
Fig. 3.
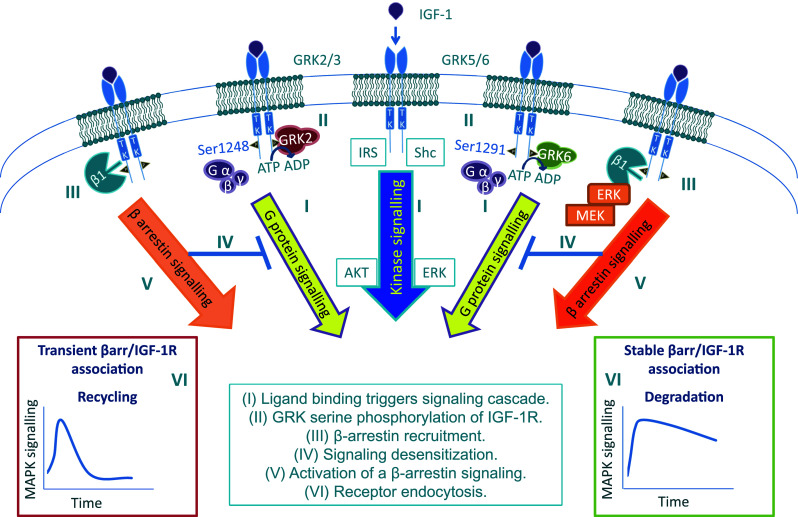
IGF-1R as a RTK/GPCR functional hybrid. Signaling: agonist-stimulation of the IGF-1R triggers the classical RTK signaling leading to downstream kinase-cascade signaling activation. In addition, agonist-stimulation of IGF-1R leads to non-canonical GPCR signaling through heterotrimeric G-proteins (G α, β, γ) (I), following which the receptors are rapidly phosphorylated by G-protein-coupled receptor kinases (GRKs) at serine residues within the C-terminus (II). Desensitization: Serine-phosphorylated receptors present high affinity binding sites to recruit the multifunctional adaptor protein β-arrestin 1 (β1) (III). Steric binding by β-arrestin to the IGF-1R C-terminal prevents further G-protein coupling, leading to the desensitization of G-protein-dependent signaling (IV). β-arrestin signaling: β-arrestin acquires an active conformation upon binding the IGF-1R and scaffolds components of the MAPK pathways leading to the activation of a second wave of IGF-1R kinase independent signaling through β-arrestin 1 (V). The dynamics of this signaling activation is determined by the strength of the β-arrestin 1/IGF-1R interaction, dependent on GRK-isoform (VI) Such β-arrestin-dependent MAPK activity has been shown to regulate multiple IGF-1R biological effects including proliferation, apoptosis, cell migration, and cancer metastasis. Endocytosis: agonist-stimulation promotes rapid endocytosis of the IGF-1R. This internalization is enabled by β-arrestin binding, connecting the receptor to the endocytic machinery efficiently. The interaction between β-arrestin and the E3 ubiquitin ligase Mdm2 promotes IGF-1R and β-arrestin ubiquitination that facilitates IGF-1R endocytosis, followed by post-endocytic sorting of internalized IGF-1R. The strength of the β-arrestin 1/IGF-1R interaction GRKs-isoform-dependent determines the fate of internalized receptor: recycling (transient β-arrestin 1 binding) or degradation (sustained β-arrestin 1 binding) (VI)