
Figure 1.
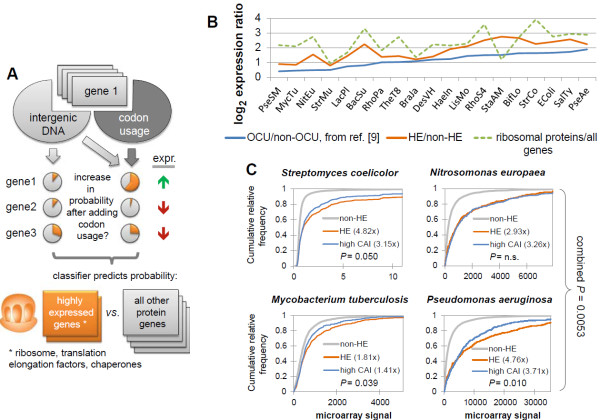
Finding highly expressed (HE) genes in prokaryotic genomes and their enrichment in particular phenotypic groups. (A) HE labels for genes were predicted by comparing the codon usage of each gene with that of a small set of known HE genes, while controlling for local background nucleotide composition determined from the neighboring intergenic DNA. (B) Comparison of average microarray signal intensity between the HE genes from this study, and the HE gene group of Supek et al. [9] in 19 diverse bacterial genomes, denoted by the UniProt species code on the x axis. The machine learning pipeline used here was derived from the methodology of Supek et al. [9] (see Additional file 1). (C) The predictions of HE genes compared favorably with those of the codon adaptation index (CAI) when evaluated against microarray data in four genomes previously claimed to lack detectable selected codon biases [21-23], representing difficult cases for predicting gene expression from codon usage. The x axes range from the minimum to the 99th percentile of microarray signal intensities. The farther away a curve is from the non-HE curve, the better the separation. Numbers in the parenthesis are the ratios of average expression in the HE (or high CAI) genes, compared with the non-HE genes. The ‘high CAI’ category was defined as containing the same number of genes as the HE category in each genome. P values are from a one-tailed Kolmogorov-Smirnov test for HE > high CAI, and were combined using Fisher’s method.