

Abstract
Neurofibromatosis type I (Nf1) is a GTPase-activating protein (GAP) that inactivates the oncoprotein Ras and plays important roles in nervous system development and learning. Alternative exon 23a falls within the Nf1 GAP domain coding sequence and is tightly regulated in favor of skipping in neurons; however, its biological function is not fully understood. Here we generated mouse embryonic stem (ES) cells with a constitutive endogenous Nf1 exon 23a inclusion, termed Nf1 23aIN/23aIN cells, by mutating the splicing signals surrounding the exon to better match consensus sequences. We also made Nf1 23aΔ/23aΔ cells lacking the exon. Active Ras levels are high in wild-type (WT) and Nf1 23aIN/23aIN ES cells, where the Nf1 exon 23a inclusion level is high, and low in Nf1 23aΔ/23aΔ cells. Upon neuronal differentiation, active Ras levels are high in Nf1 23aIN/23aIN cells, where the exon inclusion level remains high, but Ras activation is low in the other two genotypes, where the exon is skipped. Signaling downstream of Ras is significantly elevated in Nf1 23aIN/23aIN neurons. These results suggest that exon 23a suppresses the Ras-GAP activity of Nf1. Therefore, regulation of Nf1 exon 23a inclusion serves as a mechanism for providing appropriate levels of Ras signaling and may be important in modulating Ras-related neuronal functions.
INTRODUCTION
Neurofibromatosis type I (Nf1), caused by mutation of one copy of the Nf1 gene, is a common human genetic disorder affecting primarily the nervous system. The hallmark of Nf1 disease is increased susceptibility to several tumor types, including neurofibromas, malignant peripheral nerve sheath tumors (MPNSTs), and astrocytomas. However, individuals with Nf1 mutations often also exhibit other phenotypes, including mild to moderate learning disabilities (1).
Nf1 is a Ras GTPase-activating protein (Ras-GAP) that enhances the rate at which the active, GTP-bound form of Ras is converted into the inactive, GDP-bound form (Fig. 1A) (2–5). Ras-GTP signals through two main molecular pathways, namely, the Raf/MEK/extracellular signal-regulated kinase 1/2 (ERK1/2) pathway and the phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR pathway, and regulates a number of cellular functions, including proliferation, death, migration, and differentiation (6–8). By attenuating the signaling of the Ras oncogene, Nf1 acts as a tumor suppressor (9). The Ras-regulatory activity of Nf1 is mediated by its GAP domain, located in the middle of the large, 2,818-amino-acid Nf1 protein (2, 3, 10). Nf1 also increases the generation of cyclic AMP (cAMP) through mechanisms that are not well understood (Fig. 1A) (11–13).
FIG 1.

Overview of molecular functions and alternative splicing of Nf1. (A) Nf1 downregulates Ras signaling by enhancing the rate at which Ras-GTP is converted into Ras-GDP. Nf1 also promotes the generation of cAMP. GEF, guanosine nucleotide exchange factor. (B) Diagram depicting the cell-type-specific alternative splicing of Nf1 exon 23a. The alternative exon lies within the Ras-GAP domain of Nf1.
Though Nf1 is widely expressed, it is most abundant in neurons, Schwann cells, and oligodendrocytes, and it plays important roles in the nervous system (14–19). The phenotypes of mouse models suggest that mouse Nf1, whose amino acid sequence is >98% conserved with that of human Nf1, has important functions in nervous system development (20). For example, although Nf1−/− mice die at midgestation due to cardiovascular abnormalities, they also exhibit nervous system abnormalities, including enlarged sympathetic ganglia that contain increased numbers of neurons (21, 22). Also, mice with neuron-specific knockout of Nf1 exhibit abnormal cortex development, with reduced cortical thickness, increased cell density, and increased astrocyte proliferation (23). Nf1 plays a positive role in neurite extension in central nervous system (CNS) neurons. For example, primary Nf1+/− dopaminergic neurons, retinal ganglion cells, and hippocampal neurons all show decreased neurite outgrowth and growth cone areas in culture compared to those of wild-type (WT) neurons (24–26). However, Nf1 does not appear to play the same role in the peripheral nervous system, as Nf1+/− neurons from the dorsal root ganglion exhibit a normal neurite length (25).
Nf1 also plays roles in the functioning of the adult nervous system. For example, adult Nf1+/− mice show learning deficits that resemble those seen in Nf1 patients. Specifically, Nf1+/− mice perform poorly on the Morris water maze, a test of hippocampus-based spatial learning and memory, and have increased GABA-mediated inhibition that leads to decreased long-term potentiation (LTP) (27, 28). In addition, nociceptive sensory neurons from adult Nf1+/− mice show increased excitability compared to WT neurons, suggesting that Nf1 may regulate excitability, at least of specific neuron types (29, 30).
Alterations in Ras and cAMP signaling both appear to play roles in different nervous system-related phenotypes of Nf1 mutant mice. For example, the decrease in neurite growth cone area observed in CNS neurons can be rescued by pharmacologic manipulations to increase cAMP levels but not by treatments to decrease Ras signaling. This suggests that the regulation of cAMP by Nf1 is important for neurite outgrowth (25). On the other hand, the spatial learning deficits of Nf1+/− mice appear to be due at least partially to increased Ras signaling. Genetic and pharmacologic manipulations to decrease Ras signaling rescue spatial learning and LTP deficits in Nf1+/− mice, and pharmacologic inhibition of the farnesylation of Ras by treatment with lovastatin results in improvements in synaptic plasticity in human Nf1 patients (31, 32). However, cAMP and dopamine signaling also appears to play roles in at least one mouse model of Nf1 neurocognitive deficits (24, 33).
The expression of Nf1 is modulated by the regulated inclusion of three alternative exons (34–41). One of these, the 63-nucleotide (nt) in-frame cassette exon 23a (chromosome nt 79463233 to 79463295 on the mouse December 2011 GRCm38/mm10 assembly), falls within the GAP domain coding sequence of Nf1. The inclusion of Nf1 exon 23a is regulated in a developmental stage- and tissue-specific manner and is conserved across vertebrates, suggesting that the exon may be biologically important (18, 41–45). In neurons, most Nf1 mRNAs lack exon 23a and express the type 1 Nf1 protein isoform, whereas other cell types have higher levels of exon 23a inclusion and express the larger, type 2 Nf1 protein isoform (Fig. 1B) (18, 44, 46). Complex molecular mechanisms regulate Nf1 exon 23a inclusion, involving many RNA-binding proteins, including members of the Hu/ELAVL, CELF, Muscleblind, and TIA-1/TIAR protein families (47–51).
Studies in cells and mice have hinted at a possible biological role for Nf1 exon 23a. When the two isoforms of the GAP portion of Nf1 are overexpressed as truncated proteins in either mammalian or yeast cells, the Nf1 GAP domain lacking exon 23a has up to 10-fold higher Ras-GAP activity than the isoform containing exon 23a (41, 52). These findings suggest that the inclusion of exon 23a decreases the GAP activity of Nf1, but they remain to be confirmed using endogenous, full-length Nf1. In mice, deletion of Nf1 exon 23a does not increase tumor susceptibility but does result in specific learning impairments, including deficits in spatial learning, impaired contextual discrimination, and delayed acquisition of motor skills (53). The impact of deleting Nf1 exon 23a on Ras-GAP activity in mice was not reported, but based on Nf1 GAP domain cellular overexpression studies, these mice are predicted to have more Nf1 molecular activity than that of wild-type mice (41, 52, 53).
In the present study, we examined the biological importance of regulated Nf1 exon 23a inclusion in cells. Using a splicing reporter system, we identified mutations that abolish Nf1 exon 23a skipping. We then used gene targeting to create mouse embryonic stem (ES) cells with constitutive Nf1 exon 23a inclusion and with deletion of Nf1 exon 23a. Both in ES cells and in neurons derived from them, the level of active Ras increased with increased endogenous Nf1 exon 23a inclusion, but cAMP levels were unaffected. In ES cell-derived neurons, Raf/MEK/ERK1/2 signaling downstream of Ras was also elevated with increased Nf1 exon 23a inclusion. We conclude that the tight regulation of Nf1 exon 23a inclusion serves as a mechanism for controlling Ras signaling in cells.
MATERIALS AND METHODS
Plasmids.
The WT Nf1 splicing reporter (HMT-Nf1 863/499 WT) and the HuC expression vector were described previously (51). The CELF1 expression vector was a gift from Thomas Cooper at Baylor College of Medicine. The 5′SS, Py, and 23aIN splicing reporters were generated from the WT Nf1 splicing reporter by PCR mutagenesis.
Cell culture.
HeLa cells were grown in accordance with instructions from the American Type Culture Collection (Manassas, VA). CA77 cells were maintained as described previously (51). Mouse R1 ES cells were obtained from the Case Transgenic and Targeting Facility and were grown on 0.1% gelatin-coated plates in Iscove's modified Dulbecco's medium (Invitrogen) containing 20% fetal bovine serum (FBS), 50 U/ml Pen-Strep (Invitrogen), 1× minimum essential medium with nonessential amino acids (Invitrogen), 0.001% 2-mercaptoethanol, and 103 U/ml ESGRO leukemia inhibitory factor (LIF) (Millipore). ES cells were differentiated into glutamatergic neurons by retinoic acid treatment following an established protocol (54). Primary mouse cerebellar neurons were obtained and cultured as described previously (50). The Institutional Animal Care and Use Committee at Case Western Reserve University approved the mouse experiments and confirmed that they conformed to regulatory standards.
Cell transfections.
HeLa cell transfections were carried out as described previously (51), using 2 μg of splicing reporter and 1 μg of vector (pcDNA3.1HisC; Invitrogen), HuC expression plasmid, or CELF1 expression plasmid. CA77 cell transfections were carried out as described previously (51), using 2 μg of splicing reporter. Mouse cerebellar neuron transfections were carried out as described previously (50), using 2 μg of splicing reporter per transfection.
RT-PCR.
RNA was harvested from cells by use of TRIzol (Invitrogen), and reverse transcription-PCR (RT-PCR) was performed as described previously (55). RT-PCR analysis of Nf1 splicing reporter RNA was performed using the DS8 and HMT3 primers (48) and 18 to 20 PCR cycles (HeLa cells), 20 to 22 PCR cycles (CA77 cells), or 22 to 24 PCR cycles (neurons). Endogenous Nf1 RT-PCR was performed using the mouse Nf1 forward (5′-GAACCAGAGGAACCTCCTTCAGATG-3′) and mouse Nf1 reverse (5′-CATACGGCGAGACAATGGCAGGATT-3′) primers and 21 to 24 PCR cycles. Percent exon inclusion {[amount of exon included/(amount of exon included + amount of exon skipped)] × 100} was measured with a Typhoon Trio variable-mode imager (GE Healthcare), and results represent averages for at least three independent experiments.
Western blot analysis.
Western blot analyses were performed using 20 to 100 μg of total protein from HeLa cells, ES cells, or ES cell-derived neurons. Anti-Xpress (1:3,500; Invitrogen), anti-U1 70K (1:250; a gift from Susan Berget, Baylor College of Medicine), anti-γ-tubulin (1:10,000; Sigma), anti-Ras (1:200; Thermo Scientific), anti-phospho-ERK1+2 (Thr202/Tyr204) (1:250; Invitrogen), anti-phospho-Akt (pS473) (1:250; Invitrogen), anti-Akt (1:2,000; Cell Signaling), anti-p44/42 mitogen-activated protein kinases (MAPKs) (ERK1/2) (1:2,000; Cell Signaling), anti-β-tubulin isotype III (1:3,000; Sigma), anti-S6 ribosomal protein (5G10) (1:1,000; Cell Signaling), anti-phospho-S6 ribosomal protein (Ser240/244) (D68F8) XP (1:1,000; Cell Signaling), anti-neurofibromin (D) (1:200; Santa Cruz Biotechnology), anti-Nf1(1:200; gift from Nancy Ratner), anti-VGLUT1 (1:250; Synaptic Systems), and anti-HuC (1:500; Millipore) were used as primary antibodies. Goat anti-mouse IgG (1:2,000; Thermo Scientific) and goat anti-rabbit antibodies (1:5,000; Thermo Scientific) were used as secondary antibodies.
Detection of active Ras.
Active Ras (Ras-GTP) was pulled down from ES cell and ES cell-derived neuron lysates based on affinity for the glutathione S-transferase (GST)-tagged Raf1 Ras binding domain (RBD), using an active Ras pulldown and detection kit from Thermo Scientific, and Ras was detected by Western blot analysis. Mock pulldown assays were also performed using GST alone.
Immunofluorescence.
ES cells were differentiated into neurons (54), and cells were plated onto Deckglaser cover glasses. Immunofluorescence was performed as described previously (56). The primary antibody used was mouse anti-β-tubulin isotype III (1:200; Sigma), and the secondary antibody was a fluorescein isothiocyanate (FITC)-conjugated anti-mouse antibody (1:500). Images were taken using a Leica DM6000 microscope, and the number of β-tubulin III-positive cells was compared to the total number of DAPI (4′,6-diamidino-2-phenylindole)-stained cells.
Measurement of neurite length.
ES cells were differentiated into neurons (54), and cells were plated onto 6-well dishes. One day after plating, cells in each well were transfected at low efficiency with 400 ng of pmaxGFP (Lonza), using Lipofectamine 2000. On days 3 to 6 after plating, GFP-expressing live cells were imaged using a Leica DM6000 microscope, and the longest neurites, excluding the cell body, of 20 to 30 cells of each genotype were measured using Adobe Illustrator. The experiment was repeated three times.
cAMP measurement.
A Parameter cAMP assay kit (R&D Systems) was used to measure the cAMP levels of ES cell and ES cell-derived neuron lysates. Data were analyzed using ReaderFit software and were normalized to the total protein concentration in cell lysates.
Gene targeting.
Gene targeting was used to introduce mutations into the endogenous Nf1 locus in R1 ES cells that were obtained from the Case Transgenic and Targeting Facility and originally derived from the Rossant laboratory (57). The gene-targeting strategy is detailed in Fig. 3. The Case Transgenic and Targeting Facility performed the first round of gene targeting, and subsequent targeting and Cre-loxP recombination were performed in the Lou laboratory. Chromosomes were counted to ensure that there was no change in karyotype.
FIG 3.

Summary of the gene-targeting strategy used to make Nf1 exon 23a mutant ES cell lines. (A) Targeting vector used to mutate endogenous Nf1 exon 23a. It contains the 23aIN mutations (Fig. 2A), indicated in bold, a neomycin (neo) selectable marker, three loxP sites (triangles), EcoRV (RV) and EcoRI (RI) restriction sites, and sequences homologous to the endogenous Nf1 gene. (B to E) Outline of the gene-targeting and Cre-loxP recombination steps used to generate Nf1 exon 23a mutant ES cells and representative Southern blots showing mutant clones. Dotted lines represent Southern blot probes, RI and RV show the restriction enzymes used for Southern blotting, and X's represent the 23aIN mutations. RI Southern blot band sizes are as follows: WT, 15.0 kb; 23aΔ, 14.5 kb; 23aINneo, 12.4 kb; and 23aIN, 10 kb. RV Southern blot band sizes are as follows: WT, 11.1 kb; and 23aINneo, 8.6 kb.
RESULTS
Identification of mutations that increase reporter Nf1 exon 23a inclusion.
In light of in vitro studies suggesting that Nf1 exon 23a inclusion can affect the molecular activity of the exogenously expressed Nf1 Ras-GAP domain, we sought to understand the function of this tightly regulated alternative splicing event in the context of the endogenous, full-length gene in cells (41, 52). We first developed a method for manipulating Nf1 exon 23a inclusion by using a splicing reporter system that was generated in our earlier studies (51). The Nf1 splicing reporter plasmid contains human exon 23a along with portions of the surrounding introns cloned into the first intron of the human metallothionein II gene, driven by a Rous sarcoma virus promoter (Fig. 2A) (51). Human Nf1 exon 23a and mouse Nf1 exon 23a, as well as the intronic sequences immediately surrounding them, are highly conserved, as are their tissue-specific splicing patterns (37, 41, 44). Exon 23a inclusion in RNA transcribed from the reporter closely mimics that of endogenous Nf1 (51). The splicing signals surrounding Nf1 exon 23a, including the 5′ splice site (5′SS) and polypyrimidine tract (Py), deviate from the consensus sequences for optimal recognition of the exon by the spliceosome, allowing for regulation of exon inclusion. To increase inclusion, we mutated the 5′SS, Py, or both (23aIN) in the Nf1 splicing reporter to match the consensus sequences more closely (Fig. 2A). In order to avoid changing the encoded amino acid, serine, the final three nucleotides of exon 23a were mutated from TCA to TCG, even though this does not match perfectly with the consensus sequence, CAG. The polypyrimidine tract was mutated to all Us because functional studies suggest that Us may be preferred over Cs for enhancing interaction of the splicing factor U2AF65 and usage of the 3′ splice site (58–62).
FIG 2.

Mutating splicing signals to consensus sequences increases reporter Nf1 exon 23a inclusion. (A) A splicing reporter plasmid containing Nf1 exon 23a and surrounding intronic sequences was mutated so that the 5′ splice site (5′SS), polypyrimidine tract (Py), or both (23aIN) more closely match consensus sequences. Boxes indicate exons, lines represent introns, and horizontal arrows show the locations of RT-PCR primers. The RT-PCR band size was 309 bp when exon 23a was included and 246 bp when exon 23a was skipped. (B) (Top) RT-PCR showing reporter Nf1 exon 23a inclusion (Avg. % inc.) in HeLa cells. (Bottom) Western blot analysis (WB) showing expression of Xpress-tagged HuC and CELF1, with U1 70K as a loading control. *, P = 5.0 × 10−6; **, P = 7.3 × 10−4; ***, P = 5.1 × 10−4. (C) Reporter Nf1 exon 23a inclusion in primary mouse neurons as measured by RT-PCR. *, P = 1.1 × 10−4; **, P = 2.2 × 10−3; ***, P = 1.1 × 10−4. (D) Reporter Nf1 exon 23a inclusion in a rat neuron-like cell line, CA77, as measured by RT-PCR. *, P = 1.3 × 10−6; **, P = 2.2 × 10−4; ***, P = 2.4 × 10−4. Error bars represent standard errors.
To examine the molecular consequences of altering the splicing signals, we first transfected the Nf1 splicing reporters into HeLa cells and measured exon inclusion by RT-PCR. Exon 23a inclusion increased from ∼88% in the WT reporter to >99% in each of the 5′SS, Py, and 23aIN mutants (Fig. 2B, lanes 1, 5, 9, and 13). This suggests that the 23aIN mutations are effective at preventing exon skipping. We then tested whether the 23aIN mutations could block the action of members of the Hu/ELAVL and CELF protein families, which suppress Nf1 exon 23a inclusion (48, 51). In the WT reporter, exon 23a inclusion decreased dramatically upon expression of HuC or CELF1 (Fig. 2B, lanes 1 to 4). Suppression of exon 23a inclusion by HuC and CELF1 was decreased in the 5′SS and Py mutant reporters and was nearly completely abolished in the 23aIN reporter (Fig. 2B, lanes 7, 8, 11, 12, 15, and 16). Therefore, the 23aIN mutations effectively block the action of at least some negative splicing regulators.
We next transfected the Nf1 splicing reporters into primary mouse neurons, where the level of endogenous Nf1 exon 23a inclusion is very low (50). Inclusion of exon 23a was very low from the WT Nf1 reporter, while the 5′SS and Py mutant reporters showed greatly increased inclusion, and the 23aIN reporter showed almost no skipping of exon 23a (Fig. 2C). We also asked whether the mutations in the 23aIN splicing reporter are effective at increasing exon inclusion in another cell type with low levels of endogenous Nf1 exon 23a inclusion. All three mutant reporters showed close to 100% exon 23a inclusion in CA77 cells, a rat cell line with neuron-like features in which endogenous Nf1 exon 23a is mainly skipped (Fig. 2D) (51). Therefore, the 23aIN mutations are effective at abolishing skipping of exon 23a in cell types in which there is strong negative regulation of inclusion. From these experiments, we concluded that the mutations in the 23aIN splicing reporter had the desired characteristics: they abolished exon 23a skipping and prevented the action of known negative splicing regulator proteins.
Splicing signal mutations increase endogenous Nf1 exon 23a inclusion.
In order to study the biological consequences of manipulating Nf1 exon 23a inclusion, we constructed a targeting vector and used it to introduce the 23aIN mutations into the endogenous Nf1 locus in mouse ES cells (Fig. 3A). We first introduced mutations into one copy of the Nf1 gene and then used Cre-loxP recombination to remove a neomycin selectable marker (Fig. 3B and C). We then repeated gene targeting to mutate the second copy of the Nf1 gene, followed by a second round of Cre-loxP recombination (Fig. 3D and E). The placement of the loxP sites allowed for removal of either just the neomycin selectable marker or the entire Nf1 exon 23a (Fig. 3E). The end result was the creation of two ES cell lines: Nf1 23aΔ/23aΔ, in which both copies of exon 23a are deleted, and Nf1 23aIN/23IN, in which the splicing signals surrounding exon 23a are mutated to more closely match consensus sequences (Fig. 4A).
FIG 4.
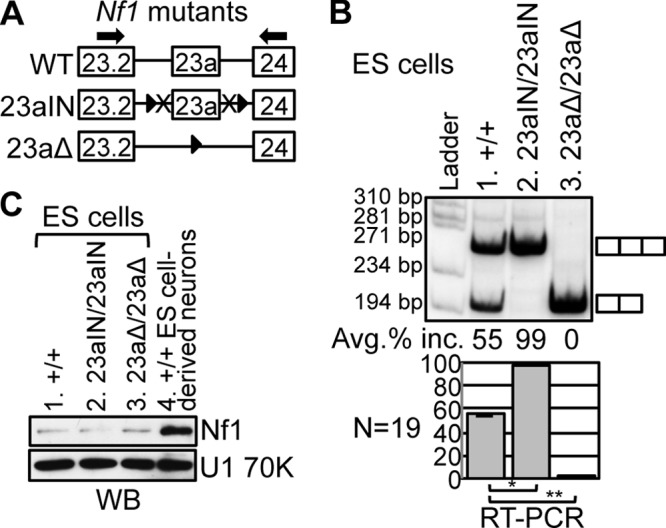
Manipulation of endogenous Nf1 exon 23a inclusion in mouse ES cells. (A) Depiction of the relevant portions of the Nf1 mutant genes in ES cell lines. See the legend to Fig. 3 for details. The Nf1 23aIN/23aIN cell line contains the 23aIN mutations depicted in Fig. 2A. In the Nf1 23aΔ/23aΔ mutant, exon 23a is replaced by a loxP site, but the remainder of the Nf1 gene remains intact. Arrows indicate locations of RT-PCR primers. The RT-PCR band size was 266 bp if exon 23a was included and 203 bp if exon 23a was skipped. (B) RT-PCR showing endogenous Nf1 exon 23a inclusion in ES cell mutants. Error bars represent standard errors. *, P = 4.5 × 10−27; **, P = 2.4 × 10−32. (C) Western blot analysis showing total Nf1 protein (∼250 kDa) expression in ES cells (lanes 1 to 3) and ES cell-derived neurons (lane 4). U1 70K (∼70 kDa) was used as a loading control.
If our strategy for manipulating exon 23a was effective, we predicted that endogenous Nf1 exon 23a inclusion would switch from ∼55% in WT cells to 0% for Nf1 23aΔ/23aΔ mutants and close to 100% for Nf1 23aIN/23aIN mutants. This is exactly what we saw when we performed RT-PCR using RNAs from the ES cell mutants (Fig. 4B). Importantly, the expression of Nf1 protein in the ES cell mutants (Fig. 4C) was the same as that in the wild type (compare lanes 2 and 3 with lane 1), indicating that we specifically altered the Nf1 isoform made without interfering with overall protein levels, which remained much lower than those in ES cell-derived neurons (lane 4). Therefore, we successfully created ES cells with manipulated endogenous Nf1 exon 23a inclusion that can be used to study the biological consequences of Nf1 alternative splicing.
Ras activity increases with increased Nf1 exon 23a inclusion in ES cells.
We next asked whether inclusion of exon 23a affects the molecular function of endogenous Nf1 in ES cells. To determine whether the inclusion of exon 23a diminishes the ability of Nf1 to inactivate Ras, we used a well-established, commercially available active Ras pulldown and detection kit (Thermo Scientific). In this assay, the GST-tagged Raf1 Ras binding domain (RBD) is used to pull down Ras-GTP, the active form of Ras, from ES cell lysates. The level of active Ras was much lower in Nf1 23aΔ/23aΔ ES cells, in which the exon was deleted, than in WT or Nf1 23aIN/23aIN cells, in which the exon was mainly included (Fig. 5A). The correlation between the level of active Ras and the level of exon 23a inclusion suggests that the inclusion of exon 23a decreases the Ras-GAP activity of endogenous Nf1 protein in ES cells.
FIG 5.
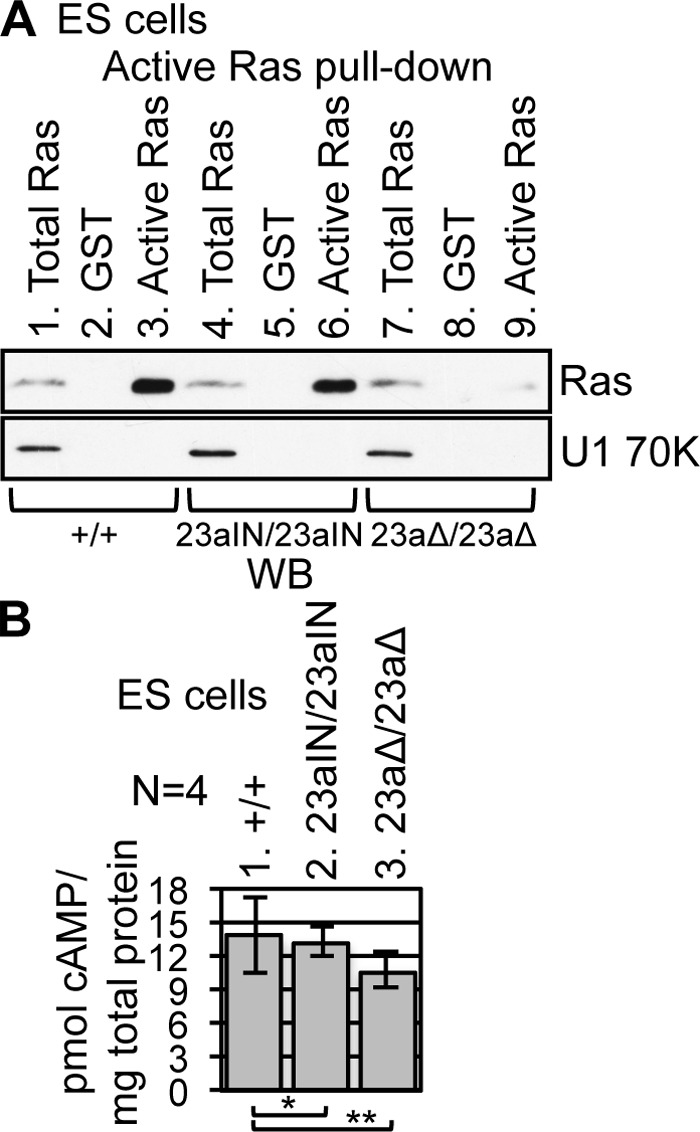
Nf1 exon 23a inclusion correlates with Ras activity in ES cells. (A) GST-Raf1 RBD was used to pull down Ras-GTP (active Ras) from ES cell lysates (lanes 3, 6, and 9), followed by Western blot analysis using anti-Ras (∼21 kDa) antibody. Mock pulldowns were performed using GST (lanes 2, 5, and 8). Samples (3%) of total ES cell lysate were loaded in lanes 1, 4, and 7. U1 70K (∼70 kDa) was used as a loading control. (B) ES cell intracellular cAMP levels were measured by enzyme-linked immunosorbent assay (ELISA). Error bars represent standard errors. *, P = 0.88; **, P = 0.43.
In addition, we measured cAMP levels in the mutant ES cells, as Nf1 is known to play a role in regulating cAMP levels (11–13). We did not observe a significant difference in intracellular cAMP levels in ES cells when Nf1 exon 23a inclusion was manipulated (Fig. 5B). These results suggest that Nf1 exon 23a inclusion specifically inhibits the Ras-GAP activity of Nf1 in ES cells without influencing its ability to regulate cAMP levels.
23aIN mutations increase Nf1 exon 23a inclusion in ES cell-derived neurons.
After our initial experiments with ES cells, we investigated the role of Nf1 alternative splicing in neurons, a cell type in which endogenous Nf1 protein expression is much higher than in ES cells (Fig. 4C), and in which WT Nf1 exon 23a is almost exclusively skipped (Fig. 6A). Using an established protocol, we differentiated the Nf1 mutant ES cell lines into CNS-like neurons to investigate how Nf1 exon 23a inclusion affects the neuronal phenotype (54). Upon neuronal differentiation, Nf1 exon 23a inclusion decreased in WT cells, from ∼55% to ∼9% (Fig. 4B and 6A). In contrast, Nf1 exon 23a inclusion remained near 100% in Nf1 23aIN/23aIN ES cell-derived neurons, indicating that the 23aIN mutations are effective at increasing exon 23a inclusion even in a cell type in which the exon is normally skipped (Fig. 6A). As expected, Nf1 23aΔ/23aΔ ES cell-derived neurons showed no Nf1 exon 23a inclusion (Fig. 6A). Exon 23a inclusion in ES cell-derived neurons did not influence the total Nf1 protein level (Fig. 6B).
FIG 6.

Manipulation of Nf1 exon 23a inclusion in ES cell-derived neurons. (A) RT-PCR showing endogenous Nf1 exon 23a inclusion in ES cell-derived neurons. Arrows indicate locations of RT-PCR primers. The RT-PCR band size was 266 bp if exon 23a was included and 203 bp if exon 23a was skipped. Error bars represent standard errors. *, P = 1.5 × 10−24; **, P = 1.5 × 10−4. (B) Western blot analysis showing Nf1 protein (∼250 kDa) expression in ES cell-derived neurons. γ-Tubulin (∼48 kDa), a ubiquitously expressed protein, was used as a loading control. (C) Western blot showing expression of the neuronal marker β-tubulin III (∼55 kDa), the glutamatergic neuron marker VGLUT1 (∼61 kDa), and the neuron-enriched protein HuC (∼39 kDa) in ES cell-derived neurons. U1 70K (∼70 kDa) was used as a loading control. (D) Immunofluorescence images demonstrating that ES cell-derived neurons express the neuronal marker β-tubulin III and show neuron-like morphology.
Nf1 exon 23a skipping is not required for neuronal differentiation.
As previous reports showed that Nf1 exon 23a switches from inclusion to skipping upon neuronal differentiation (44, 46, 50), we asked if this switch is necessary for neuronal differentiation. We examined the expression of a neuronal marker, β-tubulin III, in the Nf1 mutant ES cell-derived neurons by both Western blotting and immunofluorescence assay. We found that there was no difference in β-tubulin III expression between the three genotypes of ES cell-derived neurons and that the cells of all genotypes acquired a neuron-like morphology (Fig. 6C and D). In addition, cells of all three genotypes expressed HuC, a neuron-enriched protein, and VGLUT1, a marker of glutamatergic neurons (Fig. 6C). Therefore, Nf1 exon 23a skipping does not appear to be required for neuronal differentiation on a gross level.
Ras activity correlates with Nf1 exon 23a inclusion in ES cell-derived neurons.
To further explore the biological impact of exon 23a inclusion, we measured active Ras-GTP levels in WT and mutant ES cell-derived neurons. The Nf1 23aIN/23aIN ES cell-derived neurons, in which exon 23a was included, showed greatly increased Ras-GTP levels compared with those in WT cells (Fig. 7A, compare lanes 3 and 6). The Nf1 23aΔ/23aΔ ES cell-derived neurons, which had a level of Nf1 exon 23a inclusion similar to that in WT cells (Fig. 6A), also had levels of active Ras-GTP similar to those in WT cells (Fig. 7A, compare lanes 3 and 9). This is in contrast to the undifferentiated state, in which both Nf1 23aIN/23aIN and WT ES cells had high levels of Nf1 exon 23a inclusion and high Ras-GTP levels, and Nf1 23aΔ/23aΔ cells had lower Ras-GTP levels (Fig. 4B and 5A). Therefore, Nf1 exon 23a inclusion correlates with Ras-GTP levels in both ES cells and ES cell-derived neurons, providing compelling evidence that exon 23a inclusion decreases the Ras-GAP activity of Nf1.
FIG 7.
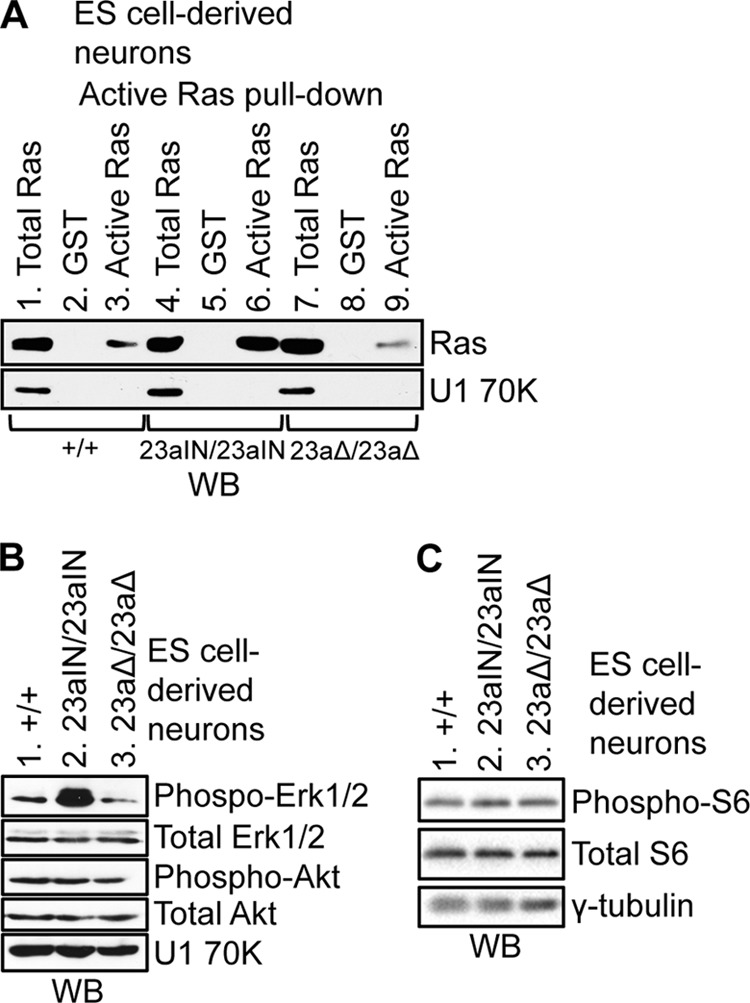
Increased endogenous Nf1 exon 23a inclusion leads to increased Ras signaling in ES cell-derived neurons. (A) GST-Raf1 RBD was used to pull down active Ras (Ras-GTP) from ES cell-derived neuron lysates (lanes 3, 6, and 9), followed by Western blot analysis using anti-Ras (∼21 kDa) antibody. Mock pulldowns were performed using GST (lanes 2, 5, and 8). Samples (3%) of total cell lysate were loaded in lanes 1, 4, and 7. U1 70K (∼70 kDa) was used as a loading control. (B) Western blot analysis showing phospho-ERK1/2 (∼42/44 kDa), total ERK1/2 (∼42/44 kDa), phospho-Akt (∼60 kDa), total Akt (∼60 kDa), and U1 70K (∼70 kDa) expression in lysates from ES cell-derived neurons. (C) Western blot analysis showing phospho-S6 (∼32 kDa) and total S6 (∼32 kDa) in lysates from ES cell-derived neurons. The loading control was γ-tubulin (∼48 kDa).
Signaling downstream of Ras increases with Nf1 exon 23a inclusion.
We next asked whether increased Ras-GTP levels in Nf1 23aIN/23aIN ES cell-derived neurons translated into a phenotype of elevated signaling downstream of Ras. As the Raf/MEK/ERK1/2 and PI3K/Akt/mTOR pathways are the most prominent pathways that are activated by Ras, we measured the levels of phospho-ERK1/2 and phospho-Akt as indicators of activation of these pathways in our cells (6–8). The level of phospho-ERK1/2 was greatly elevated in Nf1 23aIN/23aIN ES cell-derived neurons compared to that in WT and Nf1 23aΔ/23aΔ cells (Fig. 7B), indicating that Nf1 exon 23a inclusion elevates Raf/MEK/ERK1/2 signaling downstream of Ras. However, phospho-Akt levels did not change in Nf1 23aIN/23aIN cells, nor did phosphorylation of S6 downstream of mTOR (Fig. 7B and C). These results are consistent with other studies showing activation of the Raf/MEK/ERK1/2 pathway but not the PI3K/Akt/mTOR pathway in Nf1 mutant mouse brains (63, 64).
Two other pathways we investigated yielded negative results. First, there was no significant difference in intracellular cAMP level among ES cell-derived neurons with different levels of Nf1 exon 23a inclusion (Fig. 8A). Second, previous studies indicated that primary mouse Nf1+/− CNS neurons exhibit shorter neurite lengths than those of WT neurons, and this phenotype was attributed to decreased cAMP signaling rather than increased Ras signaling in these cells (24–26). Consistent with this finding, we did not observe any change in neurite length in the Nf1 23aIN/23aIN ES cell-derived neurons, which showed elevated Ras signaling but no change in cAMP levels (Fig. 8B). These results suggest that Nf1 exon 23a inclusion regulates Ras signaling but not cAMP signaling in neurons.
FIG 8.
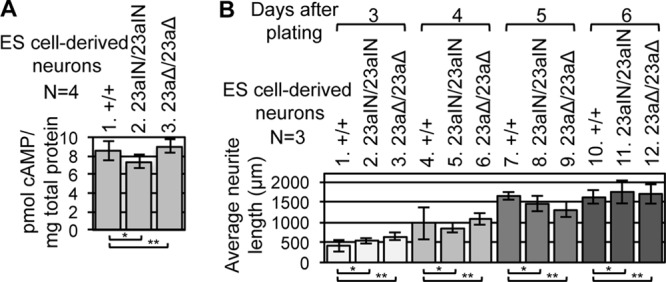
Nf1 exon 23a inclusion does not affect cAMP levels or neurite length in ES cell-derived neurons. (A) ES cell-derived neuron intracellular cAMP levels were measured by ELISA. Error bars represent standard errors. *, P = 0.38; **, P = 0.74. (B) Average longest neurite lengths for ES cell-derived neurons at different time points during differentiation. Error bars represent standard errors. A total of ≥20 neurites of each genotype were measured for each experimental repeat. For day 3, P = 0.43 (*) and P = 0.25 (**); for day 4, P = 0.74 (*) and P = 0.76 (**); for day 5, P = 0.55 (*) and P = 0.26 (**); and for day 6, P = 0.76 (*) and P = 0.83 (**).
DISCUSSION
Taken together, the results in this study support a model in which Nf1 exon 23a alternative splicing plays an important role in modulating Ras signaling in vivo (Fig. 9). When Nf1 exon 23a is skipped, as occurs in neurons, the Nf1 protein has high Ras-GAP activity, leading to low levels of active Ras-GTP and signaling downstream of Ras in cells (Fig. 9A). When Nf1 exon 23a is included, the Nf1 protein has low Ras-GAP activity, leading to high levels of Ras signaling in cells (Fig. 9B). The cAMP-regulatory activity of Nf1 is unaffected by Nf1 exon 23a inclusion (Fig. 9A and B). Therefore, the tightly regulated suppression of Nf1 exon 23a in neurons serves as a specific mechanism for increasing Ras-GAP activity in this cell type.
FIG 9.
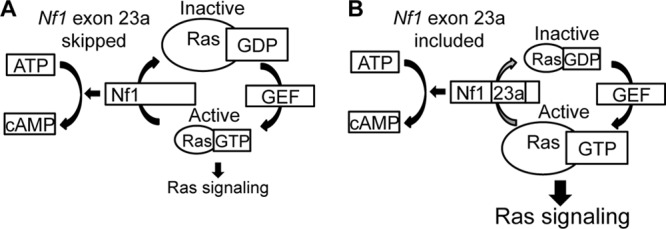
Model for regulation of endogenous Nf1 molecular activity by alternative splicing. (A) When exon 23a is excluded, the Nf1 protein has high Ras-GAP activity, leading to low levels of active Ras-GTP and low levels of signaling downstream of Ras in cells. Nf1 also promotes the generation of cAMP. (B) When exon 23a is included, the Nf1 protein has low Ras-GAP activity, leading to increased Ras-GTP and Ras signaling. The regulation of cAMP by Nf1 is unaffected by exon 23a inclusion.
In this study, we showed that strengthening the splicing signals surrounding endogenous Nf1 exon 23a prevents its skipping while leaving the amino acid sequence encoded by the exon intact (Fig. 4B and 6A). In addition to shedding light on the biological role of Nf1 exon 23a alternative splicing, the approach used here could be applied to the study of other alternative exons. The functions of many alternative exons, including Nf1 exon 23a, have traditionally been studied by deleting the exon (53, 65). While these studies provide information about the functions for which the alternative exon is required, they do not provide information on why the exons have evolved to be regulated rather than constitutively included.
Using cells with altered levels of endogenous Nf1 exon 23a inclusion, we showed that Nf1 exon 23a inclusion increases active Ras-GTP levels in both ES cells and ES cell-derived neurons and also increases phospho-ERK1/2 levels downstream of Ras in neurons (Fig. 5A and 7A and B). Earlier studies suggested that the GAP domain of Nf1 containing exon 23a has less Ras-GAP activity than the same domain lacking the exon, but these studies had some limitations (41, 52). First, they were performed in exogenous expression systems in yeast and mammalian cells, where the expression of the GAP domain was likely not at physiological levels. In addition, they used only the GAP domain of Nf1, which is a small portion of the entire protein. Therefore, it was possible that in the context of the larger protein, Nf1 exon 23a behaves differently with regard to Ras inactivation, or it could affect other functions of Nf1, such as cAMP regulation. Our system allowed us to expand upon these studies and to show for the first time that exon 23a inclusion decreases the regulation of Ras activity by the endogenous, full-length Nf1 protein in cells. Thus, Nf1 exon 23a inclusion is regulated to provide appropriate levels of Ras signaling in cells.
Precisely how Nf1 exon 23a inclusion decreases the Ras-GAP activity of Nf1 is an interesting topic for future studies. An early structural study of the type 1 isoform of the Nf1 GAP domain, which lacks exon 23a, indicated that the GAP domain consists of a series of α-helices (66). Exon 23a is located within the C-terminal portion of one of these helices, α5c, and, if unable to form an alpha-helical structure, would interrupt its continuity. It was speculated that since exon 23a is in a relatively exposed portion of the GAP domain and contains many charged residues (six lysines and two glutamates), it might function in interactions with other proteins or with other portions of the large Nf1 protein. These interactions could disrupt the function of the GAP domain, although these predictions remain to be tested.
Interestingly, the inclusion of exon 23a did not influence the regulation of cAMP levels by Nf1 (Fig. 5B and 8A). Consistent with this finding, we also did not observe changes in neurite length, which is mediated by cAMP signaling in other Nf1 mutant models (24–26), in Nf1 23aIN/23aIN ES cell-derived neurons (Fig. 8B). It appears that Nf1 exon 23a alternative splicing functions in specifically inhibiting the Ras-GAP function of Nf1 without affecting its regulation of cAMP. Perhaps the suppression of Ras signaling is particularly important in certain cell types, and the skipping of Nf1 exon 23a has evolved as a mechanism for specifically increasing Ras regulation without affecting the other molecular functions of Nf1. Since they appear to affect only Ras signaling in cells, our 23aIN mutations could serve as useful tools for teasing apart the Ras- and cAMP-related biological functions of Nf1.
In Nf1+/− mice, spatial learning and LTP deficits have been attributed in part to Ras hyperactivation and elevated ERK1/2 signaling (28, 31, 64, 67). We likewise found that both Ras activation and downstream ERK1/2 phosphorylation were increased in Nf1 23aIN/23aIN ES cell-derived neurons (Fig. 7). Therefore, it is tempting to speculate that the alternative splicing of Nf1 could be a mechanism for providing appropriate Ras signaling that is necessary for learning.
In a previous study, Nf1 23aΔ/23aΔ mice were found to exhibit a spatial learning phenotype that closely resembles that of Nf1+/− mice (27, 53). This result is somewhat surprising, as Nf1 exon 23a is already overwhelmingly skipped in the WT whole brain, and the isoform lacking the exon has increased rather than decreased molecular activity (Fig. 5A and 7A) (41, 44, 52). One possible explanation for this finding is that the inclusion of Nf1 exon 23a is important for the function of a specific population of neurons or for the function of glia, where there is evidence that WT Nf1 exon 23a is predominantly included (18). Alterations in cAMP signaling could also contribute to the learning phenotypes of Nf1 23aΔ/23aΔ mice, although we do not favor this explanation, as our data suggest that Nf1 exon 23a inclusion does not affect cAMP levels (Fig. 5B and 8A). The explanation we favor is that there is a narrow window of appropriate Ras signaling for learning and that the regulation of Nf1 exon 23a inclusion is in place to fine-tune Ras signaling for optimal learning. Supporting this idea is the finding in various mouse models that Ras signaling that is either elevated or diminished can lead to learning phenotypes (68).
In summary, we found that the inclusion of exon 23a decreases the Ras-regulatory activity of Nf1 and leads to increased ERK1/2 activation in neurons. Thus, the tight regulation of Nf1 exon 23a alternative splicing is likely in place as a mechanism for modulating Ras signaling in cells. This finding could have important implications for biological processes in which appropriate Ras signaling is particularly important, such as learning and long-term potentiation.
ACKNOWLEDGMENTS
This work was supported by the National Institutes of Health (grants NS049103 to H.L., F31 NS647242 to M.N.H., T32 GM08613 to M.N.H., and S10RR021228 and S10RR024536 [National Center for Research Resources shared-instrumentation grants for the Leica DM6000 wide-field microscope and the GE Healthcare Typhoon Trio variable-mode imager]) and the U.S. Department of Defense (grant NF060083 to H.L.).
We thank David LePage and the Case Transgenic and Targeting Facility for help with gene targeting, David Friel for assistance with neuron experiments, and Nancy Ratner for providing the anti-Nf1 antibody and Schwann cell RNA. We also thank Jo Ann Wise for a critical reading of the manuscript and Helen Salz, Ronald Conlon, Evan Deneris, and members of the Lou laboratory for helpful discussions.
Footnotes
Published ahead of print 7 April 2014
REFERENCES
- 1.Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. 2009. Neurofibromatosis type 1 revisited. Pediatrics 123:124–133. 10.1542/peds.2007-3204 [DOI] [PubMed] [Google Scholar]
- 2.Ballester R, Marchuk D, Boguski M, Saulino A, Letcher R, Wigler M, Collins F. 1990. The NF1 locus encodes a protein functionally related to mammalian GAP and yeast IRA proteins. Cell 63:851–859. 10.1016/0092-8674(90)90151-4 [DOI] [PubMed] [Google Scholar]
- 3.Martin GA, Viskochil D, Bollag G, McCabe PC, Crosier WJ, Haubruck H, Conroy L, Clark R, O'Connell P, Cawthon RM. 1990. The GAP-related domain of the neurofibromatosis type 1 gene product interacts with ras p21. Cell 63:843–849. 10.1016/0092-8674(90)90150-D [DOI] [PubMed] [Google Scholar]
- 4.Xu GF, Lin B, Tanaka K, Dunn D, Wood D, Gesteland R, White R, Weiss R, Tamanoi F. 1990. The catalytic domain of the neurofibromatosis type 1 gene product stimulates ras GTPase and complements ira mutants of S. cerevisiae. Cell 63:835–841. 10.1016/0092-8674(90)90149-9 [DOI] [PubMed] [Google Scholar]
- 5.DeClue JE, Papageorge AG, Fletcher JA, Diehl SR, Ratner N, Vass WC, Lowy DR. 1992. Abnormal regulation of mammalian p21ras contributes to malignant tumor growth in von Recklinghausen (type 1) neurofibromatosis. Cell 69:265–273. 10.1016/0092-8674(92)90407-4 [DOI] [PubMed] [Google Scholar]
- 6.Le LQ, Parada LF. 2007. Tumor microenvironment and neurofibromatosis type I: connecting the GAPs. Oncogene 26:4609–4616. 10.1038/sj.onc.1210261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gutmann DH, Parada LF, Silva AJ, Ratner N. 2012. Neurofibromatosis type 1: modeling CNS dysfunction. J. Neurosci. 32:14087–14093. 10.1523/JNEUROSCI.3242-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diggs-Andrews KA, Gutmann DH. 2013. Modeling cognitive dysfunction in neurofibromatosis-1. Trends Neurosci. 36:237–247. 10.1016/j.tins.2012.12.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu W, Mulligan LM, Ponder MA, Liu L, Smith BA, Mathew CG, Ponder BA. 1992. Loss of NF1 alleles in phaeochromocytomas from patients with type I neurofibromatosis. Genes Chromosomes Cancer 4:337–342. 10.1002/gcc.2870040411 [DOI] [PubMed] [Google Scholar]
- 10.Xu GF, O'Connell P, Viskochil D, Cawthon R, Robertson M, Culver M, Dunn D, Stevens J, Gesteland R, White R. 1990. The neurofibromatosis type 1 gene encodes a protein related to GAP. Cell 62:599–608. 10.1016/0092-8674(90)90024-9 [DOI] [PubMed] [Google Scholar]
- 11.Tong J, Hannan F, Zhu Y, Bernards A, Zhong Y. 2002. Neurofibromin regulates G protein-stimulated adenylyl cyclase activity. Nat. Neurosci. 5:95–96. 10.1038/nn792 [DOI] [PubMed] [Google Scholar]
- 12.Dasgupta B, Dugan LL, Gutmann DH. 2003. The neurofibromatosis 1 gene product neurofibromin regulates pituitary adenylate cyclase-activating polypeptide-mediated signaling in astrocytes. J. Neurosci. 23:8949–8954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.The I, Hannigan GE, Cowley GS, Reginald S, Zhong Y, Gusella JF, Hariharan IK, Bernards A. 1997. Rescue of a Drosophila NF1 mutant phenotype by protein kinase A. Science 276:791–794. 10.1126/science.276.5313.791 [DOI] [PubMed] [Google Scholar]
- 14.Hirvonen O, Lakkakorpi J, Aaltonen V, Hirvonen H, Rossi M, Karvonen SL, Yla-Outinen H, Kalimo H, Peltonen J. 1998. Developmental regulation of NF1 tumor suppressor gene in human peripheral nerve. J. Neurocytol. 27:939–952. 10.1023/A:1006905224474 [DOI] [PubMed] [Google Scholar]
- 15.Daston MM, Ratner N. 1992. Neurofibromin, a predominantly neuronal GTPase activating protein in the adult, is ubiquitously expressed during development. Dev. Dyn. 195:216–226. 10.1002/aja.1001950307 [DOI] [PubMed] [Google Scholar]
- 16.Daston MM, Scrable H, Nordlund M, Sturbaum AK, Nissen LM, Ratner N. 1992. The protein product of the neurofibromatosis type 1 gene is expressed at highest abundance in neurons, Schwann cells, and oligodendrocytes. Neuron 8:415–428. 10.1016/0896-6273(92)90270-N [DOI] [PubMed] [Google Scholar]
- 17.Gutmann DH, Wood DL, Collins FS. 1991. Identification of the neurofibromatosis type 1 gene product. Proc. Natl. Acad. Sci. U. S. A. 88:9658–9662. 10.1073/pnas.88.21.9658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gutmann DH, Geist RT, Wright DE, Snider WD. 1995. Expression of the neurofibromatosis 1 (NF1) isoforms in developing and adult rat tissues. Cell Growth Differ. 6:315–323 [PubMed] [Google Scholar]
- 19.Golubic M, Roudebush M, Dobrowolski S, Wolfman A, Stacey DW. 1992. Catalytic properties, tissue and intracellular distribution of neurofibromin. Oncogene 7:2151–2159 [PubMed] [Google Scholar]
- 20.Bernards A, Snijders AJ, Hannigan GE, Murthy AE, Gusella JF. 1993. Mouse neurofibromatosis type 1 cDNA sequence reveals high degree of conservation of both coding and non-coding mRNA segments. Hum. Mol. Genet. 2:645–650. 10.1093/hmg/2.6.645 [DOI] [PubMed] [Google Scholar]
- 21.Brannan CI, Perkins AS, Vogel KS, Ratner N, Nordlund ML, Reid SW, Buchberg AM, Jenkins NA, Parada LF, Copeland NG. 1994. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 8:1019–1029. 10.1101/gad.8.9.1019 [DOI] [PubMed] [Google Scholar]
- 22.Jacks T, Shih TS, Schmitt EM, Bronson RT, Bernards A, Weinberg RA. 1994. Tumour predisposition in mice heterozygous for a targeted mutation in Nf1. Nat. Genet. 7:353–361. 10.1038/ng0794-353 [DOI] [PubMed] [Google Scholar]
- 23.Zhu Y, Romero MI, Ghosh P, Ye Z, Charnay P, Rushing EJ, Marth JD, Parada LF. 2001. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 15:859–876. 10.1101/gad.862101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brown JA, Emnett RJ, White CR, Yuede CM, Conyers SB, O'Malley KL, Wozniak DF, Gutmann DH. 2010. Reduced striatal dopamine underlies the attention system dysfunction in neurofibromatosis-1 mutant mice. Hum. Mol. Genet. 19:4515–4528. 10.1093/hmg/ddq382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brown JA, Gianino SM, Gutmann DH. 2010. Defective cAMP generation underlies the sensitivity of CNS neurons to neurofibromatosis-1 heterozygosity. J. Neurosci. 30:5579–5589. 10.1523/JNEUROSCI.3994-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brown JA, Diggs-Andrews KA, Gianino SM, Gutmann DH. 2012. Neurofibromatosis-1 heterozygosity impairs CNS neuronal morphology in a cAMP/PKA/ROCK-dependent manner. Mol. Cell. Neurosci. 49:13–22. 10.1016/j.mcn.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silva AJ, Frankland PW, Marowitz Z, Friedman E, Laszlo GS, Cioffi D, Jacks T, Bourtchuladze R. 1997. A mouse model for the learning and memory deficits associated with neurofibromatosis type I. Nat. Genet. 15:281–284. 10.1038/ng0397-281 [DOI] [PubMed] [Google Scholar]
- 28.Costa RM, Federov NB, Kogan JH, Murphy GG, Stern J, Ohno M, Kucherlapati R, Jacks T, Silva AJ. 2002. Mechanism for the learning deficits in a mouse model of neurofibromatosis type 1. Nature 415:526–530. 10.1038/nature711 [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Nicol GD, Clapp DW, Hingtgen CM. 2005. Sensory neurons from Nf1 haploinsufficient mice exhibit increased excitability. J. Neurophysiol. 94:3670–3676. 10.1152/jn.00489.2005 [DOI] [PubMed] [Google Scholar]
- 30.Hingtgen CM. 2008. Neurofibromatosis: the role of guanosine triphosphatase activating proteins in sensory neuron function. Acta Physiol. Sin. 60:581–583 [PubMed] [Google Scholar]
- 31.Li W, Cui Y, Kushner SA, Brown RA, Jentsch JD, Frankland PW, Cannon TD, Silva AJ. 2005. The HMG-CoA reductase inhibitor lovastatin reverses the learning and attention deficits in a mouse model of neurofibromatosis type 1. Curr. Biol. 15:1961–1967. 10.1016/j.cub.2005.09.043 [DOI] [PubMed] [Google Scholar]
- 32.Mainberger F, Jung NH, Zenker M, Wahllander U, Freudenberg L, Langer S, Berweck S, Winkler T, Straube A, Heinen F, Granstrom S, Mautner VF, Lidzba K, Mall V. 2013. Lovastatin improves impaired synaptic plasticity and phasic alertness in patients with neurofibromatosis type 1. BMC Neurol. 13:131. 10.1186/1471-2377-13-131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Diggs-Andrews KA, Tokuda K, Izumi Y, Zorumski CF, Wozniak DF, Gutmann DH. 2013. Dopamine deficiency underlies learning deficits in neurofibromatosis-1 mice. Ann. Neurol. 73:309–315. 10.1002/ana.23793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Danglot G, Regnier V, Fauvet D, Vassal G, Kujas M, Bernheim A. 1995. Neurofibromatosis 1 (NF1) mRNAs expressed in the central nervous system are differentially spliced in the 5′ part of the gene. Hum. Mol. Genet. 4:915–920. 10.1093/hmg/4.5.915 [DOI] [PubMed] [Google Scholar]
- 35.Bernards A, Haase VH, Murthy AE, Menon A, Hannigan GE, Gusella JF. 1992. Complete human NF1 cDNA sequence: two alternatively spliced mRNAs and absence of expression in a neuroblastoma line. DNA Cell Biol. 11:727–734. 10.1089/dna.1992.11.727 [DOI] [PubMed] [Google Scholar]
- 36.Gutmann DH, Zhang Y, Hirbe A. 1999. Developmental regulation of a neuron-specific neurofibromatosis 1 isoform. Ann. Neurol. 46:777–782. [DOI] [PubMed] [Google Scholar]
- 37.Danglot G, Teinturier C, Duverger A, Bernheim A. 1994. Tissue-specific alternative splicing of neurofibromatosis 1 (NF1) mRNA. Biomed. Pharmacother. 48:365–372. 10.1016/0753-3322(94)90053-1 [DOI] [PubMed] [Google Scholar]
- 38.Gutmann DH, Geist RT, Rose K, Wright DE. 1995. Expression of two new protein isoforms of the neurofibromatosis type 1 gene product, neurofibromin, in muscle tissues. Dev. Dyn. 202:302–311. 10.1002/aja.1002020309 [DOI] [PubMed] [Google Scholar]
- 39.Gutman DH, Andersen LB, Cole JL, Swaroop M, Collins FS. 1993. An alternatively-spliced mRNA in the carboxy terminus of the neurofibromatosis type 1 (NF1) gene is expressed in muscle. Hum. Mol. Genet. 2:989–992. 10.1093/hmg/2.7.989 [DOI] [PubMed] [Google Scholar]
- 40.Geist RT, Gutmann DH. 1996. Expression of a developmentally-regulated neuron-specific isoform of the neurofibromatosis 1 (NF1) gene. Neurosci. Lett. 211:85–88. 10.1016/0304-3940(96)12730-0 [DOI] [PubMed] [Google Scholar]
- 41.Andersen LB, Ballester R, Marchuk DA, Chang E, Gutmann DH, Saulino AM, Camonis J, Wigler M, Collins FS. 1993. A conserved alternative splice in the von Recklinghausen neurofibromatosis (NF1) gene produces two neurofibromin isoforms, both of which have GTPase-activating protein activity. Mol. Cell. Biol. 13:487–495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gutmann DH, Cole JL, Collins FS. 1995. Expression of the neurofibromatosis type 1 (NF1) gene during mouse embryonic development. Prog. Brain Res. 105:327–335. 10.1016/S0079-6123(08)63311-7 [DOI] [PubMed] [Google Scholar]
- 43.Suzuki Y, Suzuki H, Kayama T, Yoshimoto T, Shibahara S. 1991. Brain tumors predominantly express the neurofibromatosis type 1 gene transcripts containing the 63 base insert in the region coding for GTPase activating protein-related domain. Biochem. Biophys. Res. Commun. 181:955–961. 10.1016/0006-291X(91)92029-J [DOI] [PubMed] [Google Scholar]
- 44.Mantani A, Wakasugi S, Yokota Y, Abe K, Ushio Y, Yamamura K. 1994. A novel isoform of the neurofibromatosis type-1 mRNA and a switch of isoforms during murine cell differentiation and proliferation. Gene 148:245–251. 10.1016/0378-1119(94)90695-5 [DOI] [PubMed] [Google Scholar]
- 45.Huynh DP, Nechiporuk T, Pulst SM. 1994. Differential expression and tissue distribution of type I and type II neurofibromins during mouse fetal development. Dev. Biol. 161:538–551. 10.1006/dbio.1994.1052 [DOI] [PubMed] [Google Scholar]
- 46.Metheny LJ, Skuse GR. 1996. NF1 mRNA isoform expression in PC12 cells: modulation by extrinsic factors. Exp. Cell Res. 228:44–49. 10.1006/excr.1996.0297 [DOI] [PubMed] [Google Scholar]
- 47.Barron VA, Lou H. 2012. Alternative splicing of the neurofibromatosis type I pre-mRNA. Biosci. Rep. 32:131–138. 10.1042/BSR20110060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barron VA, Zhu H, Hinman MN, Ladd AN, Lou H. 2010. The neurofibromatosis type I pre-mRNA is a novel target of CELF protein-mediated splicing regulation. Nucleic Acids Res. 38:253–264. 10.1093/nar/gkp766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fleming VA, Geng C, Ladd AN, Lou H. 2012. Alternative splicing of the neurofibromatosis type 1 pre-mRNA is regulated by the muscleblind-like proteins and the CUG-BP and ELAV-like factors. BMC Mol. Biol. 13:35. 10.1186/1471-2199-13-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhou HL, Hinman MN, Barron VA, Geng C, Zhou G, Luo G, Siegel RE, Lou H. 2011. Hu proteins regulate alternative splicing by inducing localized histone hyperacetylation in an RNA-dependent manner. Proc. Natl. Acad. Sci. U. S. A. 108:E627–E635. 10.1073/pnas.1103344108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu H, Hinman MN, Hasman RA, Mehta P, Lou H. 2008. Regulation of neuron-specific alternative splicing of neurofibromatosis type 1 pre-mRNA. Mol. Cell. Biol. 28:1240–1251. 10.1128/MCB.01509-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yunoue S, Tokuo H, Fukunaga K, Feng L, Ozawa T, Nishi T, Kikuchi A, Hattori S, Kuratsu J, Saya H, Araki N. 2003. Neurofibromatosis type I tumor suppressor neurofibromin regulates neuronal differentiation via its GTPase-activating protein function toward Ras. J. Biol. Chem. 278:26958–26969. 10.1074/jbc.M209413200 [DOI] [PubMed] [Google Scholar]
- 53.Costa RM, Yang T, Huynh DP, Pulst SM, Viskochil DH, Silva AJ, Brannan CI. 2001. Learning deficits, but normal development and tumor predisposition, in mice lacking exon 23a of Nf1. Nat. Genet. 27:399–405. 10.1038/86898 [DOI] [PubMed] [Google Scholar]
- 54.Bibel M, Richter J, Lacroix E, Barde YA. 2007. Generation of a defined and uniform population of CNS progenitors and neurons from mouse embryonic stem cells. Nat. Protoc. 2:1034–1043. 10.1038/nprot.2007.147 [DOI] [PubMed] [Google Scholar]
- 55.Lou H, Yang Y, Cote GJ, Berget SM, Gagel RF. 1995. An intron enhancer containing a 5′ splice site sequence in the human calcitonin/calcitonin gene-related peptide gene. Mol. Cell. Biol. 15:7135–7142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hinman MN, Zhou HL, Sharma A, Lou H. 2013. All three RNA recognition motifs and the hinge region of HuC play distinct roles in the regulation of alternative splicing. Nucleic Acids Res. 41:5049–5061. 10.1093/nar/gkt166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. 1993. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc. Natl. Acad. Sci. U. S. A. 90:8424–8428. 10.1073/pnas.90.18.8424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bouck J, Litwin S, Skalka AM, Katz RA. 1998. In vivo selection for intronic splicing signals from a randomized pool. Nucleic Acids Res. 26:4516–4523. 10.1093/nar/26.19.4516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coolidge CJ, Seely RJ, Patton JG. 1997. Functional analysis of the polypyrimidine tract in pre-mRNA splicing. Nucleic Acids Res. 25:888–896. 10.1093/nar/25.4.888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bouck J, Fu XD, Skalka AM, Katz RA. 1995. Genetic selection for balanced retroviral splicing: novel regulation involving the second step can be mediated by transitions in the polypyrimidine tract. Mol. Cell. Biol. 15:2663–2671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roscigno RF, Weiner M, Garcia-Blanco MA. 1993. A mutational analysis of the polypyrimidine tract of introns. Effects of sequence differences in pyrimidine tracts on splicing. J. Biol. Chem. 268:11222–11229 [PubMed] [Google Scholar]
- 62.Singh R, Valcarcel J, Green MR. 1995. Distinct binding specificities and functions of higher eukaryotic polypyrimidine tract-binding proteins. Science 268:1173–1176. 10.1126/science.7761834 [DOI] [PubMed] [Google Scholar]
- 63.Lush ME, Li Y, Kwon CH, Chen J, Parada LF. 2008. Neurofibromin is required for barrel formation in the mouse somatosensory cortex. J. Neurosci. 28:1580–1587. 10.1523/JNEUROSCI.5236-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guilding C, McNair K, Stone TW, Morris BJ. 2007. Restored plasticity in a mouse model of neurofibromatosis type 1 via inhibition of hyperactive ERK and CREB. Eur. J. Neurosci. 25:99–105. 10.1111/j.1460-9568.2006.05238.x [DOI] [PubMed] [Google Scholar]
- 65.Moroy T, Heyd F. 2007. The impact of alternative splicing in vivo: mouse models show the way. RNA 13:1155–1171. 10.1261/rna.554607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Scheffzek K, Ahmadian MR, Wiesmuller L, Kabsch W, Stege P, Schmitz F, Wittinghofer A. 1998. Structural analysis of the GAP-related domain from neurofibromin and its implications. EMBO J. 17:4313–4327. 10.1093/emboj/17.15.4313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cui Y, Costa RM, Murphy GG, Elgersma Y, Zhu Y, Gutmann DH, Parada LF, Mody I, Silva AJ. 2008. Neurofibromin regulation of ERK signaling modulates GABA release and learning. Cell 135:549–560. 10.1016/j.cell.2008.09.060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Peng S, Zhang Y, Zhang J, Wang H, Ren B. 2010. ERK in learning and memory: a review of recent research. Int. J. Mol. Sci. 11:222–232. 10.3390/ijms11010222 [DOI] [PMC free article] [PubMed] [Google Scholar]