means to increase the expression of various metalloproteins for which an apoenzyme structure is available.
To test whether the insight obtained in this work extends to other proteins and has applied potential, we tested whether mutations that stabilize apoenzymes could be predicted in a high-throughput manner using the protein design tool FoldX (64). We analyzed the structure of a second enzyme; the apoenzyme form of phosphotriesterase (1PTA), which was first solved in 1994 (65), 1 year before the holoenzyme structure (62). Three directed-evolution experiments have since been performed on bacterial PTEs to identify mutations that correlate with increased expression (60, 66, 67). Most recently, Wyganowski et al. and Tokuriki et al. performed an extensive directed-evolution experiment in which the activity was fully altered from phosphotriesterase to arylesterase. This required the accumulation of several compensatory mutations that did not affect the operational stability of the functional protein but did have significant effects on the expression of soluble PTE (20, 67). Eight mutations have been proposed to increase production of recombinant PTE: K77E, A80V, R111S, K185R, A204G, D208G, I274S, and R311S. These mutations were all made in silico in structures of apo-PTE (1PTA) and holo-PTE (1HZY). The results were interesting in that two of these mutations (A80V and R311S) were identified only as stabilizing apo-PTE. Structural analysis reveals an effect similar to that seen in TrzN (Fig. 4), in which conformational changes between the apo- and holoenzymes result in the mutations affecting each state very differently. The A80V mutation results in almost no conformational change in the holo-PTE structure, but in apo-PTE, it results in the adjacent L112 adopting a new rotamer in which it fills a hydrophobic cavity below. Similarly, the R311S mutation in holo-PTE changes a charged group to a polar group that extends into the solvent shell, but in apo-PTE, significant structural rearrangement results in R311 filling a cavity between F306 and M314, with the R311S mutation allowing F306 and M314 to collapse inward, filling the cavity and providing a stabilizing effect. This is consistent with the work of Roodveldt and Tawfik, where they demonstrated that recombinant PTE mostly exists as the apoenzyme within the cell (60).
FIG 4.
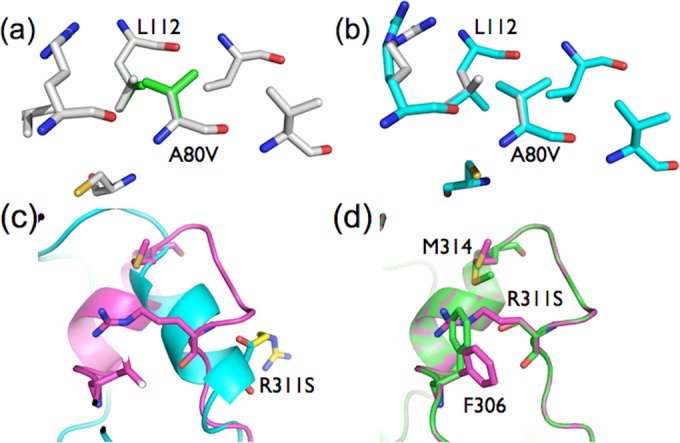
Effects of stabilizing mutations in holo-PTE and apo-PTE. (a) The A80V mutation (green) results in almost no conformational change in the holo-PTE structure (gray), only adding two methyl groups at the enzyme surface. (b) In contrast, in apo-PTE, the A80V mutation (cyan) is predicted to result in the adjacent leucine (L112) adopting a new rotamer, in which one of its methyl side chains fills a hydrophobic cavity below, which explains its predicted stabilizing effect on apo-PTE and increase in expression. (c) The R311S mutation in holo-PTE (cyan/yellow) results in the loss of a charged group on the protein surface. (d) In contrast, in apo-PTE, structural rearrangement of the apoeznyme results in R311 filling a cavity between F306 and M314. The R311S mutation allows F306 and M314 to collapse inward, filling the cavity and providing a stabilizing effect.
The observation that some other mutations (e.g., R111S, D204G, and I174S) are not predicted to increase soluble-PTE production by increasing apoenzyme stability is not surprising, since there are other possible mechanisms by which they might increase expression. Similarly, FoldX will never be completely accurate; for instance, a crystal structure reveals that the K185R mutation does stabilize PTE by introducing new salt bridges (68), but it is incorrectly modeled by FoldX (see Fig. S8 in the supplemental material).
It appears that, in the case of the two proteins examined in this work (TrzN and PTE), the stabilization of apoenzyme structure forms of metalloproteins can be an important mechanism by which evolution can maintain soluble-protein expression. Although only two examples are provided, this analysis suggests that stabilizing mutations, such as the A80V and R311S mutations in PTE, which were discovered approximately 10 to 20 years after publication of the structure of the apoenzyme (65) as a result of exhaustive directed-evolution experiments (66), might be easily predicted in other proteins by using computational algorithms similar to FoldX (64).
This work highlights the sometimes overlooked importance of apoenzyme and apoprotein stability to microbial physiology and to the overexpression and potential applications of the many catalytically useful proteins that we can obtain from prokaryotes. Heterologous expression of proteins effectively places unnatural and competing demands on cells: on one hand, they must maintain intracellular free metal ion concentrations at very low levels to prevent toxicity, while on the other hand they are forced to translate and fold unnaturally high levels of proteins that often require exogenous metal ions. Because of the complexity of metal ion homeostasis and the potential for toxicity, engineering metalloproteins to have greater stability in their apoprotein state seems to be the simplest solution to this problem.
Summary.
We used directed laboratory evolution to increase the levels of soluble, active TrzN heterologously produced by E. coli over 300-fold. The amino acid substitutions responsible for this improvement were rationalized at a molecular level, with a combination of protein crystallography and computational simulation, demonstrating that apoenzyme stabilization is the key process involved in the increase in soluble-protein production. This molecular explanation complements recent research that has established the essential role that compensatory mutations such as these play in molecular evolution. This work also makes a contribution to the goal of facile rational stabilization of protein structure for increased protein production, demonstrating that mutations that increase protein production in recombinant hosts can easily be predicted from existing apoenzyme structures.
Supplementary Material
ACKNOWLEDGMENTS
We thank Steve Kotsonis of Orica Australia Ltd. for providing the codon-optimized trzN gene. We thank the C3 Crystallisation Centre and the Australian Synchrotron. NAMD was developed by the Theoretical and Computational Biophysics Group in the Beckman Institute at the University of Illinois at Urbana. We thank T. Simonson for useful discussions.
C.J.J. thanks the ARC for funding.
Footnotes
Published ahead of print 25 April 2014
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00916-14.
REFERENCES
- 1.Thomson AJ, Gray HB. 1998. Bio-inorganic chemistry. Curr. Opin. Chem. Biol. 2:155–158. 10.1016/S1367-5931(98)80056-2 [DOI] [PubMed] [Google Scholar]
- 2.Rigby Duncan KE, Stillman MJ. 2006. Metal-dependent protein folding: metallation of metallothionein. J. Inorg. Biochem. 100:2101–2107. 10.1016/j.jinorgbio.2006.09.005 [DOI] [PubMed] [Google Scholar]
- 3.Jackson CJ, Liu JW, Coote ML, Ollis DL. 2005. The effects of substrate orientation on the mechanism of a phosphotriesterase. Org. Biomol. Chem. 3:4343–4350. 10.1039/b512399b [DOI] [PubMed] [Google Scholar]
- 4.Khurana JL, Jackson CJ, Scott C, Pandey G, Horne I, Russell RJ, Herlt A, Easton CJ, Oakeshott JG. 2009. Characterization of the phenylurea hydrolases A and B: founding members of a novel amidohydrolase subgroup. Biochem. J. 418:431–441. 10.1042/BJ20081488 [DOI] [PubMed] [Google Scholar]
- 5.Scott C, Jackson CJ, Coppin CW, Mourant RG, Hilton ME, Sutherland TD, Russell RJ, Oakeshott JG. 2009. Catalytic improvement and evolution of atrazine chlorohydrolase. Appl. Environ. Microbiol. 75:2184–2191. 10.1128/AEM.02634-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holm L, Sander C. 1997. An evolutionary treasure: unification of a broad set of amidohydrolases related to urease. Proteins 28:72–82 [PubMed] [Google Scholar]
- 7.Seibert CM, Raushel FM. 2005. Structural and catalytic diversity within the amidohydrolase superfamily. Biochemistry 44:6383–6391. 10.1021/bi047326v [DOI] [PubMed] [Google Scholar]
- 8.Wu W, Xing L, Zhou B, Lin Z. 2011. Active protein aggregates induced by terminally attached self-assembling peptide ELK16 in Escherichia coli. Microb. Cell Fact. 10:9. 10.1186/1475-2859-10-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jackson CJ, Liu JW, Carr PD, Younus F, Coppin C, Meirelles T, Lethier M, Pandey G, Ollis DL, Russell RJ, Weik M, Oakeshott JG. 2013. Structure and function of an insect alpha-carboxylesterase (alphaEsterase7) associated with insecticide resistance. Proc. Natl. Acad. Sci. U. S. A. 110:10177–10182. 10.1073/pnas.1304097110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Waldo GS. 2003. Genetic screens and directed evolution for protein solubility. Curr. Opin. Chem. Biol. 7:33–38. 10.1016/S1367-5931(02)00017-0 [DOI] [PubMed] [Google Scholar]
- 11.Crespo MD, Rubini M. 2011. Rational design of protein stability: effect of (2S,4R)-4-fluoroproline on the stability and folding pathway of ubiquitin. PLoS One 6:e19425. 10.1371/journal.pone.0019425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beadle BM, Shoichet BK. 2002. Structural bases of stability-function tradeoffs in enzymes. J. Mol. Biol. 321:285–296. 10.1016/S0022-2836(02)00599-5 [DOI] [PubMed] [Google Scholar]
- 13.Schreiber G, Buckle AM, Fersht AR. 1994. Stability and function: two constraints in the evolution of barstar and other proteins. Structure 2:945–951 [DOI] [PubMed] [Google Scholar]
- 14.Tokuriki N, Stricher F, Serrano L, Tawfik DS. 2008. How protein stability and new functions trade off. PLoS Comput. Biol. 4:e1000002. 10.1371/journal.pcbi.1000002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rutherford SL, Lindquist S. 1998. Hsp90 as a capacitor for morphological evolution. Nature 396:336–342 [DOI] [PubMed] [Google Scholar]
- 16.Tokuriki N, Tawfik DS. 2009. Chaperonin overexpression promotes genetic variation and enzyme evolution. Nature 459:668–673. 10.1038/nature08009 [DOI] [PubMed] [Google Scholar]
- 17.Bershtein S, Segal M, Bekerman R, Tokuriki N, Tawfik DS. 2006. Robustness-epistasis link shapes the fitness landscape of a randomly drifting protein. Nature 444:929–932. 10.1038/nature05385 [DOI] [PubMed] [Google Scholar]
- 18.Besenmatter W, Kast P, Hilvert D. 2007. Relative tolerance of mesostable and thermostable protein homologs to extensive mutation. Proteins 66:500–506. 10.1002/prot.21227 [DOI] [PubMed] [Google Scholar]
- 19.Bloom JD, Labthavikul ST, Otey CR, Arnold FH. 2006. Protein stability promotes evolvability. Proc. Natl. Acad. Sci. U. S. A. 103:5869–5874. 10.1073/pnas.0510098103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wyganowski KT, Kaltenbach M, Tokuriki N. 2013. GroEL/ES buffering and compensatory mutations promote protein evolution by stabilizing folding intermediates. J. Mol. Biol. 425:3403–3414. 10.1016/j.jmb.2013.06.028 [DOI] [PubMed] [Google Scholar]
- 21.Sajjaphan K, Shapir N, Wackett LP, Palmer M, Blackmon B, Tomkins J, Sadowsky MJ. 2004. Arthrobacter aurescens TC1 atrazine catabolism genes trzN, atzB, and atzC are linked on a 160-kilobase region and are functional in Escherichia coli. Appl. Environ. Microbiol. 70:4402–4407. 10.1128/AEM.70.7.4402-4407.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Udikovic-Kolic N, Scott C, Martin-Laurent F. 2012. Evolution of atrazine-degrading capabilities in the environment. Appl. Microbiol. Biotechnol. 96:1175–1189. 10.1007/s00253-012-4495-0 [DOI] [PubMed] [Google Scholar]
- 23.British Crop Protection Council. 2000. The pesticide manual, 12th ed. British Crop Protection Council, Hampshire, United Kingdom [Google Scholar]
- 24.Mulbry WW, Zhu H, Nour SM, Topp E. 2002. The triazine hydrolase gene trzN from Nocardioides sp. strain C190: cloning and construction of gene-specific primers. FEMS Microbiol. Lett. 206:75–79. 10.1111/j.1574-6968.2002.tb10989.x [DOI] [PubMed] [Google Scholar]
- 25.USDA. 2003. Agricultural chemical usage: field crops summary 2003. USDA, Washington, DC [Google Scholar]
- 26.Belluck DA, Benjamin SL, Dawson T. 1991. Groundwater contamination by atrazine and its metabolites—risk assessment, policy, and legal implications. ACS Symp. Ser. 459:254–273 [Google Scholar]
- 27.Adami G, Siviero P, Barbieri P, Piselli S, Reisenhofer E. 2001. Case study of groundwater pollution in a critical area of the Southern Friuli exposed to agricultural and landfill pressures. Ann. Chim. 91:531–540 [PubMed] [Google Scholar]
- 28.Graziano N, McGuire MJ, Roberson A, Adams C, Jiang H, Blute N. 2006. 2004 National atrazine occurrence monitoring program using the Abraxis ELISA method. Environ. Sci. Technol. 40:1163–1171. 10.1021/es051586y [DOI] [PubMed] [Google Scholar]
- 29.Hildebrandt A, Guillamon M, Lacorte S, Tauler R, Barcelo D. 2008. Impact of pesticides used in agriculture and vineyards to surface and groundwater quality (North Spain). Water Res. 42:3315–3326. 10.1016/j.watres.2008.04.009 [DOI] [PubMed] [Google Scholar]
- 30.U.S. Environmental Protection Agency. 2005. Consumer factsheet on atrazine. U.S. Environmental Protection Agency, Washington, DC [Google Scholar]
- 31.Hayes TB, Khoury V, Narayan A, Nazir M, Park A, Brown T, Adame L, Chan E, Buchholz D, Stueve T, Gallipeau S. 2010. Atrazine induces complete feminization and chemical castration in male African clawed frogs (Xenopus laevis). Proc. Natl. Acad. Sci. U. S. A. 107:4612–4617. 10.1073/pnas.0909519107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hayes TB, Collins A, Lee M, Mendoza M, Noriega N, Stuart AA, Vonk A. 2002. Hermaphroditic, demasculinized frogs after exposure to the herbicide atrazine at low ecologically relevant doses. Proc. Natl. Acad. Sci. U. S. A. 99:5476–5480. 10.1073/pnas.082121499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cooper J, Dobson H. 2007. The benefits of pesticides to mankind and the environment. Crop Protect. 26:1337–1348. 10.1016/j.cropro.2007.03.022 [DOI] [Google Scholar]
- 34.Scott C, Lewis S, Milla R, Taylor MC, Rogers A, Dumsday G, Russell RJ, Brodie J, Oakeshott JO. 2010. A free-enzyme catalyst for the bioremediation of environmental atrazine contamination. J. Environ. Manage. 91:2075–2078. 10.1016/j.jenvman.2010.05.007 [DOI] [PubMed] [Google Scholar]
- 35.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9:671–675. 10.1038/nmeth.2089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nishihara K, Kanemori M, Kitagawa M, Yanagi H, Yura T. 1998. Chaperone coexpression plasmids: differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of Japanese cedar pollen, Cryj2, in Escherichia coli. Appl. Environ. Microbiol. 64:1694–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shapir N, Pedersen C, Gil O, Strong L, Seffernick J, Sadowsky MJ, Wackett LP. 2006. TrzN from Arthrobacter aurescens TC1 is a zinc amidohydrolase. J. Bacteriol. 188:5859–5864. 10.1128/JB.00517-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Seffernick JL, Reynolds E, Fedorov AA, Fedorov E, Almo SC, Sadowsky MJ, Wackett LP. 2010. X-ray structure and mutational analysis of the atrazine chlorohydrolase TrzN. J. Biol. Chem. 285:30606–30614. 10.1074/jbc.M110.138677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W. 2010. XDS. Acta Crystallogr. D Biol. Crystallogr. 66:125–132. 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Evans PR. 2011. An introduction to data reduction: space-group determination, scaling and intensity statistics. Acta Crystallogr. D Biol. Crystallogr. 67:282–292. 10.1107/S090744491003982X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vagin A, Teplyakov A. 2010. Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66:22–25. 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 42.Murshudov GN, Skubak P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. 2011. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67:355–367. 10.1107/S0907444911001314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. 2012. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68:352–367. 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Emsley P, Lohkamp B, Scott WG, Cowtan K. 2010. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66:486–501. 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jackson CJ, Carr PD, Kim HK, Liu JW, Herrald P, Mitic N, Schenk G, Smith CA, Ollis DL. 2006. Anomalous scattering analysis of Agrobacterium radiobacter phosphotriesterase: the prominent role of iron in the heterobinuclear active site. Biochem. J. 397:501–508. 10.1042/BJ20060276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schrodinger LLC. 2010. The PyMOL molecular graphics system, version 1.5.0.4. Schrödinger LLC, Portland, OR [Google Scholar]
- 47.Li H, Robertson AD, Jensen JH. 2005. Very fast empirical prediction and rationalization of protein pKa values. Proteins 61:704–721. 10.1002/prot.20660 [DOI] [PubMed] [Google Scholar]
- 48.Brooks BR, Brooks CL, III, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, Won Y, Archontis G, Bartels C, Boresch S, Caflisch A, Caves L, Cui Q, Dinner AR, Feig M, Fischer S, Gao J, Hodoscek M, Im W, Kuczera K, Lazaridis T, Ma J, Ovchinnikov V, Paci E, Pastor RW, Post CB, Pu JZ, Schaefer M, Tidor B, Venable RM, Woodcock HL, Wu X, Yang W, York DM, Karplus M. 2009. CHARMM: the biomolecular simulation program. J. Comput. Chem. 30:1545–1614. 10.1002/jcc.21287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935 [Google Scholar]
- 50.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. 2005. Scalable molecular dynamics with NAMD. J. Comput. Chem. 26:1781–1802. 10.1002/jcc.20289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sham Y, Chu Z, Warshel A. 1997. Consistent calculations of pKa's of ionizable residues in proteins: semi-microscopic and microscopic approaches. J. Phys. Chem. B 101:4458–4472. 10.1021/jp963412w [DOI] [Google Scholar]
- 52.Simonson T, Carlsson J, Case DA. 2004. Proton binding to proteins: pK(a) calculations with explicit and implicit solvent models. J. Am. Chem. Soc. 126:4167–4180. 10.1021/ja039788m [DOI] [PubMed] [Google Scholar]
- 53.Seeliger D, de Groot BL. 2010. Protein thermostability calculations using alchemical free energy simulations. Biophys. J. 98:2309–2316. 10.1016/j.bpj.2010.01.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Aleksandrov A, Thompson D, Simonson T. 2010. Alchemical free energy simulations for biological complexes: powerful but temperamental. J. Mol. Recognit. 23:117–127. 10.1002/jmr.980 [DOI] [PubMed] [Google Scholar]
- 55.Guerois R, Nielsen JE, Serrano L. 2002. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J. Mol. Biol. 320:369–387. 10.1016/S0022-2836(02)00442-4 [DOI] [PubMed] [Google Scholar]
- 56.Tokuriki N, Stricher F, Schymkowitz J, Serrano L, Tawfik DS. 2007. The stability effects of protein mutations appear to be universally distributed. J. Mol. Biol. 369:1318–1332. 10.1016/j.jmb.2007.03.069 [DOI] [PubMed] [Google Scholar]
- 57.Jackson CJ, Weir K, Herlt A, Khurana J, Sutherland TD, Horne I, Easton C, Russell RJ, Scott C, Oakeshott JG. 2009. Structure-based rational design of a phosphotriesterase. Appl. Environ. Microbiol. 75:5153–5156. 10.1128/AEM.00629-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hadler KS, Tanifum EA, Yip SH, Mitic N, Guddat LW, Jackson CJ, Gahan LR, Nguyen K, Carr PD, Ollis DL, Hengge AC, Larrabee JA, Schenk G. 2008. Substrate-promoted formation of a catalytically competent binuclear center and regulation of reactivity in a glycerophosphodiesterase from Enterobacter aerogenes. J. Am. Chem. Soc. 130:14129–14138. 10.1021/ja803346w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Outten CE, O'Halloran TV. 2001. Femtomolar sensitivity of metalloregulatory proteins controlling zinc homeostasis. Science 292:2488–2492. 10.1126/science.1060331 [DOI] [PubMed] [Google Scholar]
- 60.Roodveldt C, Tawfik DS. 2005. Directed evolution of phosphotriesterase from Pseudomonas diminuta for heterologous expression in Escherichia coli results in stabilization of the metal-free state. Protein Eng. Des. Sel. 18:51–58. 10.1093/protein/gzi005 [DOI] [PubMed] [Google Scholar]
- 61.Wang D, Fierke CA. 2013. The BaeSR regulon is involved in defense against zinc toxicity in E. coli. Metallomics 5:372–383. 10.1039/c3mt20217h [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Benning MM, Kuo JM, Raushel FM, Holden HM. 1995. Three-dimensional structure of the binuclear metal center of phosphotriesterase. Biochemistry 34:7973–7978 [DOI] [PubMed] [Google Scholar]
- 63.Tokuriki N, Jackson CJ, Afriat-Jurnou L, Wyganowski KT, Tang R, Tawfik DS. 2012. Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme. Nat. Commun. 3:1257. 10.1038/ncomms2246 [DOI] [PubMed] [Google Scholar]
- 64.Schymkowitz J, Borg J, Stricher F, Nys R, Rousseau F, Serrano L. 2005. The FoldX web server: an online force field. Nucleic Acids Res. 33:W382–W388. 10.1093/nar/gki387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Benning MM, Kuo JM, Raushel FM, Holden HM. 1994. Three-dimensional structure of phosphotriesterase: an enzyme capable of detoxifying organophosphate nerve agents. Biochemistry 33:15001–15007 [DOI] [PubMed] [Google Scholar]
- 66.McLoughlin SY, Jackson C, Liu JW, Ollis D. 2005. Increased expression of a bacterial phosphotriesterase in Escherichia coli through directed evolution. Protein Expr. Purif. 41:433–440. 10.1016/j.pep.2005.01.012 [DOI] [PubMed] [Google Scholar]
- 67.Tokuriki N, Jackson CJ, Afriat-Jurnou L, Wyganowski KT, Tang R, Tawfik DS. 2012. Diminishing returns and tradeoffs constrain the laboratory optimization of an enzyme. Nat. Commun. 3:1257. 10.1038/ncomms2246 [DOI] [PubMed] [Google Scholar]
- 68.Jackson CJ, Foo JL, Tokuriki N, Afriat L, Carr PD, Kim HK, Schenk G, Tawfik DS, Ollis DL. 2009. Conformational sampling, catalysis, and evolution of the bacterial phosphotriesterase. Proc. Natl. Acad. Sci. U. S. A. 106:21631–21636. 10.1073/pnas.0907548106 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.