

ABSTRACT
Influenza B virus is an enveloped negative-strand RNA virus that contributes considerably to annual influenza illnesses in human. The matrix protein of influenza B virus (BM1) acts as a cytoplasmic-nuclear shuttling protein during the early and late stages of infection. The mechanism of this intracellular transport of BM1 was revealed through the identification of two leucine-rich CRM1-dependent nuclear export signals (NESs) (3 to 14 amino acids [aa] and 124 to 133 aa), one bipartite nuclear localization signal (NLS) (76 to 94 aa), and two phosphorylation sites (80T and 84S) in BM1. The biological function of the NLS and NES regions were determined through the observation of the intracellular distribution of enhanced green fluorescent protein (EGFP)-tagged signal peptides, and wild-type, NES-mutant, and NLS-mutant EGFP-BM1. Furthermore, the NLS phosphorylation sites 80T and 84S, were found to be required for the nuclear accumulation of EGFP-NLS and for the efficient binding of EGFP-BM1 to human importin-α1. Moreover, all of these regions/sites were required for the generation of viable influenza B virus in a 12-plasmid virus rescue system.
IMPORTANCE This study expands our understanding of the life cycle of influenza B virus by defining the dynamic mechanism of the nucleocytoplasmic shuttle of BM1 and could provide a scientific basis for the development of antiviral medication.
INTRODUCTION
Influenza B virus, one of the major causative agents of flu, is an enveloped negative-strand RNA virus containing eight segmented strands of genome RNA, which encodes 12 viral proteins, consisting of four membrane proteins (HA, NA, BM2, and NB) (1–3), three subunits of viral RNA polymerase (PA, PB1, and PB2) (4), nucleoprotein (NP) (5), matrix protein BM1 (6), nuclear export protein (NEP), and nonstructural protein NS1 (7).
The seventh RNA segment of the viral genome, encodes the viral proteins BM1 and BM2 which are expressed separately through the use of a stop-start pentanucleotide mechanism of regulation (1, 8). BM2 is an oligomeric membrane protein with ion channel activity (9–12), transported through trans-Golgi network (13) which participates in process of viral ribonucleoprotein complex (vRNP) incorporation into virions (14–17). In contrast to the detailed studies of BM2, our understanding of the function of BM1 in the virus life cycle has been limited, with previous studies showing that BM1 is related to mouse adaptation (18) and cold adaptation (19, 20).
Influenza A and B viruses are closely related and composed of proteins with similar functions (21). The matrix protein of influenza A virus (M1) is a multifunctional protein playing important roles in several steps of the influenza virus life cycle. Crucially, the M1 protein facilitates the nuclear export of vRNP by shuttling between the cytoplasm and nuclei of infected cells under the regulation of nuclear localization signal (NLS), nuclear export signal (NES), and phosphorylation (22–26). However, BM1 shares only ca. 30% sequence identity of amino acids with M1, showing no conservation in the functional domain that mediates the cytoplasmic-nuclear transport of M1, raising questions about its biological function and intracellular distribution.
In the present study, we found that BM1 acts as a cytoplasmic-nuclear shuttling protein during infection and identified two novel CRM1-dependent NESs, one bipartite NLS, and five phosphorylation sites in BM1. The NLS, two NESs, and phosphorylation of 80T and 84S all participated in the regulation of cytoplasmic-nuclear transport of BM1 and were all required for the rescue of recombinant influenza B virus.
MATERIALS AND METHODS
Cells and reagents.
The human embryonic kidney 293 (293T) and Madin-Darby canine kidney epithelial (MDCK) cell lines were grown in Dulbecco modified eagle medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS; Gibco) at 37°C in 5% CO2. Lipofectamine reagent was purchased from Invitrogen. dimethyl sulfoxide (DMSO), polyethyleneimine (PEI), and cycloheximide were purchased from Sigma. Leptomycin B (LMB) was purchased from Tocris Bioscience.
Plasmids and virus.
B/Lee/40 strain of influenza B virus and a 12-plasmid influenza B virus rescue system were kindly provided by Yoshihiro Kawaoka. To construct plasmids encoding enhanced green fluorescent protein (EGFP) fusion proteins, the coding sequences of full-length BM1 and truncated BM1 were either amplified by PCR or synthesized artificially and inserted between the XhoI and BamHI sites in the pEGFP-C1 vector, respectively. To construct the plasmid encoding GST–importin-α1, the open reading frame of human importin-α1 was amplified by PCR using total cDNA of human liver and then inserted in the pGEX-6P-1 vector between the BamHI and XhoI sites. The primers used to amplify the coding sequences of BM1 and human importin-α1 were as follows: BM1-1s (5′-CAACTCGAGCTATGTCGCTGTTTGGA-3′), BM1-248r (5′-CAAGGATCCTCATATAGATATTTCTTC-3′), Impα-1 (5′-TAAGGATCCATGTCCACCAACGAG-3′), and Impα-2 (5′-TAAGAGCTCCTAAAAGTTAAAGGT-3′). The plasmids encoding the M gene mutants (M131A/L133/12/14A, R76/77A, and T80A/S84A), which were used for virus rescue, and the plasmids encoding EGFP-BM1 mutants (L12/14A, M131A/L133A, M131A/L133/12/14A, R76/77A, K92/93/94A, and T80A/S84A) were generated with a site-directed mutagenesis kit (NEWPEP, Beijing, China). All of the constructs were verified by DNA sequencing.
Antibodies and reagents.
Rabbit anti-BM1 polyclonal antibody was produced by immunizing a rabbit with purified N-terminal fragment of BM1 (1 to 164 amino acids [aa]), as described previously (27). Mouse anti-BM1, anti-GFP, and anti-β-actin monoclonal antibodies were purchased from Santa Cruz Biotechnology. Goat anti-rabbit IgG conjugated to TRITC (tetramethyl rhodamine isothiocyanate) and goat anti-mouse IgG conjugated to fluorescein isothiocyanate were purchased from Zhongshan Golden Bridge Biotechnology. Complete protease inhibitor cocktail was purchased from Roche Diagnostics. Phosphatase inhibitor (5 mM Na3VO4) was purchased from Sigma.
Transfection and infection.
293T cells were grown in a 24-well cell culture plate with a cover slide in each well. Transfection of plasmids was performed using PEI or Lipofectamine reagent according to the manufacturer's instructions. At 6 h posttransfection, the medium containing the mixture of plasmids and transfection reagent was replaced with fresh DMEM supplemented with 10% FBS.
For infection, 293T or MDCK cells were incubated with influenza virus for 1 h at 37°C. The cells were then washed with phosphate-buffered saline (PBS) and incubated in DMEM with or without FBS for the indicated times at 37°C in 5% CO2.
Indirect immunofluorescence assay.
293T cells grown in a 24-well cell culture plate with cover slides were infected with influenza B virus at a multiplicity of infection (MOI) of ca. 3 to 5. The cells were fixed with 4% paraformaldehyde in PBS at the indicated time points for 1 h at room temperature. The cells were perforated with PBST (0.5% Triton X-100 in PBS) at room temperature for 10 min, incubated with 0.4% bovine serum albumin (BSA) in PBST at 37°C for 1 h, and further incubated with polyclonal anti-BM1 antibody diluted in PBST (containing 0.2% BSA) at 37°C for 1 h. After a washing step with PBST for 1 h, the cells were incubated with Cy3-labeled goat anti-rabbit antibody diluted in PBST (containing 0.2% BSA) at 37°C for 1 h and washed with PBST for 1 h. Finally, the cells were incubated with the nuclei stain DAPI (4′,6′-diamidino-2-phenylindole) for 5 min. The intracellular location of BM1 was recorded with a confocal laser scanning fluorescence microscope (Olympus LSCMFV500).
Generation of recombinant influenza B viruses with the 12-plasmid virus rescue system.
293T and MDCK cells were mixed in a 5:1 ratio and grown on a 60-mm-diameter dish in DMEM supplemented with 10% FBS until the cells reached 90% confluence. The culture medium was then replaced with fresh DMEM containing 2% FBS. After incubation for 1 h, a mixture of 1 μg of each of the 12 plasmids in the virus rescue system was transfected into the cells using Lipofectamine reagent, according to the manufacturer's instructions. Six hours later, the medium containing the mixture of plasmids and Lipofectamine reagent was replaced with DMEM supplemented with 0.1% FBS and 2 μg of TPCK-treated trypsin/μl. The cells were further incubated for 72 h at 37°C in 5% CO2, and the supernatant containing the generated recombinant virus was harvested and centrifuged at 200 × g for 10 min to remove cells.
Phos tag SDS-PAGE and Nano-LC-MS/MS analysis.
293T cells infected with mock or B/Lee/40 strain of influenza B virus were lysed in a lysis buffer (20 mM HEPES, 10% glycerol, 1% Triton X-100, 150 mM NaCl, 1 mM EDTA [pH 7.4], 5 mM Na3VO4, protease inhibitor) at 4°C for 30 min. After centrifugation at 4°C with 12,000 rpm for 10 min, the supernatant of the cell lysates were incubated at 4°C for 4 h with either monoclonal anti-BM1 antibody or an antibody isotype preconjugated to protein G-agarose beads. After three washes, the precipitated proteins conjugated on beads were treated with or without alkaline phosphatase (ALP) at 37°C for 1 h, centrifuged at 4°C with 5,000 rpm for 5 min, and separated by Phos tag SDS-PAGE as described previously (24). The gel was silver stained, and bands of interest were collected from the gel and subjected to nano-liquid chromatography-tandem mass spectrometry (Nano-LC-MS/MS) analysis conducted by the Technological Platform of the Institute of Zoology, Chinese Academy of Sciences (LCQ Deca XP Plus; Thermo).
RESULTS
BM1 shuttled between the cytoplasm and nucleus during virus infection.
293T cells were infected with mock or influenza B virus at an MOI of 3. At 5 and 12 h postinfection (hpi), the cells were fixed and stained with DAPI and anti-BM1 monoclonal antibody as described in Materials and Methods. The location of the nuclei and intracellular distribution of BM1 were imaged by confocal fluorescence microscope. At 5 hpi, more BM1 was distributed in the nucleus than in the cytoplasm of the infected cells, while at 12 hpi, BM1 was distributed predominantly in the cytoplasm of the infected cells, suggesting that BM1's nucleocytoplasmic transport is temporally regulated at early and late stages of infection (Fig. 1).
FIG 1.

BM1 shuttled between the cytoplasm and nucleus of 293T cells during infection. 293T cells were incubated with or without influenza B virus diluted in DMEM (1 μg of TPCK-trypsin/ml) at an MOI of ∼1. After a 1-h absorption at 37°C, the culture medium was replaced with DMEM (2% FBS), and the cells were further incubated at 37°C with 5% CO2. The cells were then fixed with PBS (4% paraformaldehyde) at the indicated time points postinfection. Intracellular localization of BM1 and staining with anti-BM1 polyclonal antibody were performed, followed by imaging with a confocal laser scanning fluorescence microscope (Olympus LSCMFV500).
BM1 contained two CRM1-dependent NESs and one bipartite NLS.
The amino acid sequence of BM1 was screened for candidate peptides containing the sequence motifs of conventional NESs or NLSs. Two candidate leucine-rich NESs, one candidate monopartite NLS, and one candidate bipartite NLS were discovered (Fig. 2A). According to the locations of predicted NESs and NLSs in the amino acid sequence of BM1, plasmids encoding various EGFP-tagged truncations of BM1 were constructed and transfected to 293T cells, respectively. The cells were fixed at 20 h posttransfection, stained with DAPI, and imaged by confocal fluorescence microscope to determine the location of the nuclei and the intracellular distribution of the indicated EGFP-tagged peptides. The full-length EGFP-BM1, EGFP-15-248, and EGFP-15-133, which contained the predicted NESs and the predicted NLSs, were predominantly located in the cytoplasm, whereas EGFP-15-95 and EGFP-15-123, which contained one or two of the predicted NLSs but none of the predicted NESs, were predominantly located in the nucleus (Fig. 2B). These results suggest that the predicted NLS1(76 to 94 aa) is functional and that the nuclear import activity of the NLSs are overcome by the nuclear export activity of the NESs, which results in the cytoplasmic distribution of peptides containing the predicted NESs and NLSs together. Of note, a control peptide which did not contain an NES or an NLS, EGFP-133-248, was evenly distributed between the cytoplasm and nucleus, showing similar distribution to EGFP (Fig. 3B), implying the localization of EGFP seen above was indeed the result of the presence of NES and/or NLS in the protein. The nuclear export and import activities of the candidate NESs and NLSs were further tested individually by fusing the predicted signal peptides to the C terminus of EGFP and observing the intracellular distribution of the EGFP-tagged peptides (Fig. 3A). EGFP-NLS1 (76 to 94 aa) was found to be located predominantly in the nucleus, while the distribution of EGFP-NLS2 (101 to 105 aa) was similar to the EGFP control in that it showed an even distribution between the nucleus and the cytoplasm (Fig. 3B), suggesting that only the NLS1 is functional. Taking into account the knowledge that flanking sequences of NLS2 may be required for carrying out the transport activity, the intracellular distribution of EGFP-95-105 was also tested, but was found to be no different from EGFP-101-105 or EGFP (data not shown). EGFP-NES1 (3 to 14 aa) was predominantly distributed in the cytoplasm of the transfected cells at the 20 h posttransfection and then evenly distributed between the cytoplasm and nucleus after LMB treatment for 2 h, suggesting that NES1 was a functional CRM1-dependent NES. EGFP-NES2 (124 to 133 aa) required the N-terminal flanking sequence (EGFP-115-133) for its nuclear export activity in the context of EGFP fusion protein. The predominant cytoplasmic distribution of EGFP-115-133 was also inhibited by LMB treatment, suggesting the NES2 also acts as a functional CRM1-dependent NES.
FIG 2.

Schematic diagram and intracellular distribution of WT and truncated BM1 constructs. (A) Diagram of predicted NESs and NLSs in the context of BM1. (B) Schematic representation of the constructs encoding EGFP-tagged WT or truncated BM1. (C) 293T cells were transfected with constructs encoding the EGFP tagged WT or truncated BM1, respectively. At 20 h posttransfection, cycloheximide (1 μg/ml) was added to the culture medium. After a 1-h incubation, the cells were fixed PBS (4% paraformaldehyde) and stained with DAPI. The intracellular localization of the indicated proteins was imaged by confocal laser scanning fluorescence microscopy (Olympus LSCMFV500).
FIG 3.

Verification of the biological activities of the predicted NESs and NLSs using EGFP fusion proteins. (A) The predicted NLSs and NESs peptides in BM1 were fused to the C terminus EGFP. 293T cells were transfected with constructs coding the predicted NLS1 and NLS2 (B) and NES1 and NES2 (C), fused with EGFP, respectively. At 20 h posttransfection, LMB was added to the culture medium of the indicated cells to the concentration of 11 nM. After a 3-h incubation, the cells were fixed PBS (4% paraformaldehyde) and stained with DAPI. The intracellular localization of the indicated proteins was imaged by confocal laser scanning fluorescence microscopy (Olympus LSCMFV500).
The nuclear export of BM1 was mediated by the NES1 and NES2.
To investigate the role of NES1 and NES2 in the nuclear export of BM1, several critical hydrophobic amino acids of NES1 and NES2 in the EGFP-BM1 proteins were substituted by alanine (Fig. 4A). The plasmids encoding the wild-type (WT) or mutant EGFP-BM1 were transfected to 293T cells, respectively. The cells were fixed at 20 h posttransfection and stained with DAPI. The intracellular distribution of the indicated proteins was imaged by confocal fluorescence microscope. The distribution of NES1-mutant EGFP-BM1 (L12/14A) showed no difference from the predominantly cytoplasmic distribution of WT EGFP-BM1. The NES2-mutant EGFP-BM1 (M131A/L133A) was distributed evenly between the cytoplasm and the nuclei of most of the transfected cells and predominantly in the nucleus of a small portion of the transfected cells. The NES1/2-mutant EGFP-BM1 (M131A/L133/12/14A) was predominantly distributed in the nucleus of most of the transfected cells (Fig. 4B). Taken together, these results suggest that both the NES1 and NES2 participate in the nuclear export of EGFP-BM1 and that the activity of the NES2 is higher than that of the NES1.
FIG 4.

The nuclear export of BM1 depended on NES1 and NES2. (A) The NES1 and/or NES2 were mutated in the context of full-length EGFP tagged BM1. (B) The constructs encoding the WT or mutant EGFP-BM1 were transfected into 293T cells, respectively. At 20 h posttransfection, the cells were fixed PBS (4% paraformaldehyde) and stained with DAPI. The intracellular localization of the indicated proteins was imaged by confocal laser scanning fluorescence microscopy (Olympus LSCMFV500).
The nuclear import of BM1 depended on the NLS1.
EGFP was evenly distributed in the cytoplasm and nucleus of the transfected 293T cells and was not sensitive to LMB treatment (Fig. 5B). EGFP-BM1 was distributed predominantly in the cytoplasm of transfected 293T cells; however, a nuclear accumulation of EGFP-BM1 was induced by a 2-h LMB treatment (11 nM), indicating that the nuclear accumulation of EGFP-BM1 resulted from the combination of the inhibiting effect of LMB on NES and the inherent activity of NLS-mediated nuclear import, which in turn was used to study the role of the predicted NLS1 in the context of full-length EGFP-BM1.
FIG 5.

NLS1 mediated the nuclear import of BM1. (A) The critical residues in NLS1 were substituted by alanine in the context of full-length EGFP tagged BM1. (B) The constructs encoding EGFP, the WT and mutant EGFP-BM1 were transfected into 293T cells, respectively. At 20 h posttransfection, the cells were treated with (b, d, f, and h) or without (a, c, e, and g) LMB (11 nM). After a 3-h incubation, the cells were fixed PBS (4% paraformaldehyde) and stained with DAPI. The intracellular localization of the indicated proteins was imaged by confocal laser scanning fluorescence microscopy (Olympus LSCMFV500).
The amino acid sequence of NLS1 displayed a typical bipartite NLS pattern—(R/K)2X10-12(R/K)3—in which the arginines and lysines in the C terminus and N terminus were critical for the NLS function. To determine whether the nuclear import of BM1 depended on the NLS1 identified in the present study, alanine substitutions of the predicted critical residues of NLS1, R76/77A and K93/94A, were conducted in EGFP-BM1 (Fig. 5A). Plasmids encoding WT or mutant EGFP-BM1 were transfected to 293T cells, respectively, and treated with LMB at 20 h posttransfection for 2 h. The cells were then fixed, stained with DAPI, and imaged with a confocal fluorescence microscope to determine the intracellular distribution of the indicated proteins. As mentioned above, the cytoplasmic distribution of WT EGFP-BM1 shifted to a nuclear distribution after LMB treatment. However, the predominantly cytoplasmic distribution of EGFP-BM1–R76/77A and EGFP-BM1–K93/94A was not sensitive to LMB treatment (Fig. 5B), indicating that the nuclear import of EGFP-BM1 was destroyed by alanine substitutions of the critical residues of NLS1 and also indicating that the nuclear import of EGFP-BM1 depended on the NLS1.
Identification of the phosphorylated residues in BM1.
We previously found that phosphorylation was involved in the nuclear import of M1 of influenza A virus; therefore, we investigated whether a similar regulation occurs with BM1. First, we collected BM1 from cell lysates of influenza B virus infected cells by immunoprecipitation with anti-BM1 polyclonal antibody (Fig. 6A) and determined whether BM1 was phosphorylated by Western blotting with specific monoclonal antibodies against phosphorylated serine residue and phosphorylated threonine residue. The results showed that phosphorylated serine residues but not phosphorylated threonine residues were detected in BM1 (Fig. 6B). We then used LC-MS/MS to identify candidate phosphorylation sites in BM1. Lysates of 293T cells either mock infected or infected with influenza B virus at an MOI of 3 were incubated with anti-BM1 monoclonal antibody and protein G-Sepharose. The immunoprecipitated BM1 was treated with or without ALP. An ALP-sensitive band in the SDS-PAGE gel was presumed to be phosphorylated BM1 and subjected to LC-MS/MS, which showed that the band was indeed BM1 and revealed that two phosphorylated peptides originated from BM1. One peptide contained two phosphorylated residues (80T and 84S), and the other peptide contained three phosphorylated residues (236S, 237S, and 241S) (Fig. 7).
FIG 6.

Serine-phosphorylated BM1 was detected by specific antibodies against phosphorylated serine. 293T cells were infected with (+) or without (−) influenza B virus at an MOI of 1 for 36 h. The cell lysates were incubated with anti-BM1 polyclonal antibody at 4°C overnight and then precipitated with protein G-beads. (A) The cell lysates and the precipitated proteins were analyzed by Western blotting with anti-BM1 monoclonal antibody. (B) The precipitated proteins were also detected with antibodies specific against phosphorylated serine and phosphorylated threonine, respectively.
FIG 7.

Identification of the phosphorylated residues in BM1 by MS. (A) Cell lysates of mock- or influenza B virus-infected MDCK cells were treated with or without ALP, immune precipitated with or without monoclonal anti-BM1 antibody, and subjected to gel electrophoresis. The band of candidate phosphorylated BM1 was collected from the gel and analyzed by LC-MS/MS. The candidate band was identified as BM1, and two peptides detected using MS (B and D) revealed the phosphorylated residues. (C) The peptides detected using MS (blue) and phosphorylated residues (red) are indicated in the context of BM1.
The nuclear import of BM1 was regulated by phosphorylation of 80T and 84S.
Because NLS-mediated nuclear import of a cargo protein can be regulated by phosphorylation and 80T and 84S were located in the NLS1, the role of 80T and 84S in the nuclear import of BM1 was determined. First, 80T and 84S were substituted with alanine or glutamic acid in the EGFP-tagged NLS1 peptide. In transfected 293T cells, WT EGFP-NLS1 was predominantly located in the nucleus, and the EGFP-NLS1–T80/S84A was distributed evenly between the cytoplasm and nucleus. On the other hand, the positively charged glutamic acid mutation, which mimics the charge present in phosphorylated wild-type residue rescued the function of NLS1 and showed increasing nuclear accumulation (Fig. 8). Taken together, the result indicates that phosphorylation of 80T and 84S is required for the nuclear accumulation of EGFP-NLS1. Alanine substitution of 80T and 84S was then introduced to EGFP-tagged full-length BM1 to test the effect of losing phosphorylation at these two sites on the intracellular distribution of full-length BM1. In transfected 293T cells, WT EGFP-BM1 was distributed predominantly in the cytoplasm and showed nuclear accumulation after LMB treatment, indicating the NLS-mediated nuclear import of BM1 (see Fig. 5B); however, EGFP-BM1 with alanine substitutions of 80T and 84S showed an intracellular distribution similar to that of WT EGFP-BM1 with or without LMB treatment (Fig. 9), suggesting that BM1 was still able to be imported into nucleus without the phosphorylation of 80T and 84S.
FIG 8.
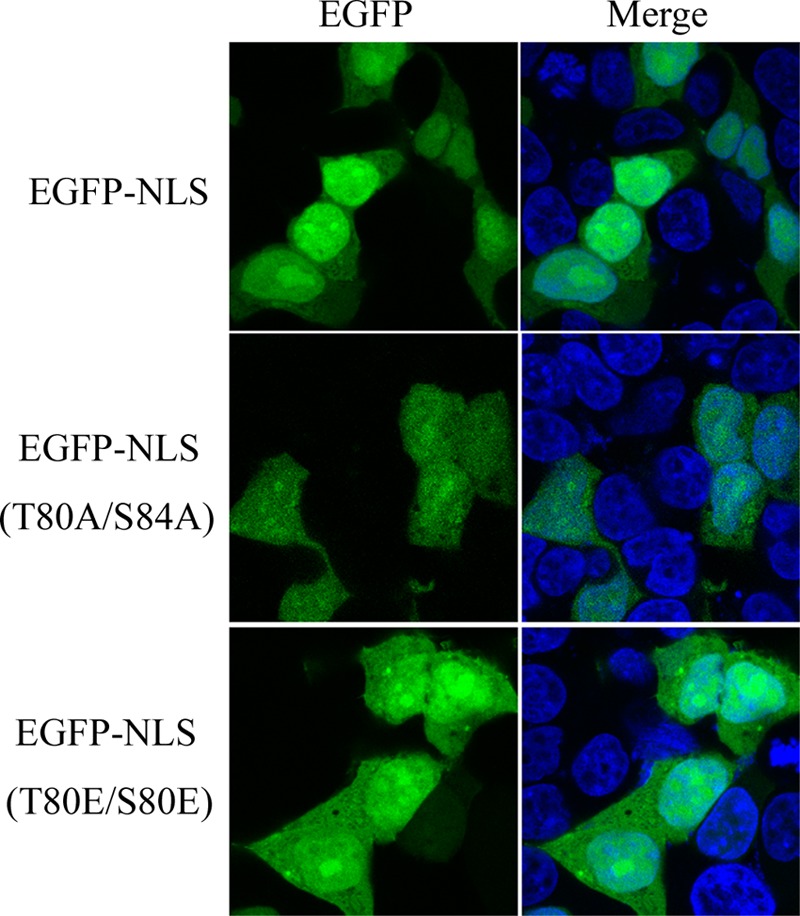
Role of phosphorylated S80 and T84 in the nuclear import of BM1. 293T cells were transfected with constructs encoding EGFP-tagged WT or mutant NLS1. At 20 h posttransfection, the cells were fixed PBS (4% paraformaldehyde) and stained with DAPI. The intracellular localization of the indicated proteins was imaged by confocal laser scanning fluorescence microscopy (Leica SP8).
FIG 9.

Role of phosphorylated S80 and T84 in the nuclear import of BM1. 293T cells were transfected with constructs encoding EGFP-tagged full-length BM1 with alanine substitution of S80 and T84. At 20 h posttransfection, LMB was added to the culture medium of the indicated cells to the concentration of 11 nM. After a 3-h incubation, the cells were fixed PBS (4% paraformaldehyde) and stained with DAPI. The intracellular localization of the EGFP-BM1–S80/T84A was imaged by confocal laser scanning fluorescence microscopy (LSCM Leica SP8). The three-dimensional images were created with the corresponding two-dimensional images taken along the Z-axis (software Imaris).
NLS-containing proteins are directly recognized by cellular nuclear import receptors of the importin-α/β family. To investigate the role of phosphorylated 80T and 84S in the binding between BM1 and human importin-α1, a modified glutathione S-transferase (GST)-pulldown assay was conducted with GST-tagged importin-α1 and WT or mutant EGFP-tagged BM1, expressed in 293T cells. GST–importin-α1 was expressed in Escherichia coli and purified with glutathione-Sepharose. Cell lysates of 293T cells transfected with the plasmids encoding EGFP-BM1, EGFP-BM1–R76/77A, or EGFP-BM1–T80A/S84A were collected, tested for the expression of the indicated proteins, and subjected to the GST-pulldown assay. The band of EGFP-BM1–T80A/S84A was weaker than the band of WT EGFP-BM1 but stronger than the band of EGFP-BM1–R76/77A (Fig. 10). Pixel-based analysis of the indicated bands showed that alanine substitution of R76/77A and T80A/S84A reduced the binding of BM1 to importin-α1 by ∼90 and ∼30%, respectively, indicating alanine substitution of 80T and 84S reduced the binding affinity between BM1 and importin-α1, but not to the extent reduced by alanine substitution of 76R and 77R. It should be noted that small amount of EGFP-BM1–R76/77A was still detected to be associated with GST–importin-α1, although 76R and 77R were shown to be critical for the nuclear import of EGFP-BM1.
FIG 10.
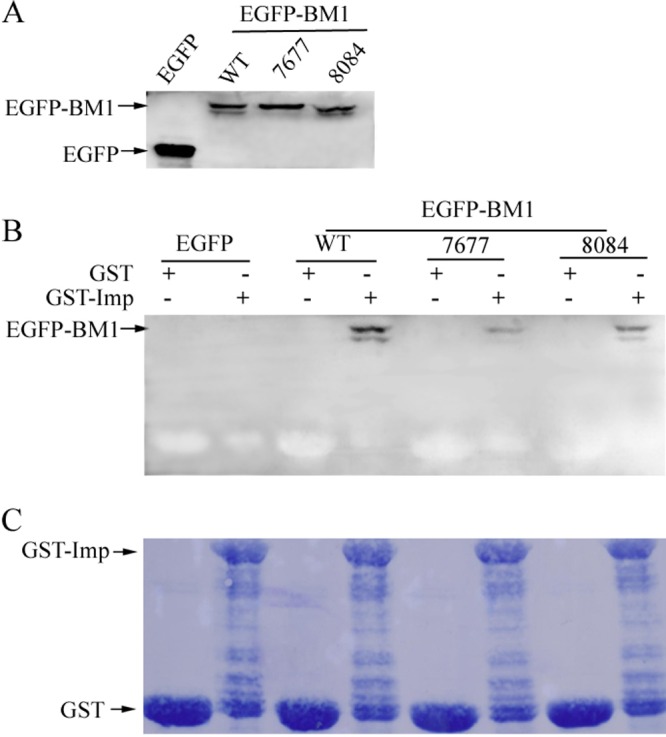
Phosphorylated S80 and T84 and the critical arginines in NLS1 were crucial for the binding of BM1 to importin-α1. (A) 293T cells were transfected with EGFP-tagged WT, R76/77A mutant, and S80/T84A mutant BM1, respectively. The cells were collected at 36 h posttransfection, and the indicated proteins in cell lysates were detected by Western blotting with monoclonal anti-GFP antibody. (B) The cell lysates containing the EGFP-tagged WT or mutant BM1 were incubated with purified GST or GST-tagged human importin-α1 at 4°C for 2 h and then precipitated with immobilized glutathione. The WT or mutant EGFP-tagged BM1 precipitated together with GST–importin-α1 were detected by Western blotting with monoclonal anti-GFP antibody. (C) The nitrocellulose blotting membrane was stained with Coomassie brilliant blue.
DISCUSSION
Influenza A and B viruses share a similar composition and organization, including a segmented viral RNA genome, a viral RNA polymerase composed of PA, PB1, and PB2, a cellular membrane derived from virion envelope scattered with neuraminidases (NA), a receptor-binding hemagglutinin (HA) and an ion channel (M2), and a layer of matrix protein (M1) beneath the envelope. The corresponding proteins of influenza A and B virus are presumed to carry out similar biological functions through homologous active domains, as demonstrated by the partially compatible viral RNA polymerases. In the present study, we show that, like M1, BM1 shuttles between the cytoplasm and nucleus of the infected cells. However, several different characteristics do exist between the mechanism of cytoplasmic-nuclear transport of M1 and BM1. First of all, M1 contains only one LMB-insensitive NES (59 to 68 aa), while there are two CRM1-dependent NESs (3 to 14aa and 133 to 124 aa) in BM1. Second, the NLS in M1 (101 to 105 aa) (23) is monopartite, but the NLS in BM1 (76 to 94 aa) has a typical bipartite NLS. It should also be noted that the peptide of 101 to 105 aa in BM1 satisfied the pattern of a monopartite NLS but was found to lack nuclear import activity.
Among the five phosphorylation sites identified in BM1 in the present study, 236S and 237S were reported in a previous study (28), but the roles of the phosphorylation of the these residues in virus life cycle were not investigated. M1 and BM1 were both phosphorylated proteins (29). Our group showed previously that a phosphorylation site in M1 (132Y) is required for the nuclear import of M1 (24). In the present study, we showed that phosphorylation of 80T and 84S, which are located in the NLS1 (76 to 94 aa) of BM1, are required for the predominantly nuclear distribution of EGFP-NLS1 peptide. However, loss of phosphorylation of 80T and 84S showed no obvious effect on the nuclear distribution of the EGFP-BM1 but did reduce the amount of full-length BM1 binding to importin-α in a immunoprecipitation assay. This suggests that the loss of phosphorylation of 80T and 84S might be compensated for by some other residues in the context of full-length BM1. Nevertheless, virus rescue was failed when 80T and 84S of BM1 were substituted with alanine. A possible explanation for this is that phosphorylation of 80T and 84S might be required for the interaction of BM1 with other viral or host proteins, which is indispensable for the rescue of influenza B virus. The rescue experiment was repeated eight times, each time the rescue of WT influenza B virus was used as a positive control. The WT influenza B virus was successfully rescued three times in eight independent experiments (3/8), while no BM1 mutant virus was rescued in the eight experiments (0/8), including NES-mutant BM1 (L12/14/133/M131A), NLS-mutant BM1 (R76/77A), and phosphorylation site mutant BM1 (T80/S84A) viruses.
M1 of influenza A virus plays multiple roles at different steps of the virus life cycle but is particularly important in the nuclear export of vRNP. As demonstrated in a recent model proposed by our group, in the late steps of influenza A virus infection, M1 is synthesized in cytoplasm and imported into nucleus where it acts as a bridge between NEP and vRNP in the NEP-M1-vRNP complex. Once complexed the NESs in NEP and M1 bind to CRM1, resulting in the subsequent nuclear export of NEP-M1-vRNP complex. However, the role of cytoplasmic-nuclear shuttling of BM1 in influenza B virus life cycle remains unclear especially given that the NEP of influenza B virus was reported to contain NES (30) and associate with vRNP directly (31), which would make BM1 redundant as abridge between NEP and vRNP. We identify here several regions that contribute to the shuttling of BM1 between the nucleus and the cytoplasm and hypothesize that BM1 and NEP may still participate in the nuclear export of vRNP collaboratively. We had hoped to reveal the role of BM1 in the nuclear export of vRNP by studying mutant influenza B viruses but, unfortunately, the rescue of recombinant influenza B virus failed with NES-mutant BM1, NLS-mutant BM1, and BM1 bearing the alanine substitutions of 80T and 84S.
ACKNOWLEDGMENTS
This study was supported by grants from the National Natural Science Foundation of China (31370200, 31101830, 81101253, and 81101254), the China Ministry of Science and Technology Project 973 (2012CB955501, 2012CB518903, and 2011CB504705), an intramural special grant for influenza virus research from the Chinese Academy of Sciences (KJZD-EW-L09-2), the Ministry of Science and Technology of China Program (2010BAD04B01-7), the Key Research Program of the Chinese Academy of Sciences (KSZD-EW-Z-005-001), the National Natural Science Foundation of China Innovative Research Group (81021003), and funding from the Animal Experimental Platform Operation of the Chinese Academy of Sciences (CZBZX-1).
We thank Xiaojuan Jia for proofreading.
Footnotes
Published ahead of print 16 April 2014
REFERENCES
- 1.Horvath CM, Williams MA, Lamb RA. 1990. Eukaryotic coupled translation of tandem cistrons: identification of the influenza B virus BM2 polypeptide. EMBO J. 9:2639–2647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Almond JW, Haymerle HA, Felsenreich VD, Reeve P. 1979. The structural and infected cell polypeptides of influenza B virus. J. Gen. Virol. 45:611–621. 10.1099/0022-1317-45-3-611 [DOI] [PubMed] [Google Scholar]
- 3.Fischer W, Pitkeathly M, Wallace B, Forrest L, Smith G, Sansom M. 2000. Transmembrane peptide NB of influenza B: a simulation, structure, and conductance study. Biochemistry 39:12708–12716. 10.1021/bi001000e [DOI] [PubMed] [Google Scholar]
- 4.Nakamura K, Kitame F, Homma M. 1981. A comparison of proteins among various influenza B virus strains by one-dimensional peptide mapping. J. Gen. Virol. 56:315. 10.1099/0022-1317-56-2-315 [DOI] [PubMed] [Google Scholar]
- 5.Ng AKL, Lam MKH, Zhang H, Liu J, Au SWN, Chan PKS, Wang J, Shaw PC. 2012. Structural basis for RNA binding and homo-oligomer formation by influenza B virus nucleoprotein. J. Virol. 86:6758–6767. 10.1128/JVI.00073-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ito T, Gorman OT, Kawaoka Y, Bean WJ, Webster RG. 1991. Evolutionary analysis of the influenza A virus M gene with comparison of the M1 and M2 proteins. J. Virol. 65:5491–5498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Briedis DJ, Lamb RA. 1982. Influenza B virus genome: sequences and structural organization of RNA segment 8 and the mRNAs coding for the NS1 and NS2 proteins. J. Virol. 42:186–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hatta M, Kohlmeier CK, Hatta Y, Ozawa M, Kawaoka Y. 2009. Region required for protein expression from the stop-start pentanucleotide in the M gene of influenza B virus. J. Virol. 83:5939–5942. 10.1128/JVI.00180-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Balannik V, Lamb RA, Pinto LH. 2008. The oligomeric state of the active BM2 ion channel protein of influenza B virus. J. Biol. Chem. 283:4895–4904. 10.1074/jbc.M110.175638 [DOI] [PubMed] [Google Scholar]
- 10.Paterson RG, Takeda M, Ohigashi Y, Pinto LH, Lamb RA. 2003. Influenza B virus BM2 protein is an oligomeric integral membrane protein expressed at the cell surface. Virology 306:7–17. 10.1016/S0042-6822(02)00083-1 [DOI] [PubMed] [Google Scholar]
- 11.Mould JA, Paterson RG, Takeda M, Ohigashi Y, Venkataraman P, Lamb RA, Pinto LH. 2003. Influenza B virus BM2 protein has ion channel activity that conducts protons across membranes. Dev. Cell 5:175–184. 10.1016/S1534-5807(03)00190-4 [DOI] [PubMed] [Google Scholar]
- 12.Odagiri T, Hong J, Ohara Y. 1999. The BM2 protein of influenza B virus is synthesized in the late phase of infection and incorporated into virions as a subviral component. J. Gen. Virol. 80(Pt 10):2573–2581 [DOI] [PubMed] [Google Scholar]
- 13.Watanabe S, Imai M, Ohara Y, Odagiri T. 2003. Influenza B virus BM2 protein is transported through the trans-Golgi network as an integral membrane protein. J. Virol. 77:10630–10637. 10.1128/JVI.77.19.10630-10637.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Imai M, Kawasaki K, Odagiri T. 2008. Cytoplasmic domain of influenza B virus BM2 protein plays critical roles in production of infectious virus. J. Virol. 82:728–739. 10.1128/JVI.01752-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jackson D, Zurcher T, Barclay W. 2004. Reduced incorporation of the influenza B virus BM2 protein in virus particles decreases infectivity. Virology 322:276–285. 10.1016/j.virol.2004.02.003 [DOI] [PubMed] [Google Scholar]
- 16.Imai M, Watanabe S, Ninomiya A, Obuchi M, Odagiri T. 2004. Influenza B virus BM2 protein is a crucial component for incorporation of viral ribonucleoprotein complex into virions during virus assembly. J. Virol. 78:11007–11015. 10.1128/JVI.78.20.11007-11015.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hatta M, Goto H, Kawaoka Y. 2004. Influenza B virus requires BM2 protein for replication. J. Virol. 78:5576–5583. 10.1128/JVI.78.11.5576-5583.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McCullers JA, Hoffmann E, Huber VC, Nickerson AD. 2005. A single amino acid change in the C-terminal domain of the matrix protein M1 of influenza B virus confers mouse adaptation and virulence. Virology 336:318–326. 10.1016/j.virol.2005.03.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Z, Aspelund A, Kemble G, Jin H. 2006. Genetic mapping of the cold-adapted phenotype of B/Ann Arbor/1/66, the master donor virus for live attenuated influenza vaccines Virology 345:416–423 [DOI] [PubMed] [Google Scholar]
- 20.Chen Z, Aspelund A, Kemble G, Jin H. 2008. Molecular studies of temperature-sensitive replication of the cold-adapted B/Ann Arbor/1/66, the master donor virus for live attenuated influenza vaccines. Virology 380:354–362. 10.1016/j.virol.2008.08.010 [DOI] [PubMed] [Google Scholar]
- 21.Steinhauer DA, Skehel JJ. 2002. Genetics of influenza viruses. Annu. Rev. Genet. 36:305–332. 10.1146/annurev.genet.36.052402.152757 [DOI] [PubMed] [Google Scholar]
- 22.Cao S, Liu X, Yu M, Li J, Jia X, Bi Y, Sun L, Gao GF, Liu W. 2012. A nuclear export signal in the matrix protein of influenza A virus is required for the efficient virus replication. J. Virol. 86:4883–4891. 10.1128/JVI.06586-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye Z, Robinson D, Wagner RR. 1995. Nucleus-targeting domain of the matrix protein (M1) of influenza virus. J. Virol. 69:1964–1970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang S, Zhao Z, Bi Y, Sun L, Liu X, Liu W. 2013. Tyrosine 132 phosphorylation of influenza A virus M1 protein is crucial for virus replication by controlling the nuclear import of M1. J. Virol. 87:6182–6191. 10.1128/JVI.03024-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martin K, Heleniust A. 1991. Nuclear transport of influenza virus ribonucleoproteins: the viral matrix protein (M1) promotes export and inhibits import. Cell 67:117–130. 10.1016/0092-8674(91)90576-K [DOI] [PubMed] [Google Scholar]
- 26.Bui M, Wills EG, Helenius A, Whittaker GR. 2000. Role of the influenza virus M1 protein in nuclear export of viral ribonucleoproteins. J. Virol. 74:1781–1786. 10.1128/JVI.74.4.1781-1786.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu X, Sun L, Yu M, Wang Z, Xu C, Xue Q, Zhang K, Ye X, Kitamura Y, Liu W. 2009. Cyclophilin A interacts with influenza A virus M1 protein and impairs the early stage of the viral replication. Cell. Microbiol. 11:730–741. 10.1111/j.1462-5822.2009.01286.x [DOI] [PubMed] [Google Scholar]
- 28.Hutchinson EC, Denham EM, Thomas B, Trudgian DC, Hester SS, Ridlova G, York A, Turrell L, Fodor E. 2012. Mapping the phosphoproteome of influenza A and B viruses by mass spectrometry. PLoS Pathog. 8:e1002993. 10.1371/journal.ppat.1002993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gregoriades A, Guzman GG, Paoletti E. 1990. The phosphorylation of the integral membrane (M1) protein of influenza virus. Virus research 16:27–41. 10.1016/0168-1702(90)90041-9 [DOI] [PubMed] [Google Scholar]
- 30.Paragas J, Talon J, O'Neill RE, Anderson DK, Garcia-Sastre A, Palese P. 2001. Influenza B and C virus NEP (NS2) proteins possess nuclear export activities. J. Virol. 75:7375–7383. 10.1128/JVI.75.16.7375-7383.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Imai M, Watanabe S, Odagiri T. 2003. Influenza B virus NS2, a nuclear export protein, directly associates with the viral ribonucleoprotein complex. Arch. Virol. 148:1873–1884. 10.1007/s00705-003-0166-x [DOI] [PubMed] [Google Scholar]