

Background: Intracellular peptides probably regulate several biological processes.
Results: pep5 derived from G1/S cyclin D2 specifically increases during the S phase of the cell cycle and, reintroduced into the cell, induces apoptosis and necrosis.
Conclusion: pep5 has potential therapeutic applications and could have biological functions.
Significance: pep5 discovery advances our understanding of limited proteolysis.
Keywords: Anticancer Drug, Apoptosis, Cell Cycle, Cell Death, Necrosis (Necrotic Death), Peptides, Proteasome
Abstract
Intracellular peptides are constantly produced by the ubiquitin-proteasome system, and many are probably functional. Here, the peptide WELVVLGKL (pep5) from G1/S-specific cyclin D2 showed a 2-fold increase during the S phase of HeLa cell cycle. pep5 (25–100 μm) induced cell death in several tumor cells only when it was fused to a cell-penetrating peptide (pep5-cpp), suggesting its intracellular function. In vivo, pep5-cpp reduced the volume of the rat C6 glioblastoma by almost 50%. The tryptophan at the N terminus of pep5 is essential for its cell death activity, and N terminus acetylation reduced the potency of pep5-cpp. WELVVL is the minimal active sequence of pep5, whereas Leu-Ala substitutions totally abolished pep5 cell death activity. Findings from the initial characterization of the cell death/signaling mechanism of pep5 include caspase 3/7 and 9 activation, inhibition of Akt2 phosphorylation, activation of p38α and -γ, and inhibition of proteasome activity. Further pharmacological analyses suggest that pep5 can trigger cell death by distinctive pathways, which can be blocked by IM-54 or a combination of necrostatin-1 and q-VD-OPh. These data further support the biological and pharmacological potential of intracellular peptides.
Introduction
Cell signaling induces modification of the protein interactome network. The ubiquitin-proteasome system is responsible for maintaining protein homeostasis in cells, and among its essential functions is its involvement with cancer biology and the immune system. The proteasome is responsible for the initial cleavage of antigenic proteins and the generation of the major histocompatibility complex class I (MHC-I)-bound antigens, which can later be trimmed within the endoplasmic reticulum by endoplasmic reticulum aminopeptidases to produce the correct N terminus to interact with the MHC-I (1, 2). In cancer biology, the ubiquitin-proteasome system is important for cell cycle progression because it targets cyclins for degradation (3). Proteasome inhibition is used clinically for the treatment of certain types of cancer and is the treatment of choice for multiple myeloma (4). Protein degradation by the proteasome generates intermediate peptides shown to contain from 2 to 21 amino acids (5, 6), which in theory suggests that cells are continually producing large quantities of peptides containing 2–21 amino acids. However, according to current knowledge, only one peptide from each cellular protein would escape postproteasome proteolytic degradation to be presented at the cell surface bound to the MHC-I, suggesting that ∼10,000 peptides cover the mammalian cell surface to allow self-recognition by the immune system (7, 8). Thus, almost the entire protein is believed to be recycled to amino acids within cells. Indeed, in yeast, mammalian cells, and flies, the injurious consequences of proteasome inhibition are rescued by amino acid supplementation, revealing the importance of the proteasome system in amino acid recycling for de novo protein synthesis (9). Further reports have suggested that peptides produced by the proteasome have a half-life of seconds (10–12). In specific circumstances, the ubiquitin-proteasome system can perform limited proteolysis, such as in the generation of the active dimeric NFκB transcriptional complexes (13).
Therefore, it is clear that the proteasome can perform both extensive and limited proteolysis, depending on the protein substrate. In our laboratory, we have shown that the proteasome could play a more extensive role in limited proteolysis than previously anticipated, generating intracellular peptides (14–16). These findings were based on a well established concept that rationally designed peptides, structurally similar to the ones produced by the proteasome, can regulate protein spatial localization within cells and control cell signal transduction (17, 18). As such, naturally occurring intracellular peptides generated by the proteasome would constitute an as yet poorly understood mechanism by which cells increase their protein network complexity and function (16). Hemopressin, the first intracellular peptide identified using this rationale (19), was shown to have cannabinoid inverse agonist action regulating food intake (20, 21), whereas the natural brain hemopressins are secreted and suggested to play an important role as novel endocannabinoids (14, 22).
Later it was shown that intracellular peptides can function in modulating signal transduction from inside the cells because peptides structurally related to proteasome products were identified by mass spectrometry, chemically synthesized, and reintroduced into cells, where they modulated both angiotensin II and β-adrenergic signal transduction (23). These peptides were used for affinity chromatography and were suggested to bind to a specific set of proteins, many involved in protein and vesicular traffic (23). In addition to the proteasome, thimet oligopeptidase (EC 3.4.24.15; EP24.15), which is an intracellular peptidase that only degrades small peptides (∼5–17 amino acids), was also shown to participate in intracellular peptide metabolism (24). By manipulating intracellular EP24.15 activity either by overexpressing the enzyme or inhibiting its activity by means of siRNA, it was possible to modulate G-protein-coupled receptor signal transduction in HEK293 and CHO-S cells (23, 25). These data suggest a previously unknown connection between intracellular peptide metabolism and signal transduction. Other signal transduction pathways could also be related to intracellular peptides because two similar peptides identified in the Wistar rat adipose tissue where shown to bind specific proteins and facilitate insulin-induced glucose uptake in 3T3-L1 adipocyte cells (26). Although the intracellular peptides have not yet been shown to directly modulate protein-protein interactions in vivo, the in vitro use of surface plasmon resonance demonstrates that at concentrations of 1–50 μm, several intracellular peptides can modulate the interactions of calmodulin and 14-3-3ϵ with proteins from the mouse brain cytoplasm or with recombinant EP24.15. One of these peptides (VFDVELL; VFD-7), shown to be a proteasome product (24), increases the free cytosolic Ca2+ concentration in a dose-dependent manner but only if introduced into HEK293 cells (27).
In the present report, we aim to obtain further information on the cell biology and therapeutic potential of intracellular peptides by investigating their possible participation in the cell cycle. To that end, we identified in extracts of HeLa cells a novel peptide fragment (WELVVLGKL; pep5) that specifically increases during the S phase of the cell cycle and is derived from the G1/S-specific cyclin D2 protein. The peptide pep5 induces cell death in HeLa and several other tumor cells and in vivo reduces by 50% the volume of the rat C6 glioblastoma. Collectively, the above results suggest that peptides generated by the proteasome and additional intracellular peptidases need further attention as novel natural modulators of cell function. These data suggest the therapeutic potential of intracellular peptides.
EXPERIMENTAL PROCEDURES
Reagents
Acetonitrile was purchased from Fisher. Mass spectrometry grade hydrochloric acid and trifluoroacetic acid were from Pierce. Hydroxylamine, glycine, sodium hydroxide, sodium phosphate, dimethyl sulfoxide (DMSO), necrostatin-1, q-VD-OPh (qVD),3 and IM-54 were obtained from Sigma. The 4-trimethylammoniumbutyryl (TMAB)-N-hydroxysuccinimide-stable isotopic labeling reagents, containing either 0, 3, or 9 atoms of deuterium (D0, D3, and D9, respectively) or 9 atoms of deuterium and three 13C atoms (D12) were synthesized as described by Che et al. (28), Morano et al. (29) and Zhang et al. (30). Fluorescamine and SB203580 were purchased from Invitrogen. All peptides were provided by Proteimax Biotechnology LTDA (São Paulo, Brazil).
Cell Lines
HeLa, MDA-MB-231, MCF-7, and C6 cells were cultured in Dulbecco's modified Eagle's medium (DMEM; Invitrogen), whereas SKRB, SK-MEL 28, MEL 85, SBCl-2, TPC-1, Nthy-ori 3-1, and KTC-2 cells were cultured in RPMI 1640 (Invitrogen) at 37 °C and 5% of CO2, containing 10% fetal bovine serum (complete medium), penicillin, and streptomycin (Invitrogen).
Cell Cycle Synchronization by Double-thymidine Block
HeLa cells were synchronized using the double-thymidine block procedure (31, 32). Thymidine (Sigma-Aldrich) was diluted in serum-free DMEM and stored at 4 °C before use. For cell cycle synchronization, HeLa cells were treated with 2 mm thymidine for 18 h and washed in phosphate-buffered saline (PBS), cell medium was replaced with complete medium, and cells were cultured for an additional 9 h at 37 °C and 5% of CO2. The above procedure was repeated once more to arrest cells in S phase. At different times (4, 10, and 16 h, at which ∼84% of cells were in S, G1, and G2/M phases, respectively) cells were harvested, analyzed by flow cytometry, and submitted to additional assays. Control cells were asynchronous.
Flow Cytometry
Cell cycle was analyzed by flow cytometry using a Guava Easy City Mini Flow Cytometry (Millipore) instrument. Cells were fixed in 70% ethanol for 1 h and incubated with propidium iodide (1 mg/ml) and RNase (10 mg/ml) for 30 min on ice, and a total of 10,000 events were analyzed in each sample.
Cell Morphology Analyses
HeLa cells (4 × 105 cells/plate) were grown in 35-mm cell culture dishes (Corning Inc.) in complete medium and treated with scrambled control peptide (SCB), pep5, cell-penetrating peptide (cpp), SCB-cpp, or pep5-cpp (50 or 100 μm) for 24 h. After that, cell morphology was visualized and photographed under an inverted phase-contrast microscope (Zeiss Axiovert 25 inverted phase contrast microscope) at ×400 magnification.
Apoptosis Assay
Cells were treated with pep5-cpp, cpp alone, and ΔN or ΔC-pep5-cpp for 10 or 30 min. After incubation, cells were harvested and washed in cold phosphate-buffered saline (PBS) and resuspended in 100 μl of 1× annexin-binding buffer (Molecular Probes). Alexa Fluor® 488 annexin V (5 μl) was added with 1 μl of 100 μg/ml PI working solution to each 100 μl of cell suspension. The samples were incubated at room temperature for 15 min, and after the incubation period, 400 μl of 1× annexin-binding buffer were added, mixed gently, and kept on ice. As soon as possible, the samples were analyzed by flow cytometry, measuring the fluorescence emission at 530 and >575 nm.
Surgery
C6 rat glioma (Rattus norvegicus) (33) cells were grown in DMEM containing 10% fetal bovine serum and antibiotics (penicillin/streptomycin). Cells in the exponential phase of growth were used, and a suspension was prepared in sterile saline at a concentration of 5 × 105 cells/4–5 μl. Adult male Wistar rats of 250–350 g (n = 5) were anesthetized with an intramuscular injection of ketamine/xylazine (10 mg/1.5 mg/100 g of body weight) to provide deep anesthesia and analgesia. The rats were placed on a stereotaxic surgical table, a midline incision was made, and a burr hole was drilled 0.48 mm anterior and 3 mm lateral to the bregma. The C6 cell suspension was slowly injected into the striatum using a Hamilton syringe at a depth of 5.4 mm to the bone surface, and the needle was left in situ for 3 min before its removal. After 14 days, Alzet osmotic pumps containing either pep5-cpp or ΔN-pep5-cpp (100 μm) diluted in artificial cerebrospinal fluid were surgically implanted and attached to Alzet brain infusion kits. Artificial cerebrospinal fluid was chosen as a vehicle solution in order to mimic more closely the composition of the interstitial fluid within the brain. The peptide concentrations were chosen based on our previous results using cell lines. The pump infusion rate was 0.5 μl/h with a duration of 2 weeks. After a further 14 days, the rats were killed by transcardiac perfusion with 4% formaldehyde in 0.1 m phosphate buffer, pH 7.4.
After perfusion, the brains were removed, fixed in 4% formaldehyde, and cryoprotected with 30% sucrose in PBS. The frozen tissue sections (30 μm) were obtained on a freezing microtome. The brain slices were stained with H&E, and tumor area was analyzed under a microscope. The largest areas of tumor were measured using image analysis software (Image ProPlus). The volume was calculated using the formula, V = π/6 × L × W × H. This procedure was approved by the Ethical Commission for Animal Experimentation of the Biomedical Sciences Institute (University of São Paulo), protocol number 116/08.
Peptide Extraction and Quantification
HeLa cells (1 × 106 cells/plate) were grown in 10-cm cell culture plates (Corning Inc.) in complete medium. Fifteen plates of cells were used per group, and each group was characterized by flow cytometry to be in specific phases of the cell cycle, meaning asynchronous, G1, S, or G2/M. Complete medium was removed, and cells were washed three times with PBS, scraped from the plates, and centrifuged at 830 × g for 5 min. The pellet was resuspended in 10 ml of 80 °C water and incubated in an 80 °C water bath for 20 min. The cell lysates were stored at −80 °C overnight. To extract peptides, samples were acidified with 10 μl of ice-cold 0.1 m HCl to a final concentration of 10 mm HCl and sonicated three times with 20 pulses (4 Hz) on ice. The samples were centrifuged at 2,500 rpm in a microcentrifuge for 40 min at 4 °C, and the supernatant was collected for peptide extraction. The supernatant was ultracentrifuged at 34,000 rpm for 60 min at 4 °C, transferred to a centrifugal filter device for the separation of molecules of less than 5,000 Da (Millipore), and centrifuged again at 2,500 rpm for 1 h at 4 °C. The peptide quantification reaction was performed at pH 6.8 to ensure that only the amino groups of peptides and not free amino acids reacted with fluorescamine, as described previously (23, 34). Briefly, 2.5 μl of peptide samples were mixed with 25 μl of 0.2 m phosphate buffer (pH 6.8) and 12.5 μl of a 0.3 mg/ml acetone fluorescamine solution. After vortexing for 1 min, 110 μl of water was added, and fluorescence was measured with a SpectraMax M2e plate reader (Molecular Devices) at an excitation wavelength of 370 nm and an emission wavelength of 480 nm. A peptide mixture of known composition and concentration was used as the standard reference for determining the peptide concentration in the experimental samples.
Isotopic Labeling and Mass Spectrometry for Peptide Identification
The labeling procedure has been previously described in detail (35, 36). In brief, each group of cells within an experiment was labeled with one of the isotopic TMAB-N-hydroxysuccinimide labels. The S, G2/M, or G1 groups of characterized cells were labeled with D3-, D9-, or D12-TMAB, respectively (Run 1), whereas the control asynchronous cells were labeled with D0-TMAB. This labeling was altered (reverse labeling) between experiments (Run 2). These two experiments above were performed independently. A total of 50 μg of the peptide extract was combined with 250 μl of 0.4 m phosphate buffer, pH 9.5, and 15 mg of TMAB label per group. After the addition of TMAB, the mixture was incubated at room temperature for an additional 60 min. To quench any remaining labeling reagent, 30 μl of 2.5 m glycine was added to the reaction. The sample was dried in a vacuum centrifuge and analyzed by liquid chromatography/mass spectrometry (LC-MS/MS) on a Synapt G1 mass spectrometer with a nanoACQUITY UltraPerformance LC System (Waters Co.). The MS spectra were analyzed using the MassLynx software (Waters) to identify groups of peaks representing peptides labeled with the different isotope tags. Quantification was performed by determining the relative intensity of each isotopic peak (37). To identify the peptides, the MS/MS data were analyzed using the Mascot search engine (Matrix Science Ltd.).
Peptide Synthesis
Peptides 3 and 5, whose concentrations were altered in different phases of the cell cycle, and a control scrambled peptide were synthesized with cpp (YGRKKRRQRRR) covalently bound to either their C or N terminus (38–40). In all assays, peptide purity was greater than 95%. All synthesized peptides were provided by Proteimax Biotechnology LTDA.
Caspase Assays
Caspase 3/7, 8, and 9 activities were evaluated in extracts of HeLa cells previously treated for 10 min with either the proapoptotic pep5-cpp or the control peptide (100 μm), using a commercial luminescent assay kit (Caspase-Glo® assay, Promega). The relative luminescence was measured in a SpectraMax luminometer (Molecular Devices).
Phospho-MAPK Array
A human phospho-MAPK array kit (Proteome Profiler) was used to simultaneously test pep5-cpp effects on the phosphorylation of diverse MAPK signaling pathways, according to instructions provided by the manufacturer. Briefly, all arrays were incubated with 200 μg of HeLa cell extracts treated previously for 10 min with either pep5-cpp or the control peptide (100 μm).
Pharmacological Treatments
HeLa cells (4 × 105 cells/plate) were pretreated with necrostatin-1 (nec-1), qVD, SB203580, or IM-54 (30 μm) for 1 h, and then pep5-cpp (75 μm) was added to the medium containing one or more inhibitors. After 4 h of treatment with pep5-cpp, cells were harvested and washed in PBS and analyzed by flow cytometry.
Proteasome Activity Assays
HeLa cells treated or not with pep5-cpp (50–200 μm) were washed twice with PBS and lysed on ice for 30 min. Cells extracts were centrifuged at 13,000 rpm for 10 min, and total protein concentration was determined by the Bradford assay (41) using bovine serum albumin as a standard. The proteasome substrate GloTM Chymotrypsin-like (Suc-LLVY-GloTM, Promega) was used to determine the proteasome activity, as recommended by the manufacturer.
Statistics
Values are expressed as means ± S.E. Statistical analyses were conducted by Student's unpaired t test for independent samples or analysis of variance followed by Tukey's or Bonferroni's test to compare more than two groups, using GraphPad Prism version 5.0. p values of <0.05 were considered significant.
RESULTS
The initial hypothesis of our work was that the relative concentration of specific intracellular peptides, similarly to that of certain specific proteins, could vary throughout the cell cycle, and perhaps even contribute to its control. Therefore, we began by synchronizing HeLa cells using the double-thymidine block procedure (31, 32) to further isolate and characterize intracellular peptides from each individual phase of the cell cycle. As a result of the double-thymidine block, analyses by flow cytometry suggested that ∼84% of HeLa cells were arrested in each phase of the cell cycle (G1, S, or G2/M) when compared with the asynchronous cell group (Fig. 1). The intracellular peptide content was then extracted from asynchronous, G1, S, or G2/M HeLa cells, labeled with specific TMAB isotopes, and analyzed by mass spectrometry. In these analyses, 19 peptides were sequenced and identified in order of appearance (Table 1; MS and MS/MS data are shown in the supplemental material). pep3, a fragment from the 40 S ribosomal protein S21, and pep5, a fragment of G1/S-specific cyclin D2 (Table 1), were observed to clearly fluctuate along the cell cycle (Fig. 2) and were further investigated with respect to their possible biological activity on regulating the progression of the cell cycle and/or cell survival. pep3, pep5, and SCB were chemically synthesized either free or covalently bound to a cpp at their C or N terminus. The cpp was necessary to allow cell penetration and consequent investigation of the possible intracellular function of these peptides. Moreover, the cpp used herein has been extensively described to transport its cargo into the cells (37, 38).
FIGURE 1.

Synchronization of HeLa cells using 2 mm thymidine. The percentage of the cells in each phase was determined by flow cytometry analysis. DNA content frequency histograms represent cells in S, G2/M, and G1, respectively, 4, 10, and 16 h after double thymidine block or asynchronous growing cells in the control culture. Fluorescence intensity was measured after adding propidium iodide to the cells. The histograms and the graph (mean and S.E. (error bars)) shown here are representative of three independent experiments. A, control (asynchronous cells); B–D, cells synchronized in S, G2/M, and G1 phases.
TABLE 1.
Summary of protein precursors and sequence of the peptides identified in HeLa cells along the progression of the cell cycle
Protein name, precursor protein of the respective peptide sequence; pep#, peptide identity, according to the order in which it was sequenced; Sequence, the respective peptide sequence identified by MS/MS analyses (all of the MS and MS/MS spectra are presented in the supplemental material); z, the peptide charge state; #T, the number of tags in the identified peptide; Obs M, observed mass; Theor M, theoretical mass of the peptide; ppm, mass accuracy; Ratio, relative level of each peptide in the specific cell cycle phase indicated (G1, S, or G2)/control asynchronous cells. Except for peptides 16 and 18, all data presented are mean ± S.E.; n = 4. MS and MS/MS data are given in the supplemental material.
| Protein name | pep# | Sequence | z | #T | Obs M | Theor M | ppm | Ratio |
||
|---|---|---|---|---|---|---|---|---|---|---|
| G1/CT | S/CT | G2/CT | ||||||||
| Peptidyl-prolyl cis-trans isomerase A | 1 | ADKVPKTAENFR | 4 | 3 | 1,374.7 | 1,374.7 | −13.4 | 1.19 ± 0.24 | 2.11 ± 2.13 | 1.75 ± 1.17 |
| 40 S ribosomal protein S21 | 2 | KADGIVSKNF | 3 | 3 | 1,077.5 | 1,077.6 | −52.4 | 0.71 ± 0.21 | 0.69 ± 0.13 | 0.83 ± 0.08 |
| 40 S ribosomal protein S21 | 3 | AKADGIVSKNF | 3 | 3 | 1,148.6 | 1148.6 | −45,0 | 1.52 ± 0.41 | 1.95 ± 0.14 | 1.88 ± 0.32 |
| 40 S ribosomal protein S21 | 4 | GSGSKGKGGEIQPVSV | 3 | 3 | 1,485.7 | 1485.8 | −62.7 | 1.39 ± 0.51 | 1.57 ± 1.16 | 1.29 ± 0.96 |
| G1/S-specific cyclin D2 | 5 | WELVVLGKL | 2 | 2 | 1,055.5 | 1055.6 | −164.2 | 1.27 ± 0.67 | 2.16 ± 0.40 | 1.22 ± 0.47 |
| TOX high mobility group box family member 3 | 6 | QITSPIPAIGS | 3 | 1 | 1,082.6 | 1082.6 | 10.4 | 0.70 ± 0.30 | 0.58 ± 0.06 | 1.24 ± 0.62 |
| 40 S ribosomal protein S21 | 7 | ADGIVSKNF | 2 | 2 | 949.4 | 949.5 | −73.1 | 0.98 ± 0.27 | 0.94 ± 0.32 | 0.85 ± 0.17 |
| Polypyrimidine tract-binding protein 1 | 8 | RIIVENL | 3 | 1 | 855.5 | 855.5 | −64.4 | 1.15 ± 0.12 | 1.19 ± 0.04 | 1.61 ± 0.44 |
| Activated RNA polymerase II transcriptional coactivator p15 | 9 | KEQISDIDDAVRKL | 4 | 3 | 1,628.8 | 1,628.9 | −45.0 | 1.60 ± 0.39 | 1.94 ± 0.12 | 1.84 ± 0.39 |
| PPIA_HUMAN, peptidyl-prolyl cis-trans-isomerase A | 10 | AVDGEPLGRVSF | 2 | 1 | 1,245.6 | 1,245.6 | −52,4 | 0.71 ± 0.28 | 0.69 ± 0.33 | 0.83 ± 0.18 |
| Potassium channel subfamily T member 1 | 11 | SILLNPGPRHILA | 2 | 1 | 1,399.4 | 1,399.8 | −286.5 | 0.84 ± 0.06 | 0.71 ± 0.19 | 0.76 ± 0.16 |
| Dipeptidyl peptidase 1 | 12 | DPFNPFELTNH | 2 | 1 | 1,329.8 | 1,329.6 | 162.9 | 0.96 ± 0.08 | 0.33 ± 0.46 | 0.66 ± 0.93 |
| Hemopexin | 13 | LTKGGYTL | 2 | 2 | 851.4 | 851.5 | −115,6 | 0.61 ± 0.09 | 0.57 ± 0.63 | 0.41 ± 0.35 |
| Cytochrome c oxidase subunit 5A, mitochondrial | 14 | GISTPEELGLDKV | 2 | 2 | 1,356.6 | 1,356.7 | −52.2 | 0.62 ± 0.14 | 0.49 ± 0.20 | 0.51 ± 0.12 |
| Cofilin-1 | 15 | GGSAVISLEGKPL | 2 | 2 | 1,226.6 | 1,226.7 | −59.7 | 1.14 ± 0.01 | 1.07 ± 0.23 | 1.03 ± 0.17 |
| Lysosomal protective protein | 16 | APDQDEIQR | 2 | 1 | 1,070.5 | 1,070.5 | −39.2 | 0.46 | 0.73 | 0.71 |
| Protocadherin α-3 | 17 | GEGLPKTDL | 2 | 2 | 928.4 | 928.5 | −86.7 | 0.82 ± 0.16 | 0.83 ± 0.04 | 0.97 ± 0.08 |
FIGURE 2.

MS spectra representing the semiquantitative analysis of peptides from synchronized HeLa cell extracts (S, G2/M, and G1) or controls. HeLa cells were synchronized with 2 mm thymidine, and the peptides were labeled with isotopes TMAB (D0, D3, D9, and D12). A and B, representative MS spectra of two peptides (pep5 and pep3) that reproducibly changed during the HeLa cell cycle. The data shown here are representative of two independent experiments.
First, HeLa cells were treated with each of these peptides at a specific concentration (100 μm), and after 24 h, they were analyzed by flow cytometry (Fig. 3). The control peptides SCB and pep3, either free or bound to the cpp, or the cpp alone had no effect on cell cycle progression or cell death (Fig. 3). pep5 had no effect on cell cycle progression, whereas it killed ∼90% of cells when cpp was linked to its C terminus and ∼40% when cpp was linked to its N terminus (Fig. 3). A dose-response curve of pep5-cpp (1–100 μm) was performed in HeLa cells, showing cell death activity at concentrations above 50 μm (Fig. 4, A and B). A time course was also performed, indicating that pep5-cpp (50–100 μm) starts to induce significant cell death in HeLa cells after 2 h of incubation (Fig. 4C). HeLa cell morphology was observed following the treatments with SCB, pep5, cpp, SCB-cpp, or pep5-cpp (50 or 100 μm; 24 h). These results corroborate the suggestion that pep5-cpp at either 50 or 100 μm is inducing large cell death (Fig. 5).
FIGURE 3.

Effect of intracellular peptides in HeLa cells. HeLa cells were treated with peptide 3 or 5 free or fused to a cpp, cpp alone, or SCB for 24 h. After that, the cells were fixed in 70% ethanol and analyzed by propidium iodide staining and flow cytometry. Results were considered significant when p was ≤0.001 (***). The data shown here are representative of three independent experiments performed in triplicate. NT, not treated; pep-cpp, cpp fused to the C terminus of the peptides; cpp-pep, cpp fused to the N terminus of the peptides. The histograms show the difference among the groups treated with pep5 bound or not to the cpp in the N or C terminus. Error bars, S.E.
FIGURE 4.

Effect of pep5-cpp in HeLa cells. A, HeLa cells were treated with peptide 5, cpp alone, or SCB for 24 h. B, cells were incubated with pep5-cpp at different concentrations (1, 10, 25, 50, 75 and 100 μm) for 24 h. C, cells were treated with pep5-cpp and SCB (control) for 10 h at 50 and 100 μm. After all tests, the cells were fixed in 70% ethanol and analyzed by propidium iodide staining and flow cytometry. Results were considered significant when p was ≤0.001 (***) or ≤0.05 (#). The data shown here are representative of three experiments performed in triplicate. NT, not treated; pep-cpp, cpp fused to the C terminus of the peptides; cpp-pep, cpp fused to the N terminus of the peptides. Error bars, S.E.
FIGURE 5.

Morphology of HeLa cells treated with pep5-cpp. HeLa cells were treated or not with cpp, SCB, pep5, or pep5-cpp (50 and 100 μm) for 24 h. The cells were treated and photographed under an optical microscope after 24 h of treatment. The pictures show the difference in morphology of the cells treated or not with pep5-cpp (50 and 100 mm). NT, not treated. Scale bar, 50 μm.
Next we performed several modifications in the pep5 sequence to investigate the relationship between structure and activity. In order to identify the minimal active sequence, we started by deleting 1, 2, or 3 amino acids from either the N or C terminus. Removal of a single amino acid from the N terminus (ΔN-pep5-cpp) totally abolished the induction of cell death by pep5-cpp (Fig. 6). The potency of pep5-cpp was only slightly reduced by the removal of two or three amino acids from its C terminus, whereas it was totally abolished after the removal of four amino acids from the C terminus (Fig. 6A). Additional experiments were performed, substituting specific hydrophobic amino acids (Leu or Val) from the original sequence by Ala, which inactivated the cell death activity of pep5-cpp (Fig. 6B). pep5-cpp was also acetylated at the N terminus, and the results suggested that the cell death activity is retained, although with reduced potency compared with the original sequence (Fig. 6C). Table 2 summarizes the pep5-cpp sequences investigated herein. Taken together, these data suggest a strong structural specificity of pep5-cpp in inducing cell death.
FIGURE 6.

Effect of modifications in the original sequence of pep5-cpp. A, effect of deletions in pep5 sequence. Cells were treated with pep5-cpp, SCB (control), and ΔN- or ΔC-pep5-cpp (pep5 with deletions) for 24 h (100 μm) and then analyzed by flow cytometry. NT, not treated, SCB control; ΔN-pep5-cpp, deletion of one amino acid at the N terminus; A, B, C, and D, C terminus deletions of 1, 2, 3, and 4 amino acids, respectively. B, analysis of substitutions in the original sequence of pep5. Cells were treated with pep5-cpp, SCB, and a modified pep5-cpp for 24 h (100 μm) and then analyzed by flow cytometry. A, B, and C, pep5 with one or more substitutions, in the original sequence, for alanine. C, cells were treated with pep5-cpp (100 μm) and the acetylated pep5-cpp at different concentrations (1, 10, 50, and 100 μm) for 24 h and then analyzed by flow cytometry. Ac-pep5-cpp, acetylated pep5. The data shown here are representative of three experiments performed in triplicate, and the results were considered significant when p was <0.001 (***). Error bars, S.E.
TABLE 2.
Amino acid sequences of pep5-cpp investigated herein
ΔN, one or more amino acids deleted from N terminus; ΔC, one or more amino acids deleted from C terminus; A, B, and C, amino acids substitutions for the minimal active sequence (ΔC3-pep5-cpp*) of pep5-cpp. Ac-pep5-cpp, ΔC3-pep5-cpp acetylated in the N terminus.
| Amino acid sequences from pep5-cpp | Abbreviation |
|---|---|
| WELVVLGKL-YGRKKRRQRRR | pep5-cpp |
| ELVVLGKL-YGRKKRRQRRR | ΔN-pep5-cpp |
| LVVLGKL-YGRKKRRQRRR | ΔN2-pep5-cpp |
| VVLGKL-YGRKKRRQRRR | ΔN3-pep5-cpp |
| WELVVLGK-YGRKKRRQRRR | ΔC1-pep5-cpp |
| WELVVLG-YGRKKRRQRRR | ΔC2-pep5-cpp |
| WELVVL-YGRKKRRQRRR | ΔC3-pep5-cpp* |
| WELVV-YGRKKRRQRRR | ΔC4-pep5-cpp |
| WELV-YGRKKRRQRRR | ΔC5-pep5-cpp |
| WEL-YGRKKRRQRRR | ΔC6-pep5-cpp |
| WE-YGRKKRRQRRR | ΔC7-pep5-cpp |
| WELVVA-YGRKKRRQRRR | A |
| WEAVVL-YGRKKRRQRRR | B |
| WEAVVA-YGRKKRRQRRR | C |
| Ac-WELVVL-YGRKKRRQRRR | Ac-pep5-cpp |
To investigate whether the cell death induction caused by pep5-cpp was specific to HeLa cells, we investigated the effect of this peptide in additional tumor cell lines (Fig. 7). With distinctive efficacy, pep5-cpp (100 μm) induced cell death in all tumor cell lines investigated here (Fig. 7). The percentage of cell death caused by pep5-cpp was 81.5 ± 1.2% in SKRB (human breast cancer cell line), 94.7 ± 2.1% in SK-MEL-28 (human skin melanoma cell line), 84.6 ± 4.4% in SBCl-2 (human melanoma), 93.8 ± 1.7% in MEL-85 (human melanoma), 52.9 ± 9.7% in C6 (rat glial tumor), 82.1 ± 8.9% in TPC-1, and 84.9 ± 1.0% in KTC-2 (human thyroid tumor cell lines). The normal human thyroid cell line Nthy-ori 3-1 was also tested, and pep5-cpp killed 45.3 ± 3.0% of cells. Despite differences in efficacy, pep5-cpp was observed to induce cell death in all cell lines investigated (Fig. 7). Moreover, a dose-response curve was performed to investigate the effect of pep5-cpp in two distinctive human breast adenocarcinoma cell lines, MDA-MB-231 and MCF-7 (Fig. 8). These results suggest that MDA-MB-231 cells (Fig. 8A) are more sensitive than MCF-7 cells (Fig. 8B) to the cell death effects of pep5-cpp.
FIGURE 7.
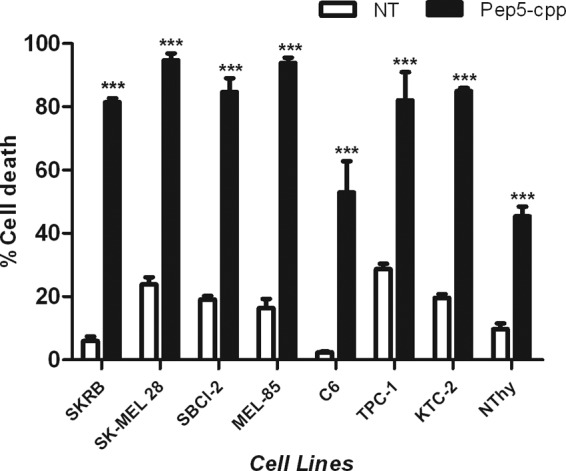
Effect of pep5-cpp in several different cell lines. A, cells were treated or not with pep5-cpp for 24 h (100 μm) and then analyzed by flow cytometry. NT, not treated. The data shown here are representative of three experiments performed in triplicate, and the results were considered significant when p was <0.001 (***). Error bars, S.E.
FIGURE 8.
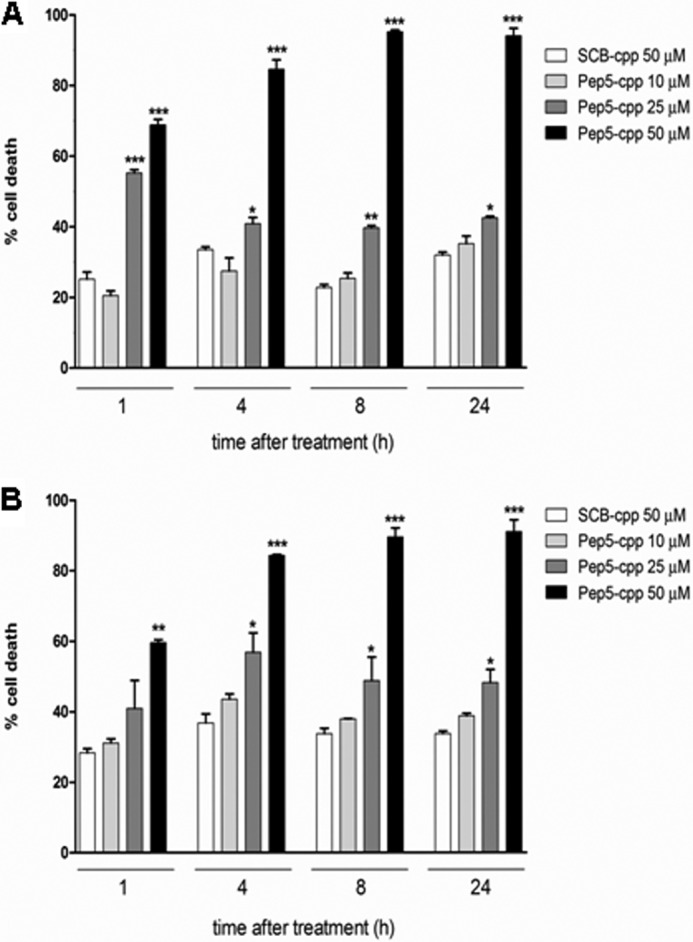
Dose-response and time course of the cell death activity of pep5-cpp in MDA-MB-231 and MCF-7 tumor cell lines. The MDA-MB-231 (A) or MCF-7 (B) cells were treated with pep5-cpp (1–50 μm) for different times (1–24 h). NT, not treated. The data shown here are representative of three experiments performed in triplicate, and the results were considered significant when p was <0.001 (***). Error bars, S.E.
In vivo infusion of pep5-cpp (100 μm), but not ΔN-pep5-cpp (100 μm), caused a significant decrease in C6 tumor growth (Fig. 9). Whereas the average tumor volume in the ΔN-pep5-cpp-treated animals was 6.705 ± 1.20 mm3, in the animals treated with pep5-cpp, it was only 3.404 ± 0.844 mm3 (Fig. 9A), suggesting that pep5-cpp reduced the tumor volume by 49% (p = 0.0275, n = 5).
FIGURE 9.

In vivo effects of ΔN-pep5-cpp or pep5-cpp in the rat C6 glioma. Male Wistar rats (n = 5) with 14-day pre-established tumors were treated with ΔN-pep5-cpp or pep5-cpp (100 μm) for an additional 14 days, and after infusion, the animals were killed with an overdose of ketamine-xylazine (Parke-Davis). After perfusion, the brains were removed, fixed in 4% formaldehyde, and cryoprotected with 30% sucrose in PBS. The frozen tissue sections (30 μm) were obtained on a freezing microtome. The brain slices were stained with H&E, and tumor area was analyzed under a microscope. The largest areas of tumor were measured using image analysis software (Image ProPlus). The volume was calculated using the formula, V = π/6 × L × W × H. A, tumor volume. B–D, H, and I, ΔN-pep5-cpp. E–G, J, and K, pep5-cpp. B–G, arrows indicate tumor borders; scale bar, 400 μm. J and K, arrows indicate phagocytic macrophage-like cells. Scale bar (H–K), 20 μm. Analysis was done using the one-tailed t test, and the results were considered significantly different (p = 0.0275). Error bars, S.E.
Next, we attempted to investigate the possible cell death-inducing mechanism of pep5-cpp. HeLa cells were treated with pep5-cpp or the cpp alone (50 μm) for 30 min and analyzed by flow cytometry in the presence of annexin V (Molecular Probes). Results suggest that pep5-cpp, but not cpp alone, significantly induces apoptosis and necrosis in HeLa cells (Fig. 10). After a 30-min treatment with pep5-cpp, apoptotic cells corresponded to ∼30% of the total population, and necrotic cells corresponded to ∼60%, whereas ∼10% of the cells remained viable (Fig. 10A). Similar results were obtained using either pep5-cpp or its minimal active sequence (ΔC-pep5-cpp), which has three amino acids deleted from the C terminus (Fig. 10B). HeLa cells treated for 10 min with a lower dose of pep5-cpp (50 μm) showed no difference between apoptosis versus necrosis (Fig. 10C). Those cells treated with pep5-cpp (100 μm, 10 min) showed a significant increase of caspase 3/7 and 9 activities, whereas the activity of caspase 8 remained unaltered (Fig. 11). Treatment of HeLa cells with the control SCB peptide caused no cell death or caspase activation (Fig. 11).
FIGURE 10.

Mechanism of cell death induced by pep5-cpp. Cells were treated with cpp, pep5-cpp, or modified pep5-cpp (10–50 μm) for 30 min (A and B) or 10 min (C) and incubated with annexin V and propidium iodide (PI). After the incubation period (15 min), the samples were analyzed by flow cytometry. The population should separate into at least three groups: viable cells (V) with only a low level of fluorescence, apoptotic cells (A) with a substantially higher green fluorescence, and necrotic cells (N) with higher red fluorescence intensity. NT, not treated; ΔN- or ΔC-pep5-cpp, pep5 with deletions. The data shown here are representative of three experiments performed in triplicate, and the results were considered significant when p was <0.001 (***) or <0.05 (*). Error bars, S.E.
FIGURE 11.
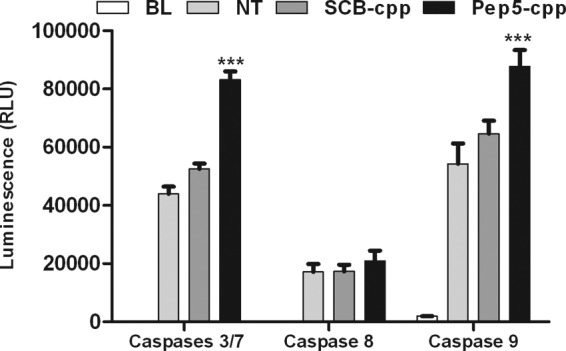
Caspase assay after treatment with pep5-cpp. Cells were treated with peptide 5 and SCB (scrambled) for 10 min (100 μm). After treatment, the substrates were added for the detection of different caspase activities. The relative luminescence was read using a luminometer (Spectramax, Molecular Devices). BL, blank; NT, not treated; SCB, control; Pep5, pep5-cpp. The data shown here are representative of three experiments performed in triplicate, and the results were considered significant when p was <0.001 (***). RLU, relative light units. Error bars, S.E.
Next, the mitogen-activated protein kinase (MAPK) pathway was evaluated in a standard array assay, which simultaneously investigates the phosphorylation of several kinases, including extracellular signal-regulated kinases (ERKs), c-Jun N-terminal or stress-activated protein kinases (JNK/SAPK), ERK/big MAP kinase 1 (BMK1), and the p38 kinases (42). HeLa cells were treated with pep5-cpp (100 μm, 10 min), the cell extracts were analyzed in duplicates according to the manufacturer's instructions, and the results were compared with untreated HeLa cells. Specific kinases or their substrates, such as ERK1/2, HSP27, p38 α/γ, and p70 S6 kinase had increased phosphorylation upon treatment with pep5-cpp, when compared with the untreated control group. However, substrates, such as Akt2 and GSK-3β, had decreased phosphorylation after treatment with pep5-cpp (Fig. 12A). Additional experiments were similarly conducted to compare the effect of cell death inducer pep5-cpp with the cell death-inactive ΔN-pep5-cpp; the latter peptide only lacks the original N-terminal amino acid residue from pep5-cpp. These results further suggest the structural specificity of pep5-cpp to inhibit Akt2 and to activate ERK2, HSP27, and p38γ phosphorylation (Fig. 12B).
FIGURE 12.
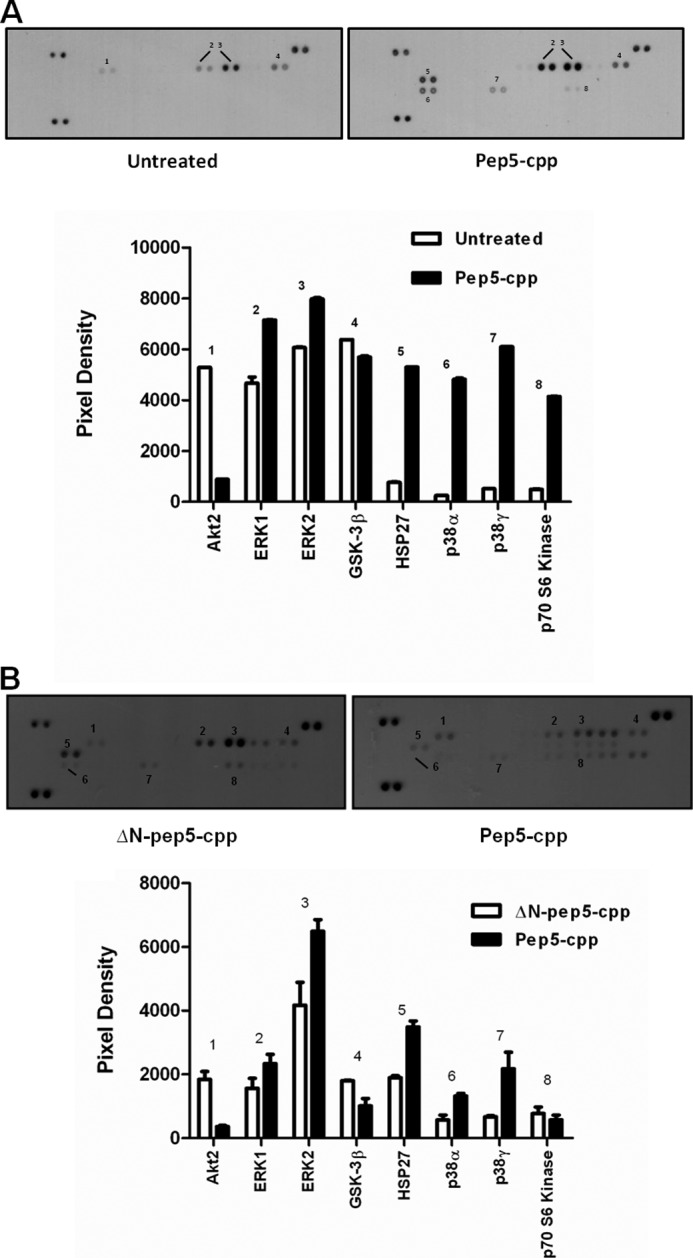
Effect of pep5-cpp in the phosphorylation of specific human MAPKs or their substrates. HeLa cells were treated or not with peptide 5 (A) and ΔN-pep5-cpp (B) for 10 min (100 μm). All arrays were incubated with 200 μg of lysate. ΔN-pep5, pep5-cpp without one amino acid in the N terminus. The assayed MAPKs and targets included Akt1, Akt2, Akt3, Akt pan, CREB, ERK1, ERK2, GSK-3α/β, GSK-3β, HSP27, JNK1, JNK2, JNK3, JNK pan, MKK3, MKK6, MSK2, p38α, p38β, p38δ, p38γ, p53, p70 S6K, RSK1, RSK2, and TOR. Error bars, S.E.
The ability of pep5 to affect the chymotrypsin activity of the proteasome was evaluated both in whole cells and in cell extracts using a standard commercial assay. In HeLa cells treated with pep5-cpp for 24 h, the proteasome activity was significantly reduced (Fig. 13A). pep5 also showed inhibitory activity of the chymotrypsin activity of the proteasome in cell extracts (Fig. 13B).
FIGURE 13.

Proteolytic activities of the proteasome in HeLa cells treated or not with pep5-cpp. Proteasome chymotrypsin activity was measured in aliquots of cell lysates containing 20 μg of protein as described under “Experimental Procedures.” A, whole HeLa cells were treated with pep5-cpp (100 μm) for 24 h, and the proteasome activity was measured in the cell extracts. B, the proteasome activity was measured in the HeLa cell extracts in the presence or absence of pep5 at the indicated concentrations. Lactacystin (10 μm) was used as a positive control. NT, not treated; BL, blank. RLU, relative light units. Error bars, S.E.
Furthermore, pharmacological treatments using distinctive inhibitors of cell death were conducted (Fig. 14). Necrostatin-1, an inhibitor of necroptosis (43); qVD, a potent caspase inhibitor that protects cells from capase-dependent apoptosis (44); SB203580, a selective inhibitor of p38 mitogen-activated protein kinase, which inhibits also the phosphorylation and activation of protein kinase B (PKB, also known as Akt1) (45, 46); and/or IM-54, a cell-permeable, potent, and selective inhibitor of oxidative stress-induced necrosis (47) were evaluated (30 μm, 1 h of pretreatment). Used alone, only the IM-54 significantly reduced the cell death induction caused by pep5-cpp. On the other hand, only in combination were nec-1 and qVD able to reduce the cell death induced by pep5-cpp in HeLa cells. Nec-1, qVD, or SB203580 treatment alone was not effective in blocking pep5-cpp-induced cell death (Fig. 14).
FIGURE 14.
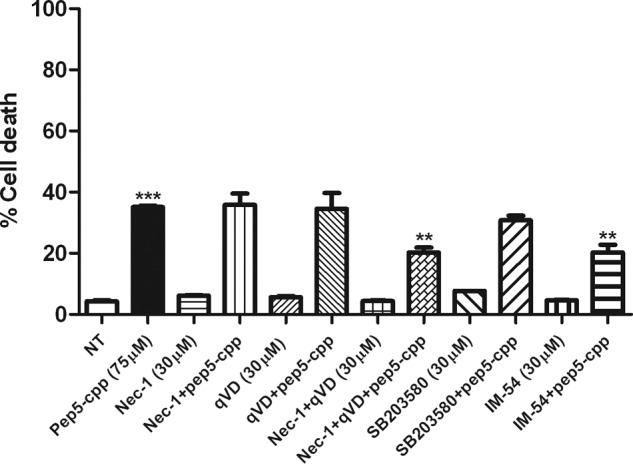
Effect of specific inhibitors of cell death on pep5-cpp activity. HeLa cells were pretreated with nec-1, qVD, SB203580, or IM-54 (30 μm, 1 h), and then pep5-cpp (75 μm) was added to the medium containing one or more inhibitors. After treatment with pep5-cpp (∼4 h), the cells were analyzed by flow cytometry. The data shown here are representative of three experiments performed in triplicate, and the results were considered significant when p was <0.01 (**) or <0.001 (***). Error bars, S.E.
DISCUSSION
In the present study, several novel intracellular peptides were identified in HeLa cells. The relative levels of some of these intracellular peptides appear to fluctuate throughout the cell cycle, which resembles that which is already well established for proteins controlling cell cycle progression. Whereas the broad biological significance of these findings remains elusive, to our knowledge this is the first report to describe fluctuation in the relative levels of intracellular peptides during the cell cycle progression of HeLa cells.
The relative level of the peptide WELVVLGKL (pep5) increases 2-fold in the S phase of HeLa cell cycle, compared with asynchronous cells. Reintroduction of pep5 in HeLa or several additional tumor cell lines induces cell death, suggesting its functionality. In a previous study in HEK293T cells, the concentration of the intracellular peptide VFD-7 was determined to be ∼16 μm (27). It is possible that the minimal effective pharmacological dose of pep5 that induces cell death in MDA-MB-231 cells (∼25 μm) could be occurring within cells. Differences in the kinetics of administration and/or coupling to a transport mechanism could be related to the distinctive efficacy of pep5 seen in the tumor cell lines investigated in this study. A high structural specificity was observed for pep5, based on the finding that a single amino acid removal, modification, or substitution in the minimal active sequence (WELVVL) reduced or abolished pep5 cell death activity. Therefore, it is also possible that differences in the efficacy of pep5 observed between cell lines are due to variation in the activity of proteases and/or peptidases. Indeed, it has been shown previously that oligopeptidases (24, 25) as well as the proteasome (15, 48) can metabolize a number of intracellular peptides.
Pharmacological examination conducted in HeLa cells suggests that IM-54, a potent and selective inhibitor of oxidative stress-induced necrosis (47), efficiently blocked the cell death activity of pep5. IM-54 selectively inhibits oxidative stress-induced necrosis, as observed in ischemia-reperfusion injury caused by heart attack, and does not inhibit caspase-dependent apoptosis (47). IM-54 is suggested to directly interact with mitochondrial protein(s), which may therefore play a critical role in induction of necrosis. Some studies have demonstrated that necrosis induced by H2O2 treatment in HL60 cells also is inhibited by IM-54 (47, 49). Together, these data suggest that pep5 can trigger cell death through the oxidative stress pathway.
Other pathways may also be involved in the induction of cell death by pep5. The simultaneous inhibition of necroptosis and caspase activity by a combination of nec-1 and qVD inhibitors efficiently blocked pep5-cpp cell death activity. Neither of these inhibitors individually reduced cell death induced by pep5-cpp. Mixed type cell death modes containing features of both forms of cell death have been reported and named “aponecrosis.” This form of cell death may represent aborted or partially executed apoptotic programs, which occur in the context of a final necrotic outcome (50, 51). The existence of necrotic cell death pathways regulated by an intrinsic death program distinct from that of apoptosis has also been proposed and named necroptosis (52). In apoptosis, caspases 3/7 are effector caspases involved in both the intrinsic and extrinsic pathways. Caspase 8 is involved in the extrinsic apoptotic pathway, and it is activated by ligand binding to membrane-associated death receptors, whereas caspase 9 participates in the intrinsic apoptotic pathway, and it is activated by mitochondrial perturbation, which causes cytochrome c release (53–56).
The phosphorylation levels of only six MAPKs or their substrates were affected by pep5 among the 26 proteins evaluated, including Akt2 inhibition and p38γ and p38α activation. It is known that Akt2-deficient mice have an increase in apoptotic cell death in the aorta, whereas the knockdown of all three isoforms of Akt (Akt1/Akt2/Akt3) induces apoptosis in several human tumor cell lines (57). On the other hand, Akt2 overexpression causes an increase in adhesion, invasion, and metastasis in ovarian and human breast cancer cells (58, 59). Other reports show that Akt2 knockdown in non-small cell lung cancer simultaneously causes cleavage of one antiapoptotic protein, cytochrome c release, and caspase activation (60). In asynchronous HeLa cells, pep5 was not able to induce cytochrome c release (data not shown). pep5 has a similar effect in HeLa cells, inhibiting Akt2 and activating caspase 9. Moreover, mitogen-activated protein kinases such as p38γ and p38α were activated by treatment of HeLa cells with pep5, suggesting that pep5 induces a cross-talk in proapoptotic pathways. Some studies suggest that p38γ is regulated by hypoxia, decreasing the levels of cyclin D1 in PC12 cells, and by ionizing radiation in NIH-3T3 cells, inducing cell cycle arrest (61, 62). Furthermore, when p38α is inhibited, it blocks a checkpoint in G2/M after ultraviolet radiation in murine and human cells (63). Here, when HeLa cells were treated with the specific p38 inhibitor SB203580, no inhibition of pep5-cpp cell death activity was observed. Therefore, the present results do not suggest that Akt2 inhibition or p38γ/p38α activation are key effectors of pep5-cpp cell death activity in HeLa cells. One exciting possibility is that endogenous pep5 alters cell-specific signaling pathways without causing cell death.
pep5-cpp induced cell death in both C6 rat glioma cells and in the rat glioblastoma in vivo. Glioblastoma is the most malignant astrocytoma in humans. Several drugs have been developed to treat gliomas, such as inhibitors of key oncogenic signaling pathways, apoptosis-inducing drugs, and DNA-damaging drugs (64–67). The group of animals treated with pep5-cpp showed a decrease of ∼50% of the tumor volume when compared with the control group. It was interesting to note that pep5-cpp induced an increase in macrophage-like cells in the tumor area, probably due to induction of cell death. These data suggest that pep5-cpp also could be a new pharmacological tool in glioblastoma treatment.
pep5 was shown to inhibit the chymotrypsin activity of the proteasome, suggesting the possibility of an inhibitory feedback mechanism over the proteasome during cyclin D2 degradation. Cell cycle, gene expression, and apoptosis are events regulated by the cleavage of several proteins by the ubiquitin-proteasome system, including transcription factors, cyclins, and cyclin-dependent kinase inhibitors (68, 69). Several transcription factors, such as NF-κB, are involved in the control of the immune response, cell proliferation, and programmed cell death. In the development of malignancy, these proteins can alter the regulation of other factors, contributing to tumorigenesis (70–72). Cyclins are degraded by the proteasome in order to permit the exit of cells from mitosis and their entry into another round of the cell cycle (73). Five main classes of mammalian cyclins have been described. Cyclins C, D (1, 2, and 3), and E act during the G1 phase and regulate the transition from G1 to S phase. In contrast, cyclins A and B (1 and 2) have activity during the S and G2 phases and are regulators of entry into mitosis (74–76). The expression levels of cyclin-dependent kinase inhibitors, including p21, p27, and p57, also are degraded by the ubiquitin-proteasome system, and this control is an important event for cell regulation (77). In tumor cells, there is uncontrolled cell growth and nearly constant proliferation and division, which requires extensive protein degradation. For this reason, proteasome inhibitors have been used as anti-tumor agents, helping to regulate the uncontrolled cell growth and inducing apoptosis in several tumor cells (78–81).
The sequence of pep5 is present only in cyclin D2 and is not found in the other cyclins (Fig. 15). There is total homology of the pep5 sequence within cyclin D2 from a wide range of organisms, including humans, rats, bovines, mice, and chickens, suggesting its biological significance. Cyclin D2 must be degraded by the proteasome during G1/S transition to allow cell cycle progression (82), which suggests that pep5 can be endogenously generated by the proteasome. The proteasome has been shown to regulate the levels of most intracellular peptides (14, 15), and pep5 was seen by bioinformatics analysis to be compatible with generation by the proteasome, followed by aminopeptidase activity (data not shown). Therefore, it is possible that proteasome inhibitors could alter levels of pep5, which could contribute to cell death.
FIGURE 15.

Sequence alignment for cyclins D. Sequences (in FASTA format) or UniProt identifiers of the three human cyclins D (CCND1, CCND2, and CCND3). Note the similarity in amino acids among the D1, D2, and D3 in humans (*). The sequence underlined in green corresponds to pep5 identity, which is only present in cyclin D2.
In the maintenance of mitochondrial protein stability, proteases degrade proteins into peptides that are exported from the mitochondrial matrix into the intermembrane space by ABC transporters and reach the cytosol by passive diffusion. Haynes et al. (83) demonstrated by genetic analysis of Caenorhabditis elegans that under stress of mitochondrial protein misfolding, signal peptides generated by the mitochondrial ATP-dependent proteolytic complex can activate the cell genome, providing a mechanism of intracellular communication among mitochondria, cytosol, and nucleus. As a consequence, the organelle can react against the loss of thermodynamic stability and the propensity of proteins to aggregate, inducing the expression of a nuclear encoded protein gene termed ubiquitin-like ClpXP, which is activated by a homeobox containing the transcription factor bZIP. bZIP is believed to be activated by a pathway that involves different peptides produced by ATP-dependent Clp proteases. The combined expression of these proteins using intracellular signal peptides represents the cell response toward the damage produced by irreversible aggregates and protein misfolding affecting cell functions and survival (83). These findings corroborate previous evidence that endogenous intracellular peptides are probably functional and of biological significance (84).
In summary, these findings suggest that pep5 is a novel bioactive intracellular peptide. There is an exciting possibility that pep5 could serve as a novel prototype molecule in pharmacology and therapeutics.
Supplementary Material
Acknowledgments
We thank Andrea S. Heimann (Proteimax Biotechnology LTDA) for scientific advice and peptide design and supply. We also thank Professor Edna T. Kimura for generously providing NThy-ori 3-1, KTC-2, and TPC-1 cell lines and for assistance with flow cytometry; Adilson Kleber Ferreira, Ph.D., for providing additional tumor cell lines; Marcella Faria, Ph.D., and Professor Hugo A. Armelin for critical discussions and suggestions and for providing the p38 inhibitor; and Joao Gustavo Pessini Amarante Mendes for providing the qVD inhibitor. We also thank Professor Lloyd D. Fricker for outstanding and helpful discussions during manuscript preparation.
This work was supported by Brazilian National Research Council (CNPq) Grant 559698/2009-7 (Rede GENOPROT) and by the Pro-Reitoria de Pesquisa, University of São Paulo, through the Support Center for Research in Proteolysis and Cell Signaling (NAPPS; Grant 2012.1.17607.1.2).

This article contains supplemental data.
- qVD
- quinolyl-Val-Asp-OPh
- TMAB
- 4-trimethylammoniumbutyryl
- cpp
- cell-penetrating peptide
- SCB
- scrambled control peptide.
REFERENCES
- 1. Paz P., Brouwenstijn N., Perry R., Shastri N. (1999) Discrete proteolytic intermediates in the MHC class I antigen processing pathway and MHC I-dependent peptide trimming in the ER. Immunity 11, 241–251 [DOI] [PubMed] [Google Scholar]
- 2. Rock K. L., Goldberg A. L. (1999) Degradation of cell proteins and the generation of MHC class I-presented peptides. Annu. Rev. Immunol. 17, 739–779 [DOI] [PubMed] [Google Scholar]
- 3. Hershko A. (2005) The ubiquitin system for protein degradation and some of its roles in the control of the cell division cycle. Cell Death Differ. 12, 1191–1197 [DOI] [PubMed] [Google Scholar]
- 4. Goldberg A. L. (2012) Development of proteasome inhibitors as research tools and cancer drugs. J. Cell Biol. 199, 583–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Kisselev A. F., Akopian T. N., Goldberg A. L. (1998) Range of sizes of peptide products generated during degradation of different proteins by archaeal proteasomes. J. Biol. Chem. 273, 1982–1989 [DOI] [PubMed] [Google Scholar]
- 6. Kisselev A. F., Akopian T. N., Woo K. M., Goldberg A. L. (1999) The sizes of peptides generated from protein by mammalian 26 and 20 S proteasomes. Implications for understanding the degradative mechanism and antigen presentation. J. Biol. Chem. 274, 3363–3371 [DOI] [PubMed] [Google Scholar]
- 7. Rammensee H. G. (2002) Survival of the fitters. Nature 419, 443–445 [DOI] [PubMed] [Google Scholar]
- 8. Geier E., Pfeifer G., Wilm M., Lucchiari-Hartz M., Baumeister W., Eichmann K., Niedermann G. (1999) A giant protease with potential to substitute for some functions of the proteasome. Science 283, 978–981 [DOI] [PubMed] [Google Scholar]
- 9. Suraweera A., Münch C., Hanssum A., Bertolotti A. (2012) Failure of amino acid homeostasis causes cell death following proteasome inhibition. Mol. Cell 48, 242–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reits E., Neijssen J., Herberts C., Benckhuijsen W., Janssen L., Drijfhout J. W., Neefjes J. (2004) A major role for TPPII in trimming proteasomal degradation products for MHC class I antigen presentation. Immunity 20, 495–506 [DOI] [PubMed] [Google Scholar]
- 11. Reits E., Griekspoor A., Neijssen J., Groothuis T., Jalink K., van Veelen P., Janssen H., Calafat J., Drijfhout J. W., Neefjes J. (2003) Peptide diffusion, protection, and degradation in nuclear and cytoplasmic compartments before antigen presentation by MHC class I. Immunity 18, 97–108 [DOI] [PubMed] [Google Scholar]
- 12. Beninga J., Rock K. L., Goldberg A. L. (1998) Interferon-γ can stimulate post-proteasomal trimming of the N terminus of an antigenic peptide by inducing leucine aminopeptidase. J. Biol. Chem. 273, 18734–18742 [DOI] [PubMed] [Google Scholar]
- 13. Kravtsova-Ivantsiv Y., Cohen S., Ciechanover A. (2009) Modification by single ubiquitin moieties rather than polyubiquitination is sufficient for proteasomal processing of the p105 NF-κB precursor. Mol. Cell 33, 496–504 [DOI] [PubMed] [Google Scholar]
- 14. Gelman J. S., Dasgupta S., Berezniuk I., Fricker L. D. (2013) Analysis of peptides secreted from cultured mouse brain tissue. Biochim. Biophys. Acta 1834, 2408–2417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fricker L. D., Gelman J. S., Castro L. M., Gozzo F. C., Ferro E. S. (2012) Peptidomic analysis of HEK293T cells: effect of the proteasome inhibitor epoxomicin on intracellular peptides. J. Proteome Res. 11, 1981–1990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ferro E. S., Hyslop S., Camargo A. C. (2004) Intracellullar peptides as putative natural regulators of protein interactions. J. Neurochem. 91, 769–777 [DOI] [PubMed] [Google Scholar]
- 17. Burns-Hamuro L. L., Ma Y., Kammerer S., Reineke U., Self C., Cook C., Olson G. L., Cantor C. R., Braun A., Taylor S. S. (2003) Designing isoform-specific peptide disruptors of protein kinase A localization. Proc. Natl. Acad. Sci. U.S.A. 100, 4072–4077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Churchill E. N., Qvit N., Mochly-Rosen D. (2009) Rationally designed peptide regulators of protein kinase C. Trends Endocrinol. Metab. 20, 25–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Rioli V., Gozzo F. C., Heimann A. S., Linardi A., Krieger J. E., Shida C. S., Almeida P. C., Hyslop S., Eberlin M. N., Ferro E. S. (2003) Novel natural peptide substrates for endopeptidase 24.15, neurolysin, and angiotensin-converting enzyme. J. Biol. Chem. 278, 8547–8555 [DOI] [PubMed] [Google Scholar]
- 20. Heimann A. S., Gomes I., Dale C. S., Pagano R. L., Gupta A., de Souza L. L., Luchessi A. D., Castro L. M., Giorgi R., Rioli V., Ferro E. S., Devi L. A. (2007) Hemopressin is an inverse agonist of CB1 cannabinoid receptors. Proc. Natl. Acad. Sci. U.S.A. 104, 20588–20593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dodd G. T., Mancini G., Lutz B., Luckman S. M. (2010) The peptide hemopressin acts through CB1 cannabinoid receptors to reduce food intake in rats and mice. J. Neurosci. 30, 7369–7376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gomes I., Grushko J. S., Golebiewska U., Hoogendoorn S., Gupta A., Heimann A. S., Ferro E. S., Scarlata S., Fricker L. D., Devi L. A. (2009) Novel endogenous peptide agonists of cannabinoid receptors. FASEB J. 23, 3020–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cunha F. M., Berti D. A., Ferreira Z. S., Klitzke C. F., Markus R. P., Ferro E. S. (2008) Intracellular peptides as natural regulators of cell signaling. J. Biol. Chem. 283, 24448–24459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Berti D. A., Morano C., Russo L. C., Castro L. M., Cunha F. M., Zhang X., Sironi J., Klitzke C. F., Ferro E. S., Fricker L. D. (2009) Analysis of intracellular substrates and products of thimet oligopeptidase in human embryonic kidney 293 cells. J. Biol. Chem. 284, 14105–14116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Russo L. C., Castro L. M., Gozzo F. C., Ferro E. S. (2012) Inhibition of thimet oligopeptidase by siRNA alters specific intracellular peptides and potentiates isoproterenol signal transduction. FEBS Lett. 586, 3287–3292 [DOI] [PubMed] [Google Scholar]
- 26. Berti D. A., Russo L. C., Castro L. M., Cruz L., Gozzo F. C., Heimann J. C., Lima F. B., Oliveira A. C., Andreotti S., Prada P. O., Heimann A. S., Ferro E. S. (2012) Identification of intracellular peptides in rat adipose tissue: insights into insulin resistance. Proteomics 12, 2668–2681 [DOI] [PubMed] [Google Scholar]
- 27. Russo L. C., Asega A. F., Castro L. M., Negraes P. D., Cruz L., Gozzo F. C., Ulrich H., Camargo A. C., Rioli V., Ferro E. S. (2012) Natural intracellular peptides can modulate the interactions of mouse brain proteins and thimet oligopeptidase with 14-3-3ϵ and calmodulin. Proteomics 12, 2641–2655 [DOI] [PubMed] [Google Scholar]
- 28. Che F. Y., Biswas R., Fricker L. D. (2005) Relative quantitation of peptides in wild-type and Cpe(fat/fat) mouse pituitary using stable isotopic tags and mass spectrometry. J. Mass Spectrom. 40, 227–237 [DOI] [PubMed] [Google Scholar]
- 29. Morano C., Zhang X., Fricker L. D. (2008) Multiple isotopic labels for quantitative mass spectrometry. Anal. Chem. 80, 9298–9309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhang R., Sioma C. S., Thompson R. A., Xiong L., Regnier F. E. (2002) Controlling deuterium isotope effects in comparative proteomics. Anal. Chem. 74, 3662–3669 [DOI] [PubMed] [Google Scholar]
- 31. Gallo C. J., Koza R. A., Herbst E. J. (1986) Polyamines and HeLa-cell DNA replication. Biochem. J. 238, 37–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zhou J. Y., Ma W. L., Liang S., Zeng Y., Shi R., Yu H. L., Xiao W. W., Zheng W. L. (2009) Analysis of microRNA expression profiles during the cell cycle in synchronized HeLa cells. BMB Rep. 42, 593–598 [DOI] [PubMed] [Google Scholar]
- 33. Benda P., Lightbody J., Sato G., Levine L., Sweet W. (1968) Differentiated rat glial cell strain in tissue culture. Science 161, 370–371 [DOI] [PubMed] [Google Scholar]
- 34. Udenfriend S., Stein S., Böhlen P., Dairman W., Leimgruber W., Weigele M. (1972) Fluorescamine: a reagent for assay of amino acids, peptides, proteins, and primary amines in the picomole range. Science 178, 871–872 [DOI] [PubMed] [Google Scholar]
- 35. Castro L. M., Berti D. A., Russo L. C., Coelho V., Gozzo F. C., Oliveira V., Ferro E. S. (2010) Similar intracellular peptide profile of TAP1/β2 microglobulin double-knockout mice and C57BL/6 wild-type mice as revealed by peptidomic analysis. AAPS J. 12, 608–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Che F. Y., Zhang X., Berezniuk I., Callaway M., Lim J., Fricker L. D. (2007) Optimization of neuropeptide extraction from the mouse hypothalamus. J. Proteome Res. 6, 4667–4676 [DOI] [PubMed] [Google Scholar]
- 37. Wardman J., Fricker L. D. (2011) Quantitative peptidomics of mice lacking peptide-processing enzymes. Methods Mol. Biol. 768, 307–323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wang G., Sun W., Luo Y., Fang N. (2010) Resolving rotational motions of nano-objects in engineered environments and live cells with gold nanorods and differential interference contrast microscopy. J. Am. Chem. Soc. 132, 16417–16422 [DOI] [PubMed] [Google Scholar]
- 39. Ziegler A., Nervi P., Dürrenberger M., Seelig J. (2005) The cationic cell-penetrating peptide CPP(TAT) derived from the HIV-1 protein TAT is rapidly transported into living fibroblasts: optical, biophysical, and metabolic evidence. Biochemistry 44, 138–148 [DOI] [PubMed] [Google Scholar]
- 40. Wender P. A., Mitchell D. J., Pattabiraman K., Pelkey E. T., Steinman L., Rothbard J. B. (2000) The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: peptoid molecular transporters. Proc. Natl. Acad. Sci. U.S.A. 97, 13003–13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bradford M. M. (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 72, 248–254 [DOI] [PubMed] [Google Scholar]
- 42. Zarubin T., Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18 [DOI] [PubMed] [Google Scholar]
- 43. Degterev A., Huang Z., Boyce M., Li Y., Jagtap P., Mizushima N., Cuny G. D., Mitchison T. J., Moskowitz M. A., Yuan J. (2005) Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat. Chem. Biol. 1, 112–119 [DOI] [PubMed] [Google Scholar]
- 44. Caserta T. M., Smith A. N., Gultice A. D., Reedy M. A., Brown T. L. (2003) Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Apoptosis 8, 345–352 [DOI] [PubMed] [Google Scholar]
- 45. Cuenda A., Rouse J., Doza Y. N., Meier R., Cohen P., Gallagher T. F., Young P. R., Lee J. C. (1995) SB 203580 is a specific inhibitor of a MAP kinase homologue which is stimulated by cellular stresses and interleukin-1. FEBS Lett. 364, 229–233 [DOI] [PubMed] [Google Scholar]
- 46. Lali F. V., Hunt A. E., Turner S. J., Foxwell B. M. (2000) The pyridinyl imidazole inhibitor SB203580 blocks phosphoinositide-dependent protein kinase activity, protein kinase B phosphorylation, and retinoblastoma hyperphosphorylation in interleukin-2-stimulated T cells independently of p38 mitogen-activated protein kinase. J. Biol. Chem. 275, 7395–7402 [DOI] [PubMed] [Google Scholar]
- 47. Sodeoka M., Dodo K. (2010) Development of selective inhibitors of necrosis. Chem. Rec. 10, 308–314 [DOI] [PubMed] [Google Scholar]
- 48. Gelman J. S., Sironi J., Berezniuk I., Dasgupta S., Castro L. M., Gozzo F. C., Ferro E. S., Fricker L. D. (2013) Alterations of the intracellular peptidome in response to the proteasome inhibitor bortezomib. PLoS One 8, e53263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dodo K., Katoh M., Shimizu T., Takahashi M., Sodeoka M. (2005) Inhibition of hydrogen peroxide-induced necrotic cell death with 3-amino-2-indolylmaleimide derivatives. Bioorg. Med. Chem. Lett. 15, 3114–3118 [DOI] [PubMed] [Google Scholar]
- 50. Wang X., Ryter S. W., Dai C., Tang Z. L., Watkins S. C., Yin X. M., Song R., Choi A. M. (2003) Necrotic cell death in response to oxidant stress involves the activation of the apoptogenic caspase-8/bid pathway. J. Biol. Chem. 278, 29184–29191 [DOI] [PubMed] [Google Scholar]
- 51. Ryter S. W., Kim H. P., Hoetzel A., Park J. W., Nakahira K., Wang X., Choi A. M. (2007) Mechanisms of cell death in oxidative stress. Antioxid. Redox Signal. 9, 49–89 [DOI] [PubMed] [Google Scholar]
- 52. Kitanaka C., Kuchino Y. (1999) Caspase-independent programmed cell death with necrotic morphology. Cell Death Differ. 6, 508–515 [DOI] [PubMed] [Google Scholar]
- 53. Hengartner M. O. (2000) The biochemistry of apoptosis. Nature 407, 770–776 [DOI] [PubMed] [Google Scholar]
- 54. Jin Z., El-Deiry W. S. (2005) Overview of cell death signaling pathways. Cancer Biol. Ther. 4, 139–163 [DOI] [PubMed] [Google Scholar]
- 55. Peter M. E., Krammer P. H. (2003) The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ. 10, 26–35 [DOI] [PubMed] [Google Scholar]
- 56. Li P., Nijhawan D., Budihardjo I., Srinivasula S. M., Ahmad M., Alnemri E. S., Wang X. (1997) Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade. Cell 91, 479–489 [DOI] [PubMed] [Google Scholar]
- 57. Koseoglu S., Lu Z., Kumar C., Kirschmeier P., Zou J. (2007) AKT1, AKT2 and AKT3-dependent cell survival is cell line-specific and knockdown of all three isoforms selectively induces apoptosis in 20 human tumor cell lines. Cancer Biol. Ther. 6, 755–762 [DOI] [PubMed] [Google Scholar]
- 58. Shen Y. H., Zhang L., Ren P., Nguyen M. T., Zou S., Wu D., Wang X. L., Coselli J. S., LeMaire S. A. (2013) AKT2 confers protection against aortic aneurysms and dissections. Circ. Res. 112, 618–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Arboleda M. J., Lyons J. F., Kabbinavar F. F., Bray M. R., Snow B. E., Ayala R., Danino M., Karlan B. Y., Slamon D. J. (2003) Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of β1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 63, 196–206 [PubMed] [Google Scholar]
- 60. Lee M. W., Kim D. S., Lee J. H., Lee B. S., Lee S. H., Jung H. L., Sung K. W., Kim H. T., Yoo K. H., Koo H. H. (2011) Roles of AKT1 and AKT2 in non-small cell lung cancer cell survival, growth, and migration. Cancer Sci. 102, 1822–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Conrad P. W., Freeman T. L., Beitner-Johnson D., Millhorn D. E. (1999) EPAS1 trans-activation during hypoxia requires p42/p44 MAPK. J. Biol. Chem. 274, 33709–33713 [DOI] [PubMed] [Google Scholar]
- 62. Wang X., McGowan C. H., Zhao M., He L., Downey J. S., Fearns C., Wang Y., Huang S., Han J. (2000) Involvement of the MKK6-p38γ cascade in γ-radiation-induced cell cycle arrest. Mol. Cell Biol. 20, 4543–4552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bulavin D. V., Higashimoto Y., Popoff I. J., Gaarde W. A., Basrur V., Potapova O., Appella E., Fornace A. J., Jr. (2001) Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature 411, 102–107 [DOI] [PubMed] [Google Scholar]
- 64. Ferguson S., Lesniak M. S. (2007) Convection enhanced drug delivery of novel therapeutic agents to malignant brain tumors. Curr. Drug Deliv. 4, 169–180 [DOI] [PubMed] [Google Scholar]
- 65. Wen P. Y., Kesari S., Drappatz J. (2006) Malignant gliomas: strategies to increase the effectiveness of targeted molecular treatment. Expert Rev. Anticancer Ther. 6, 733–754 [DOI] [PubMed] [Google Scholar]
- 66. Jiang H., Alonso M. M., Gomez-Manzano C., Piao Y., Fueyo J. (2006) Oncolytic viruses and DNA-repair machinery: overcoming chemoresistance of gliomas. Expert Rev. Anticancer Ther. 6, 1585–1592 [DOI] [PubMed] [Google Scholar]
- 67. Kim L., Glantz M. (2006) Chemotherapeutic options for primary brain tumors. Curr. Treat. Options Oncol. 7, 467–478 [DOI] [PubMed] [Google Scholar]
- 68. Hershko A. (1997) Roles of ubiquitin-mediated proteolysis in cell cycle control. Curr. Opin. Cell Biol. 9, 788–799 [DOI] [PubMed] [Google Scholar]
- 69. Chun K. T., Mathias N., Goebl M. G. (1996) Ubiquitin-dependent proteolysis and cell cycle control in yeast. Prog. Cell Cycle Res. 2, 115–127 [DOI] [PubMed] [Google Scholar]
- 70. Murray R. Z., Norbury C. (2000) Proteasome inhibitors as anti-cancer agents. Anticancer Drugs 11, 407–417 [DOI] [PubMed] [Google Scholar]
- 71. Spataro V., Norbury C., Harris A. L. (1998) The ubiquitin-proteasome pathway in cancer. Br. J. Cancer 77, 448–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Karin M., Cao Y., Greten F. R., Li Z. W. (2002) NF-kappaB in cancer: from innocent bystander to major culprit. Nat. Rev. Cancer 2, 301–310 [DOI] [PubMed] [Google Scholar]
- 73. Glotzer M., Murray A. W., Kirschner M. W. (1991) Cyclin is degraded by the ubiquitin pathway. Nature 349, 132–138 [DOI] [PubMed] [Google Scholar]
- 74. Sherr C. J. (1994) G1 phase progression: cycling on cue. Cell 79, 551–555 [DOI] [PubMed] [Google Scholar]
- 75. King R. W., Jackson P. K., Kirschner M. W. (1994) Mitosis in transition. Cell 79, 563–571 [DOI] [PubMed] [Google Scholar]
- 76. Nurse P. (1994) Ordering S phase and M phase in the cell cycle. Cell 79, 547–550 [DOI] [PubMed] [Google Scholar]
- 77. Nakayama K. (1998) Cip/Kip cyclin-dependent kinase inhibitors: brakes of the cell cycle engine during development. BioEssays 20, 1020–1029 [DOI] [PubMed] [Google Scholar]
- 78. Imajoh-Ohmi S., Kawaguchi T., Sugiyama S., Tanaka K., Omura S., Kikuchi H. (1995) Lactacystin, a specific inhibitor of the proteasome, induces apoptosis in human monoblast U937 cells. Biochem. Biophys. Res. Commun. 217, 1070–1077 [DOI] [PubMed] [Google Scholar]
- 79. Adams J., Palombella V. J., Sausville E. A., Johnson J., Destree A., Lazarus D. D., Maas J., Pien C. S., Prakash S., Elliott P. J. (1999) Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 59, 2615–2622 [PubMed] [Google Scholar]
- 80. Drexler H. C. (1997) Activation of the cell death program by inhibition of proteasome function. Proc. Natl. Acad. Sci. U.S.A. 94, 855–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Sunwoo J. B., Chen Z., Dong G., Yeh N., Crowl Bancroft C., Sausville E., Adams J., Elliott P., Van Waes C. (2001) Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-κB, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin. Cancer Res. 7, 1419–1428 [PubMed] [Google Scholar]
- 82. Chen B. B., Glasser J. R., Coon T. A., Zou C., Miller H. L., Fenton M., McDyer J. F., Boyiadzis M., Mallampalli R. K. (2012) F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood 119, 3132–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Haynes C. M., Yang Y., Blais S. P., Neubert T. A., Ron D. (2010) The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol. Cell 37, 529–540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ferro E. S., Rioli V., Castro L. M., Fricker L. D. (2014) Intracellular peptides: from discovery to function. EuPA Open Proteomics 3, 143–151 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.