

Abstract

RNA polymerase I (Pol I) is a dedicated polymerase that transcribes the 45S ribosomal (r) RNA precursor. The 45S rRNA precursor is subsequently processed into the mature 5.8S, 18S, and 28S rRNAs and assembled into ribosomes in the nucleolus. Pol I activity is commonly deregulated in human cancers. On the basis of the discovery of lead molecule BMH-21, a series of pyridoquinazolinecarboxamides have been evaluated as inhibitors of Pol I and activators of the destruction of RPA194, the Pol I large catalytic subunit protein. Structure–activity relationships in assays of nucleolar stress and cell viability demonstrate key pharmacophores and their physicochemical properties required for potent activation of Pol I stress and cytotoxicity. This work identifies a set of bioactive compounds that potently cause RPA194 degradation that function in a tightly constrained chemical space. This work has yielded novel derivatives that contribute to the development of Pol I inhibitory cancer therapeutic strategies.
Introduction
Ribosomal (r) DNA is the most highly transcribed genomic region and occurs in a dedicated subcellular compartment, the nucleolus.1,2 Transcription of rRNA is mediated by RNA polymerase I (Pol I) that transcribes the multicopy rDNA gene to a long 45S rRNA precursor.3 The 45S rRNA precursor is processed through multiple steps to the 18S, 5.8S, and 28S mature rRNAs requisite for the assembly of the ribosomes. Pol I transcription is initiated by binding of a multisubunit preinitiation complex to rDNA promoter, which stochastically recruits the Pol I holocomplex.4 The Pol I holocomplex is composed of 14 subunits in eukaryotes, of which the subunits RPA194, RPA135, and RPA12 form the catalytically active site. Destabilization of the rDNA helix, or loss of the protein framework, will effectively stall transcription.5
The rate of rRNA transcription is tightly controlled by external signaling pathways that cause the assembly and binding of the preinitiation complex. Deregulation of rRNA synthesis is highly frequent in human cancers.6−8 This is due to activation of extracellular and intracellular signaling pathways and oncogenes such as Myc, Neu, Akt/PKB, and mTOR that promote the preinitiation complex assembly and hence increase the rate of rRNA transcription. Conversely, loss-of-function of tumor suppressors p53, pRB, ARF, and PTEN leads to activation of Pol I transcription.7 Cancer cells have a high degree of dependency on protein synthesis in general due to their increased needs for proteins requisite for their high proliferation rates and to compensate for their proteotoxic environment, misfolding, and errors in protein synthesis.9 These presumably create a setting in which cancer cells acquire dependency on increased rRNA synthetic rates, which are supported by the convergence of cancer cell deregulated pathways. Therefore, inhibitors of Pol I transcription may provide novel approaches toward cancer therapies.
Despite the key impact of Pol I contributing to cancer cell characteristics, its therapeutic exploitation has been minimal. Compound 1 (CX-5461) is a recently described small molecule that inhibits Pol I preinitation complex (Figure 1).10−12 We have recently presented the discovery of an anticancer small molecule, 12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide, N-[2(dimethylamino)ethyl]-12-oxo (compound 2, BMH-21) (Figure 1).13 It has a distinct mode of inhibition of Pol I compared to compound 1, an inhibitor of Pol I preinitiation complex assembly.10 Our studies demonstrated that compound 2 intercalates with GC-rich rDNA, inhibits Pol I, and causes proteasome-mediated degradation of RPA194.14 Compound 2 also showed broad and potent anticancer activity in NCI60 cancer cell lines and reduced tumor burden in mouse xenograft assays. These studies have provided proof-of-principle confirmation that Pol I targeting is a feasible approach for cancer control.12,14
Figure 1.

RNA Pol inhibitors 1 and 2 and the inactive analogue 3.
Compound 2 was identified by screening commercially available libraries, with few readily available analogues. The goal of this study was to assess its structure–activity relationships (SARs) with respect to Pol I transcription stress and RPA194 degradation and their association with cytotoxicity.
Results
Chemistry. Synthesis of Pyridoquinazolinecarboxamide Derivatives
Compound 2 inhibits Pol I transcription and causes nucleolar stress as measured by nucleolar protein relocalization and expression at submicromolar concentrations.14 A structurally similar small molecule, BMH-21a1 (3) (Figure 1), that lacks an amino group in the N,N-dimethylamino carboxamide arm was devoid of all activities.14 This was indicative that the N,N-dimethylamino carboxamide arm was relevant for the biological activity of compound 2. To test this hypothesis and to explore other active moieties, we conducted SAR analysis. For this purpose, synthesis of the desired compounds was achieved as shown in Scheme 1, where the acid 6, synthesized from compounds 4 and 5, was coupled to a variety of amines to give the desired products with generic structure 7 (Scheme 1) and as detailed in the Experimental Section.
Scheme 1. General Scheme for the Synthesis To Provide Generic Structure 7.

Evaluation of RPA194 Degradation and Nucleolar Stress in U2OS Cancer Cell Line
We have shown that the Pol I inhibitory activity of compound 2 is reflected as nucleolar stress, that is, translocation of nucleolar marker proteins, such as nucleolin (NCL) to the nucleoplasm, and degradation of RPA194, the large catalytic subunit of Pol I.14 We hence used these as readouts to assess the biological activity of the derivatives. For this purpose U2OS osteosarcoma cells were seeded on glass coverslips incubated with 2 log titration of the compounds for 3 h, fixed, and immunostained for RPA194 and NCL. As shown in representative images in Figure 2A, RPA194 expression was affected to a variable degree by the compounds. At 0.5 μM concentration shown, compounds 8, 9, 10, 11, 12, and 13 led to an almost complete loss of expression of RPA194, similar to compound 2, while compounds 14, 15, and 36 had no effect. Similarly, derivatives 8, 9, 10, 11, 12, and 13 decreased the nucleolar localization of NCL and led to its translocation to the nucleoplasm (Figure 2B). Quantitative image analysis was conducted for expression of RPA194 and NCL over the full concentration range, and IC50 was determined (Figure 3 and Tables 1–4). Compound 2 IC50 values for degradation of RPA194 and translocation of NCL were very similar (0.05 ± 0.04 μM and 0.07 ± 0.06 μM, respectively) and showed its submicromolar activity. Quantitative image analysis for these RPA194 and NCL phenotypes was then employed to assess the bioactivity of the compounds.
Figure 2.

Effect of compounds on expression and localization of RPA194 and NCL. Immunofluorescence staining of U2OS cells treated with the indicated compounds (0.5 μM) for 3 h. Cells were stained for (A) RPA194 (red) and (B) NCL (green) and counterstained for DNA (blue). Scale bars, 10 μm.
Figure 3.

Quantitative image analysis of expression and localization of RPA194 and NCL by the derivatives. U2OS cells were treated with the compounds at 0, 0.1, 0.5, 1, and 5 μM and incubated for 3 h. Cells were fixed and stained for (A) RPA194 and (B) NCL and counterstained for DNA and imaged using epifluorescence. Quantitative image analysis for RPA194 degradation (A) and loss of NCL nucleolar intensity was conducted based on two biological replicates, and the fold change to control is shown. Error bars represent SEM.
Table 1. Compound 2 Core Modifications.

cLogD calculated at pH 7.4 by ACD Labs Percepta.
pKa calculated by ACD Labs Percepta.
RPA IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments except when noted with ∗.
NCL IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments except when noted with ∗.
Table 4. Compound 2 Two-Carbon Linker Variations.
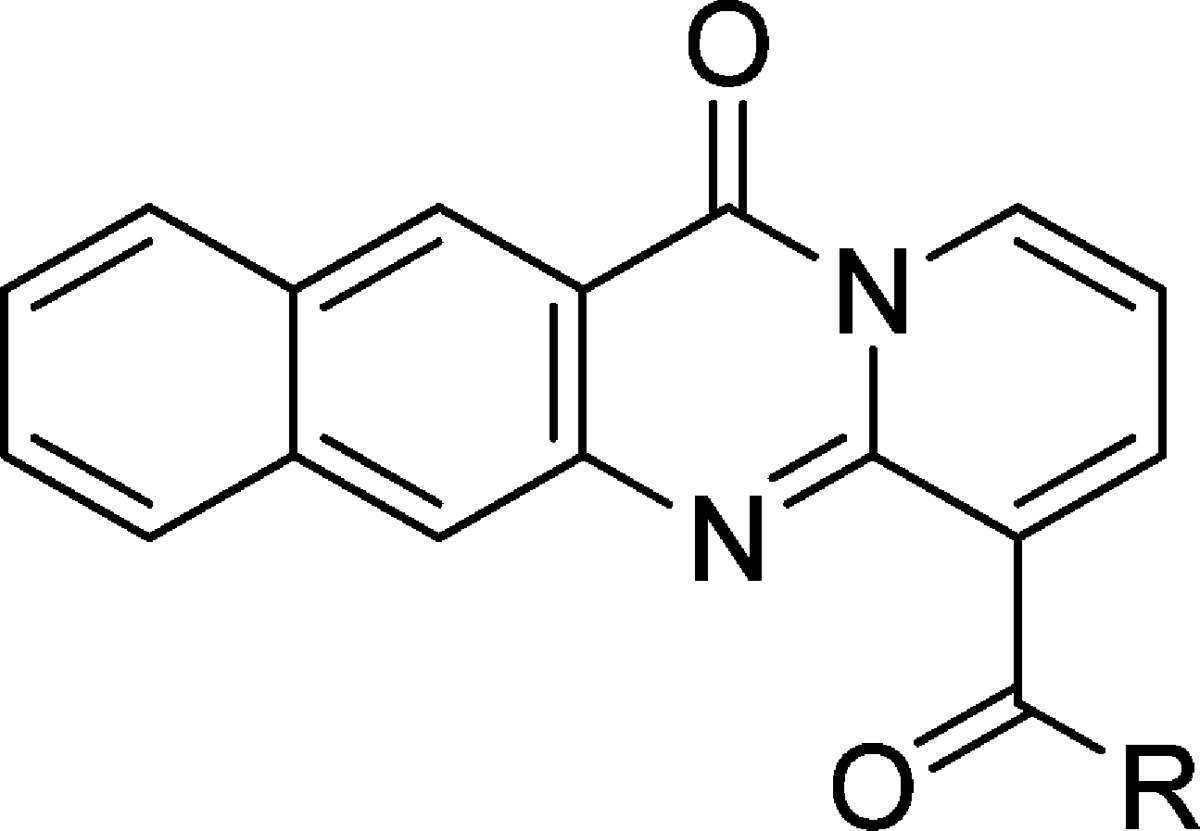
cLogD calculated at pH 7.4 by ACD Labs Percepta.
pKa calculated by ACD Labs Percepta.
RPA IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments except when noted with ∗.
NCL IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments except when noted with ∗.
Human liver microsome T1/2 in minutes.
SAR Analysis
As shown in Table 1, significant changes to the structure of compound 2 were poorly tolerated. As expected, truncation of the intercalator portion of the molecules (compounds 15 and 16) reduced the activity by over 40-fold by reducing π-stacking contacts with the DNA bases. Therefore, attention was focused on the amide side chain. Conversion of the amide to an ester resulted in a significant loss in potency of compound 17 as did alkylation of the amide NH (compounds 18 and 19), which rendered the molecule either inactive or caused >400-fold decrease in potency. Replacement of the pendent basic amine with less basic heterocycles such as imidazole (compound 20) and pyridine (compounds 21 and 22) was also not tolerated. Other heterocycles and nonbasic functional groups (compounds 23, 24, 25) were also not tolerated. All these led to >200-fold decrease in potency or rendered the molecule inactive.
The SAR based on the early analogue, compound 3, suggested that a basic amine was critical and that only conservative changes to compound 2 were to be tolerated. Compounds 14, 26, 27, and 28 in Table 2 show the effect of changing the distance of the basic amine from the tetracycle (IC50 of 0.66, 0.43, 2.01, and 33.85 μM for the RPA194 assay, respectively), and once again, the two-carbon linker present in compound 2 appeared optimal.
Table 2. Compound 2 Other Linkers.
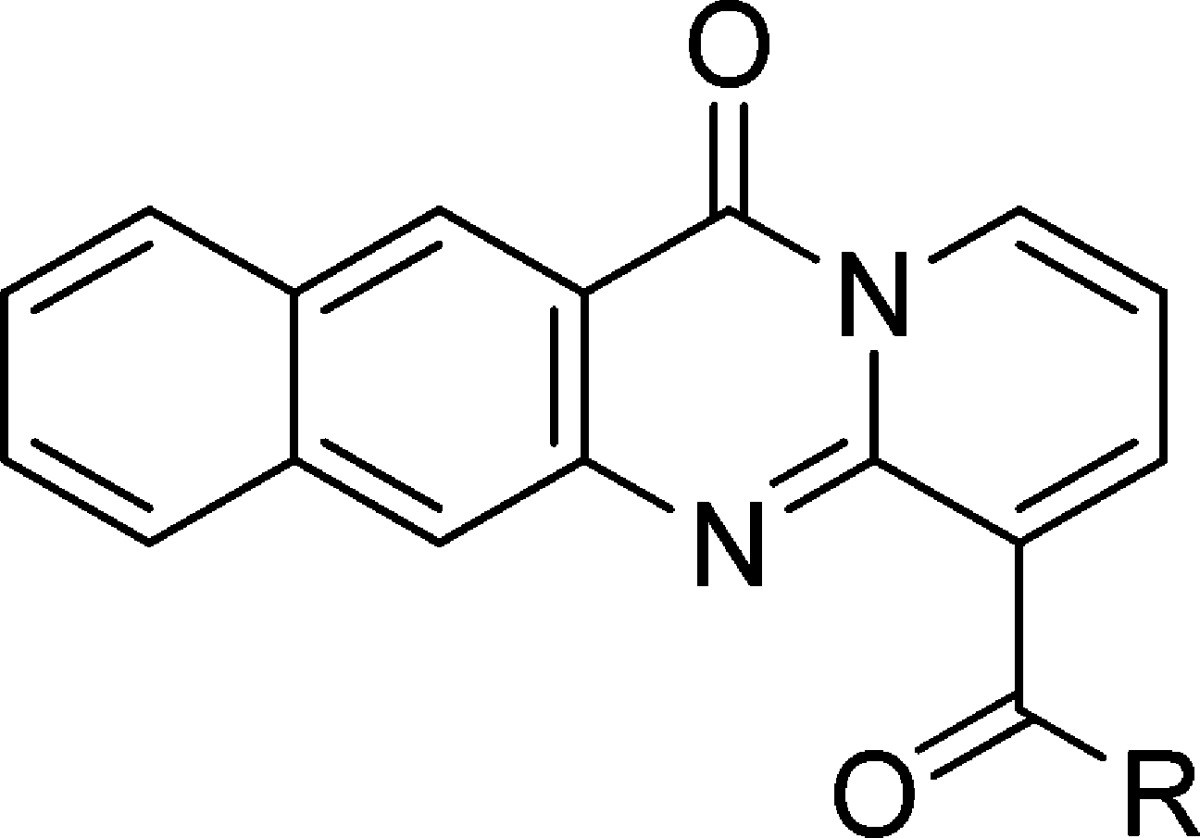
cLogD calculated at pH 7.4 by ACD Labs Percepta.
pKa calculated by ACD Labs Percepta.
RPA IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments.
NCL IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments.
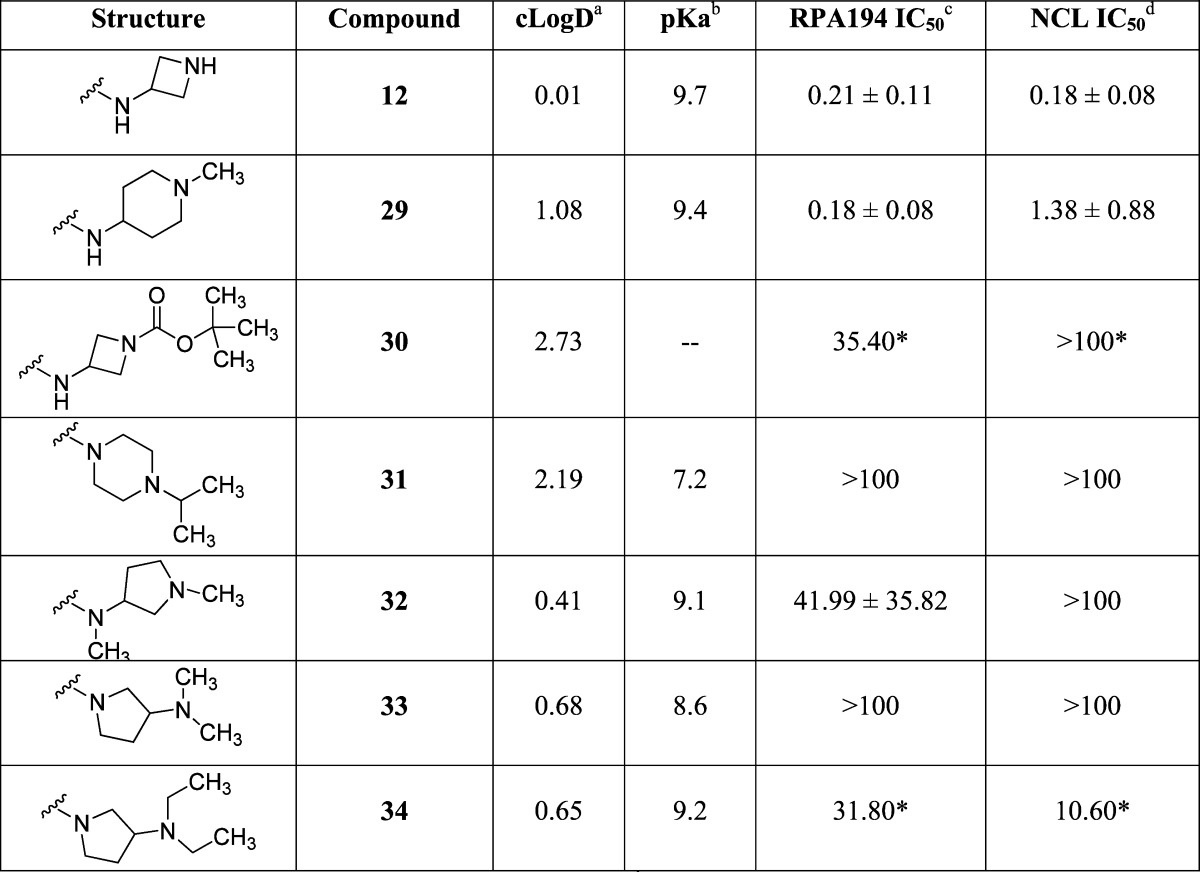
Several cyclic constraints to the two-carbon linker were explored as shown in Table 3, and compounds 12 and 29 retained most of the activity (IC50 of 0.21 and 0.18 μM, respectively), but no constraint was found that improved potency. Compounds 30, 31, 32, 33, 34 were inactive. More subtle changes to the two-carbon linker were explored as shown in Table 4. Wrapping the terminal methyl groups of compound 2 into rings or to larger ethyl group provided compounds with similar potency (compounds 8, 9, and 10 IC50 of 0.18, 0.09, and 0.14 μM, respectively), compared to isopropyl (compound 35 IC50 of 3.04 μM) alkyl chains, which was poorly tolerated. Conversion of the piperidine in compound 8 to a morpholine (compound 36 IC50 of 1.25 μM) or piperazine (compound 37 IC50 of 1.09 μM) reduced potency, as did appending a methoxyethyl (compound 38 IC50 of 0.73 μM). Addition of methyl groups into the linker (compounds 11 and 13, IC50 of 0.11 and 0.04 μM) was tolerated with compound 13 representing the most potent variant prepared. In general, there was good agreement between the RPA194 and NCL assays, with the NCL assay showing slightly more sensitivity toward changes in the structure.
Table 3. Compound 2 Linker Constraints.

cLogD calculated at pH 7.4 by ACD Labs Percepta.
pKa calculated by ACD Labs Percepta.
RPA IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments except when noted with ∗.
NCL IC50 represents mean ± SEM (μM); assays represent duplicate independent biological experiments except when noted with ∗.
The changes in RPA194 expression observed by immunofluorescence were confirmed for representative derivatives by Western blotting analysis of cell lysates (Figure 4). Similar to compound 2, compounds 8 and 11 caused degradation of RPA194, whereas other tested compounds were less potent in doing so (compounds 14, 15, 36) or were inactive (compound 18). The translocation of NCL to the nucleoplasm by compound 2 does not affect the expression of the protein.14 Accordingly, the expression of NCL was largely unaffected except at very high (10 μM) concentrations of compound 2 and other active derivatives (Figure 4).
Figure 4.

Protein expression analyses for RPA194 and NCL. U2OS cells were treated with the compounds at 0, 0.1, 1, and 10 μM and incubated for 3 h. Protein was extracted using RIPA lysis buffer, and Western blotting was conducted for RPA194, NCL, and GAPDH as control.
Derivatives That Cause Nucleolar Stress Decrease U2OS Cancer Cell Viability
Selected derivatives were then tested for their effect on viability of U2OS cells. Cells were cultured for 48 h in the presence of the compounds, and viability was determined using WST1 assay. As shown in Figure 5, derivatives that elicited potent degradation of RPA194 (8, 9, 13) caused profound loss of cell viability similar to compound 2, whereas compounds 14, 15, 18, and 36 had little or no activity. This suggested that loss of viability was coupled with loss of RPA194 and nucleolar stress as demonstrated for compound 2.14
Figure 5.

Cell viability assay. U2OS cells were treated with the compounds at 0, 0.5, and 5 μM and incubated for 48 h. Cell viability was determined using WST-1 assay. N = 2 biological repeats. Error bars represent SEM.
Physicochemical Characterization
The derivatives were analyzed with respect to their physicochemical properties using ACD Labs Percepta prediction software. The pKa of the derivatives varied between 5.3 and 9.96. All active derivatives were highly basic and had pKa values between 8.6 and 9.7 and were closely comparable to that of the parent compound with predicted pKa at 8.6 (Tables 1–4). cLogD estimates at pH 7.4 showed variation between −0.15 and 3.7 where the active derivatives possessed values between 0.59 and 1.59 (Tables 1–4). Finally, given that the previously described inactive analogue, compound 3, was distinguished by lack of the terminal amine and the prediction that this nitrogen is protonated (ref (14)), we assessed the predicted protonation of the derivatives at pH 7.4. All derivatives with predicted lack or low degree of protonation, based on pKa values and predictions using ChemAxon Marvin analysis program (not shown), were biologically inactive (Tables 1–4). We conclude that the main physicochemical determinants for biologically active derivatives included the protonation of the terminal amine and a closely defined basic pKa resembling that of the parent.
Metabolism Studies
Compound 2 has a remarkably good PK profile with good exposure and a half-life of 2.5 h after 25 mg/kg ip dose.14 By use of an in vitro human liver microsomal stability assay, compound 2 had an in vitro half-life of 57 min. Several of the active analogues prepared were tested in this paradigm (Table 4), and despite modification of the terminal methyl metabolic soft spots, none of the analogues tested significantly improved the in vitro stability. While identification of the major metabolites of compound 2 showed N-dealkylation as one of several major metabolites, modifications that would be predicted to slow dealkylation (compounds 8, 9, and 10) did not improve stability, despite a range of calculated cLogD values (Table 4).
Discussion and Conclusion
A series of compound 2 variants were prepared and evaluated as potential novel anticancer agents that act via the repression of Pol I activity. The activity of compound 2 is due to its ability to intercalate to GC-rich rDNA sequences,14 which makes it very different from other four-ring anthracyclines that tend to cause DNA damage.15 Their intercalation modalities are also quite distinct from the anthracyclines intercalating perpendicular to the DNA helix, whereas compound 2 intercalates in a near-parallel fashion.14 While our previous modeling has suggested some molecular determinants for this activity, the high sensitivity of the pendent compound 2 chain as exemplified in Table 4 suggests that there may be other components to the 2–DNA complex that lead to its biological activity. Notably, all near equipotent derivatives retained a predicted protonation of the terminal amine and had a basic pKa close to that of the parent at 8.6. These findings indicated that the overall charge of the molecule was critical as well as maintaining the length and basic charge close to the end of the carboxamide arm. Considering molecular modeling of compound 2 in ref (14), these findings suggest that compound 2 intercalates with acidic DNA complemented with additional electrostatic interactions. It also raises the possibility that derivatives with more highly charged moieties may change the nature of the intercalation or that those with larger molecular sizes alter the DNA intercalation cavity. This further implies that such molecules could perturb other DNA metabolic processes.
The key marks used here to define compound 2 activity were based on biological assays measuring the integrity of the nucleolus, expression and localization of nucleolar proteins, and cell viability. Nucleolar stress, characterized by nucleolar protein relocalization, such as NCL and loss of integrity of the nucleolus is a hallmark of Pol I transcription stress and its inhibition5 and is rapidly and profoundly activated by compound 2. Degradation of RPA194, the large catalytic subunit of Pol I, is a distinctive and unique response to compound 2.14 Hence, these markers function as definitive and sensitive cellular indicators for compound 2 activity. As shown here, the parent molecule activated RPA194 degradation and NCL translocation at IC50 of 50–70 nM. Assays for degradation of RPA194 and NCL translocation by the derivatives were highly consistent with each other, indicating their usefulness in further cell-based screens for Pol I transcription inhibitors.
As determination of the derivative activities were based on cellular assays, the changes in potency could reflect their permeability. Large changes in potency of the derivatives were observed across a wide range of physiochemical properties, as illustrated by their cLogD values shown in the Tables 1–4. The compound cLogD ranged between −0.15 and 3.7, and very potent compounds existed at either end (compound 12 cLogD of 0.01, compound 8 cLogD of 1.53), suggesting that permeability alone did not determine the phenotypes.
The structure–activity relationships described herein have defined key structural elements in the compound 2 structure such as the four-ring tetracycle, secondary amide bond and two-carbon link to a basic amine, and charge. Variations around this theme have led led to very potent novel analogues such as compounds 8, 9, 12 and 13, which retain the desirable potency and DMPK properties compared to compound 2. These compounds share close potency for both RPA194 degradation and NCL localization change as reflection of Pol I transcription stress. Given that Pol I transcription is an emerging therapeutic target, development of such bioactive compounds holds substantial promise.
Experimental Section
Synthesis
General Methods
All commercially available reagents and solvents were used without further purification unless otherwise stated. Automated flash chromatography was performed on an ISCO CombiFlash Rf or Biotage Isolera using Biotage Flash cartridges with peak detection at 254 nm. Reverse phase purification was accomplished using a Gilson 215 liquid handler equipped with a Phenomenex C18 column (150 mm × 20 mm i.d., 5 μm). Peak collection was triggered by UV detection at 214 or 254 nm. 1H NMR spectra were recorded on a Bruker 400 instrument operating at 400 MHz with tetramethylsilane or residual protonated solvent used as a reference. Analytical LC/MS was performed using Agilent 1260 equipped with autosampler (Agilent Poroshell 120 C18 column (50 mm × 4.6 mm i.d., 3.5 μm); 0.1% TFA in water/acetonitrile gradient; UV detection at 215 and 254 nm) and electrospray ionization. All final compounds showed purity greater than 95% at 215 and 254 nm using this method.
12-Oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxylic Acid (6a)
A mixture of 3-amino-2-naphthoic acid (5) (2.00 g, 10.68 mmol), 2-chloronicotinic acid (4) (1.68 g, 10.68 mmol), and hydrochloric acid (0.9 mL, 29.13 mmol) in ethanol (70 mL) was stirred at 80 °C for 66 h (for convenience). After cooling, the reddish-orange suspension was filtered, washed with ethanol, and air-dried to give 12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxylic acid (1.56 g, 5.37 mmol, 50.3% yield) as a yellow-orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.16 (s, 1 H), 9.03 (dd, J = 7.20, 1.64 Hz, 1 H), 8.63 (dd, J = 6.95, 1.64 Hz, 1 H), 8.49 (s, 1 H), 8.34 (d, J = 8.34 Hz, 1 H), 8.19 (d, J = 8.08 Hz, 1 H), 7.76 (t, J = 7.07 Hz, 1 H), 7.64 (t, J = 6.95 Hz, 1 H), 7.18 (t, J = 7.07 Hz, 1 H). MS [M + 1] = 291.
11-Oxopyrido[2,1-b]quinazoline-6-carboxylic Acid (6b)
A mixture of 2-aminobenzoic acid (250 mg, 1.82 mmol), 2-chloronicotinic acid (287.2 mg, 1.82 mmol), and hydrochloric acid (0.3 mL, 9.85 mmol) in ethanol (20 mL) was stirred at 80 °C for 48 h (for convenience). After cooling, the reddish-orange suspension was filtered, washed with ethanol, and air-dried to give 11-oxopyrido[2,1-b]quinazoline-6-carboxylic acid (107 mg, 0.45 mmol, 24.5% yield) as a pale yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.50 (dd, J = 7.07, 1.52 Hz, 3 H), 9.18 (dd, J = 7.45, 1.64 Hz, 3 H), 8.56 (dd, J = 8.21, 1.39 Hz, 3 H), 8.17 (ddd, J = 8.46, 7.20, 1.52 Hz, 3 H), 8.03 (s, 2 H), 8.01 (s, 1 H), 7.80 (ddd, J = 8.15, 7.26, 1.01 Hz, 4 H), 7.72 (t, J = 7.20 Hz, 3 H). MS [M + 1] = 241.
Method A: Synthesis of Amide Analogues (7). N-[2-(Dimethylamino)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (2)
To a solution of 12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxylic acid (50.mg, 0.17 mmol) and TBTU (82.9 mg, 0.26 mmol) in DMF (1 mL) was added DIPEA (90 μL, 0.52 mmol). After the contents were stirred at room temperature for 15 min, N,N-dimethylethylenediamine (28.4 μL, 0.26 mmol) was added and stirring continued for 16 h (for convenience). The reaction mixture was added to 100 mL of cold water with stirring. The solid was collected by filtration and dried under vacuum to give N-[2-(dimethylamino)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (36 mg, 0.10 mmol, 58.0 % yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.50 (br s, 1 H), 9.10 (s, 1 H), 8.91 (d, J = 5.81 Hz, 1 H), 8.55 (d, J = 5.56 Hz, 1 H), 8.28–8.34 (m, 2 H), 8.12 (d, J = 8.34 Hz, 1 H), 7.73 (t, J = 7.45 Hz, 1 H), 7.61 (t, J = 7.33 Hz, 1 H), 7.05 (t, J = 7.07 Hz, 1 H), 3.56 (d, J = 5.05 Hz, 2 H), 2.59 (t, J = 5.94 Hz, 2 H), 2.40 (s, 6 H). 1H NMR (400 MHz, CDCl3) δ ppm 11.70 (br s, 1 H), 9.10 (s, 1 H), 8.94 (dd, J = 7.33, 1.77 Hz, 1 H), 8.73 (dd, J = 6.82, 1.77 Hz, 1 H), 8.29 (s, 1 H), 8.12 (d, J = 8.59 Hz, 1 H), 8.00 (d, J = 8.34 Hz, 1 H), 7.66 (t, J = 7.58 Hz, 1 H), 7.52–7.60 (m, 1 H), 6.89 (t, J = 7.07 Hz, 1 H), 3.66–3.77 (m, 2 H), 2.71 (t, J = 6.06 Hz, 2 H), 2.49 (s, 6 H). MS [M + 1] = 361.
12-Oxo-N-[2-(1-piperidyl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (8)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 1-(2-aminoethyl)piperidine (24.6 μL, 0.17 mmol) according to method A to give 12-oxo-N-[2-(1-piperidyl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (25 mg, 0.062 mmol, 36.2 % yield) as an orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.48 (br s, 1 H), 9.12 (s, 1 H), 8.96 (dd, J = 7.33, 1.77 Hz, 1 H), 8.74 (dd, J = 6.95, 1.64 Hz, 1 H), 8.44 (s, 1 H), 8.13 (d, J = 8.34 Hz, 1 H), 8.00 (d, J = 8.34 Hz, 1 H), 7.68 (dd, J = 8.08, 7.07 Hz, 1 H), 7.53–7.61 (m, 1 H), 6.90 (t, J = 7.07 Hz, 1 H), 3.75 (q, J = 5.81 Hz, 2 H), 2.73 (t, J = 6.19 Hz, 2 H), 2.61 (br s, 4 H), 1.78 (dt, J = 11.24, 5.75 Hz, 4 H), 1.54–1.59 (m, 2 H). MS [M + 1] = 401.
12-Oxo-N-(2-pyrrolidin-1-ylethyl)-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (9)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-pyrrolidin-1-ylethanamine (21.8 μL, 0.17 mmol) according to method A to give 12-oxo-N-(2-pyrrolidin-1-ylethyl)-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (32.4 mg, 0.084 mmol, 48.7% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.65 (br s, 1 H), 9.11 (s, 1 H), 8.96 (dd, J = 7.20, 1.64 Hz, 1 H), 8.71–8.77 (m, 1 H), 8.37 (s, 1 H), 8.13 (d, J = 8.34 Hz, 1 H), 8.02 (d, J = 7.83 Hz, 1 H), 7.64–7.72 (m, 1 H), 7.54–7.63 (m, 1 H), 6.87–6.94 (m, 1 H), 3.79–3.88 (m, 2 H), 2.96–3.03 (m, 2 H), 2.89 (m, 2 H), 2.78 (m, 2 H), 2.00 (m, 4 H). MS [M + 1] = 387.
N-[2-(Diethylamino)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (10)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N′,N′-diethylethane-1,2-diamine (24.3 μL, 0.17 mmol) according to method A to give N-[2-(diethylamino)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (18.2 mg, 0.047 mmol, 27% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.56–11.65 (m, 1 H), 9.12 (s, 1 H), 8.94–8.99 (m, 1 H), 8.75 (d, J = 7.07 Hz, 1 H), 8.41 (s, 1 H), 8.14 (d, J = 8.08 Hz, 1 H), 8.01 (d, J = 7.83 Hz, 1 H), 7.68 (t, J = 7.58 Hz, 1 H), 7.51–7.62 (m, 1 H), 6.91 (t, J = 7.07 Hz, 1 H), 3.77 (br s, 2 H), 2.83 (m, 6 H), 1.21 (t, J = 7.20 Hz, 6 H). MS [M + 1] = 389.
N-[2-(Dimethylamino)-1-methylethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (11)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N1,N1-dimethylpropane-1,2-diamine (22.2 μL, 0.17 mmol) according to method A to give N-[2-(dimethylamino)-1-methyl-ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide hydrochloride (38.7 mg, 0.103 mmol, 60% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.12 (s, 1 H), 8.82 (td, J = 6.63, 1.64 Hz, 1 H), 8.31–8.35 (m, 1 H), 8.12 (d, J = 8.59 Hz, 1 H), 8.03 (d, J = 8.08 Hz, 1 H), 7.62–7.68 (m, 1 H), 7.53–7.58 (m, 1 H), 6.74–6.80 (m, 1 H), 3.47 (br s, 1 H), 2.91 (br s, 1 H), 2.43–2.49 (m, 1 H), 2.39 (s, 3 H), 2.20 (s, 3 H), 1.96 (br s, 3 H). MS [M + 1] = 375.
N-(Azetidin-3-yl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (12)
A solution of 30 (76 mg, 0.17 mmol) in chloroform (1 mL) was treated with trifluoroacetic acid (1.5 mL, 19.6 mmol) at room temperature for 24 h (for convenience). The solvents were removed in vacuo to give N-(azetidin-3-yl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide trifluoroacetate (76.1 mg, 0.17 mmol, 99% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.14–9.20 (m, 1 H), 9.10 (d, J = 5.81 Hz, 1 H), 8.67–8.74 (m, 1 H), 8.46 (s, 1 H), 8.20–8.27 (m, 1 H), 8.12 (s, 1 H), 7.92 (s, 1 H), 7.75 (d, J = 7.83 Hz, 1 H), 7.65 (d, J = 6.82 Hz, 1 H), 7.11 (t, J = 7.20 Hz, 1 H), 4.67 (dd, J = 7.07, 4.55 Hz, 1 H), 4.53 (d, J = 7.33 Hz, 2 H), 2.83 (br.s., 3 H). MS [M + 1] = 345.
N-[2-(Dimethylamino)propyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (13)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-(dimethylamino)propan-1-amine (21.1 mg, 0.21 mmol) according to method A to give N-[2-(dimethylamino)propyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (52 mg, 0.139 mmol, 80.6 % yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.70 (br s, 1 H), 9.09 (s, 1 H), 8.94 (dd, J = 7.33, 1.77 Hz, 1 H), 8.73 (dd, J = 6.82, 1.77 Hz, 1 H), 8.27 (s, 1 H), 8.12 (d, J = 8.59 Hz, 1 H), 7.99 (d, J = 8.59 Hz, 1 H), 7.66 (dd, J = 7.96, 6.95 Hz, 1 H), 7.53–7.58 (m, 1 H), 6.89 (t, J = 7.07 Hz, 1 H), 3.77 (dt, J = 14.02, 5.62 Hz, 1 H), 3.40–3.48 (m, 1 H), 2.95–3.05 (m, 1 H), 2.51 (s, 6 H), 1.15 (d, J = 6.32 Hz, 3 H). MS [M + 1] = 375.
N-[4-(Dimethylamino)butyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (14)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N′,N′-dimethylbutane-1,4-diamine (24.5 μL, 0.17 mmol) according to method A to give N-[4-(dimethylamino)butyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (22.1 mg, 0.057 mmol, 33% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.35–11.41 (m, 1 H), 9.13 (s, 1 H), 8.98 (dd, J = 7.20, 1.64 Hz, 1 H), 8.76 (dd, J = 6.95, 1.64 Hz, 1 H), 8.26 (s, 1 H), 8.14 (d, J = 8.59 Hz, 1 H), 8.06 (d, J = 8.59 Hz, 1 H), 7.66–7.72 (m, 1 H), 7.54–7.63 (m, 1 H), 6.92 (t, J = 7.07 Hz, 1 H), 3.62–3.73 (m, 2 H), 2.46–2.52 (m, 2 H), 2.32 (s, 6 H), 1.59–1.91 (m, 4 H). MS [M + 1] = 389.
N-[2-(Dimethylamino)ethyl]-11-oxopyrido[2,1-b]quinazoline-6-carboxamide (15)
This compound was synthesized from 6b (40 mg, 0.17 mmol) and N,N-dimethylethylenediamine (14.7 mg, 0.17 mmol) according to method A to give N-[2-(dimethylamino)ethyl]-11-oxo-pyrido[2,1-b]quinazoline-6-carboxamide (19.8 mg, 0.064 mmol, 38% yield) as a yellow-orange solid. 1H NMR (400 MHz, MeOD) δ ppm 9.32 (d, J = 7.33 Hz, 1 H), 8.91 (d, J = 7.58 Hz, 1 H), 8.51 (d, J = 7.58 Hz, 1 H), 8.07 (d, J = 7.07 Hz, 1 H), 7.99–8.03 (m, 2 H), 7.71 (t, J = 7.45 Hz, 1 H), 3.98 (t, J = 5.81 Hz, 2 H), 3.53 (t, J = 5.81 Hz, 2 H), 3.06 (s, 6 H). MS [M + 1] = 311.
N-[2-(Diethylamino)ethyl]-11-oxopyrido[2,1-b]quinazoline-6-carboxamide (16)
This compound was synthesized from 6b (40 mg, 0.17 mmol) and N′,N′-diethylethane-1,2-diamine (19.4 mg, 0.17 mmol) according to method A to give N-[2-(diethylamino)ethyl]-11-oxo-pyrido[2,1-b]quinazoline-6-carboxamide (25.2 mg, 0.075 mmol, 44.7% yield) as a yellow solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.44 (br s, 1 H), 9.06 (dd, J = 7.07, 1.77 Hz, 1 H), 8.83 (dd, J = 6.82, 1.77 Hz, 1 H), 8.45–8.51 (m, 1 H), 7.87–7.94 (m, 2 H), 7.54 (ddd, J = 8.08, 5.68, 2.40 Hz, 1 H), 7.04 (t, J = 7.07 Hz, 1 H), 3.71 (q, J = 5.81 Hz, 2 H), 2.69–2.84 (m, 6 H), 1.13 (t, J = 7.20 Hz, 6 H). MS [M + 1] = 339.
2-(Dimethylamino)ethyl 12-Oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxylate (17)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-dimethylaminoethanol (41.6 μL, 0.41 mmol) according to method A to give 2-(dimethylamino)ethyl 12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxylate (47 mg, 0.13 mmol, 47.2 % yield) as an orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.10 (s, 1 H), 8.87 (dd, J = 7.45, 1.64 Hz, 1 H), 8.36 (s, 1 H), 8.11 (d, J = 8.34 Hz, 1 H), 8.02 (d, J = 8.34 Hz, 1 H), 7.78 (dd, J = 6.69, 1.64 Hz, 1 H), 7.64 (ddd, J = 8.27, 6.76, 1.14 Hz, 1 H), 7.52–7.57 (m, 1 H), 6.72 (dd, J = 7.33, 6.57 Hz, 1 H), 4.58 (t, J = 5.81 Hz, 2 H), 2.80 (t, J = 5.68 Hz, 2 H), 2.38 (s, 6 H). MS [M + 1] = 362.
N-[2-(Dimethylamino)ethyl]-N-methyl-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (18)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N,N,N′-trimethylethylenediamine (21.3 μL, 0.17 mmol) according to method A to give N-[2-(dimethylamino)ethyl]-N-methyl-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide trifluoroacetate (18.8 mg, 0.050 mmol, 29.1% yield) as an orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.14 (s, 1 H), 8.93 (d, J = 7.33 Hz, 1 H), 8.28 (s, 1 H), 8.14 (d, J = 8.34 Hz, 1 H), 8.04 (d, J = 8.84 Hz, 1 H), 7.93 (d, J = 6.82 Hz, 1 H), 7.67–7.74 (m, 1 H), 7.58–7.64 (m, 1 H), 6.97 (t, J = 7.07 Hz, 1 H), 4.12 (t, J = 5.94 Hz, 2 H), 3.57 (t, J = 6.19 Hz, 2 H), 3.12 (s, 6 H), 3.10 (s, 3 H). MS [M + 1] = 375.
N-[2-(Dimethylamino)ethyl]-N-ethyl-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (19)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N,N-dimethyl-N′-ethylethylenediamine (27.1 μL, 0.17 mmol) according to method A to give N-[2-(dimethylamino)ethyl]-N-ethyl-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide trifluoroacetate (36.1 mg, 0.093 mmol, 54% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.13 (s, 1 H), 8.87 (d, J = 7.33 Hz, 1 H), 8.21 (s, 1 H), 8.13 (d, J = 8.34 Hz, 1 H), 8.03 (d, J = 8.59 Hz, 1 H), 7.65–7.71 (m, 1 H), 7.56–7.62 (m, 2 H), 6.84 (t, J = 7.07 Hz, 1 H), 4.06 (m, 2 H), 3.62 (t, J = 6.69 Hz, 2 H), 3.39 (q, J = 7.24 Hz, 2 H), 3.12 (s, 6 H), 1.20 (t, J = 7.07 Hz, 3 H). MS [M + 1] = 389.
N-(2-Imidazol-1-ylethyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (20)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-imidazol-1-ylethanamine (16.6 μL, 0.17 mmol) according to method A to give N-(2-imidazol-1-ylethyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (28.5 mg, 0.074 mmol, 43% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.57 (br s, 1 H), 9.08 (s, 1 H), 8.98 (dd, J = 7.07, 1.77 Hz, 1 H), 8.73 (dd, J = 7.07, 1.77 Hz, 1 H), 8.10 (t, J = 8.34 Hz, 3 H), 7.94 (s, 1 H), 7.66–7.72 (m, 3 H), 7.55–7.60 (m, 2 H), 7.22 (s, 1 H), 7.16 (s, 1 H), 6.91 (t, J = 7.07 Hz, 2 H), 4.32–4.37 (m, 2 H), 4.01–4.07 (m, 2 H). MS [M + 1] = 384.
12-Oxo-N-[2-(3-pyridyl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (21)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 3-(2-aminoethyl)pyridine (20.5 μL, 0.17 mmol) according to method A to give 12-oxo-N-[2-(3-pyridyl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (55 mg, 0.14 mmol, 81% yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.39 (br s, 1 H), 9.12 (s, 1 H), 8.96 (dd, J = 7.33, 1.77 Hz, 1 H), 8.75 (dd, J = 7.07, 1.77 Hz, 1 H), 8.26 (s, 1 H), 8.13 (d, J = 8.59 Hz, 1 H), 8.03 (d, J = 8.59 Hz, 1 H), 7.64–7.72 (m, 1 H), 7.53–7.61 (m, 1 H), 6.91 (t, J = 7.07 Hz, 1 H), 3.64–3.74 (m, 2 H), 2.50–2.58 (m, 2 H). MS [M + 1] = 395.
12-Oxo-N-[2-(2-pyridyl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (22)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-(2-aminoethyl)pyridine (20.6 μL, 0.17 mmol) according to method A to give 12-oxo-N-[2-(2-pyridyl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (40.7 mg, 0.103 mmol, 60% yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.39 (br s, 1 H), 9.12 (s, 1 H), 8.96 (dd, J = 7.33, 1.77 Hz, 1 H), 8.75 (dd, J = 7.07, 1.77 Hz, 1 H), 8.26 (s, 1 H), 8.13 (d, J = 8.59 Hz, 1 H), 8.03 (d, J = 8.59 Hz, 1 H), 7.64–7.72 (m, 1 H), 7.53–7.61 (m, 1 H), 6.91 (t, J = 7.07 Hz, 1 H), 3.64–3.74 (m, 2 H), 2.50–2.58 (m, 2 H). MS [M + 1] = 395.
12-Oxo-N-[2-(2-oxoimidazolidin-1-yl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (23)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 1-(2-aminoethyl)imidazolidin-2-one (19.6 μL, 0.17 mmol) according to method A to give 12-oxo-N-[2-(2-oxoimidazolidin-1-yl)ethyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (6.5 mg, 0.016 mmol, 9.4% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.54 (br s, 1 H), 9.12 (s, 1 H), 8.98 (dd, J = 7.20, 1.64 Hz, 1 H), 8.74 (d, J = 7.07 Hz, 1 H), 8.43 (s, 1 H), 8.12 (dd, J = 12.63, 8.08 Hz, 2 H), 7.66–7.71 (m, 1 H), 7.57 (dd, J = 15.79, 8.72 Hz, 2 H), 7.02 (s, 1 H), 6.91 (t, J = 7.33 Hz, 1 H), 3.80–3.88 (m, 2 H), 3.61–3.69 (m, 4 H), 3.47 (m, 2 H). MS [M + 1] = 402.
N-[2-(1H-Indol-3-yl)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (24)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and tryptamine (27.6 mg, 0.17 mmol) according to method A to give N-[2-(1H-indol-3-yl)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (22.9 mg, 0.053 mmol, 30.7% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.43–11.50 (m, 1 H), 9.04 (s, 1 H), 8.91–8.95 (m, 1 H), 8.77 (dd, J = 6.82, 1.77 Hz, 1 H), 8.06 (s, 2 H), 7.83 (d, J = 7.33 Hz, 1 H), 7.74 (d, J = 8.08 Hz, 1 H), 7.60–7.67 (m, 1 H), 7.54 (s, 1 H), 7.47 (s, 1 H), 7.34 (s, 2 H), 7.19–7.26 (m, 3 H), 7.16 (d, J = 2.02 Hz, 2 H), 6.90 (t, J = 7.20 Hz, 1 H), 4.08 (d, J = 6.06 Hz, 2 H), 3.27 (m, 2 H). MS [M + 1] = 433.
N-(2-Methylsulfonylethyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (25)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-methylsulfonylethanamine (21.2 mg, 0.17 mmol) according to method A to give N-(2-methylsulfonylethyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (2.1 mg, 0.0053 mmol, 3.1% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.93 (br s, 1 H), 9.12 (s, 1 H), 9.00 (dd, J = 7.45, 1.64 Hz, 1 H), 8.73 (dd, J = 6.95, 1.64 Hz, 1 H), 8.49 (s, 1 H), 8.12 (t, J = 8.84 Hz, 2 H), 7.67–7.72 (m, 1 H), 7.56–7.62 (m, 1 H), 6.92 (t, J = 7.07 Hz, 1 H), 4.16–4.22 (m, 2 H), 3.49–3.54 (m, 2 H), 3.07 (s, 3 H). MS [M + 1] = 396.
N-[3-(Dimethylamino)propyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (26)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N,N-dimethyl-1,3-propanediamine (21.7 μL, 0.17 mmol) according to method A to give N-[3-(dimethylamino)propyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (18.1 mg, 0.048 mmol, 28.1 % yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.39 (br s, 1 H), 9.12 (s, 1 H), 8.96 (dd, J = 7.33, 1.77 Hz, 1 H), 8.75 (dd, J = 7.07, 1.77 Hz, 1 H), 8.26 (s, 1 H), 8.13 (d, J = 8.59 Hz, 1 H), 8.03 (d, J = 8.59 Hz, 1 H), 7.64–7.72 (m, 1 H), 7.53–7.61 (m, 1 H), 6.91 (t, J = 7.07 Hz, 1 H), 3.64–3.74 (m, 2 H), 2.50–2.58 (m, 2 H), 2.33 (s, 6 H), 1.98 (quin, J = 7.14 Hz, 2 H). MS [M + 1] = 375.
N-[2-(1-Methylpyrrolidin-2-yl)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (27)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-(2-aminoethyl)-1-methylpyrrolidine (22 mg, 25 μL, 0.17 mmol) according to method A to give N-[2-(1-methylpyrrolidin-2-yl)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (18 mg, 0.045 mmol, 26% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.59 (br s, 1 H), 9.10 (s, 1 H), 8.98 (dd, J = 7.33, 1.77 Hz, 1 H), 8.70 (dd, J = 6.82, 1.77 Hz, 1 H), 8.28 (s, 1 H), 8.11 (t, J = 7.71 Hz, 2 H), 7.70 (dd, J = 8.59, 7.33 Hz, 1 H), 7.56–7.62 (m, 1 H), 6.92 (t, J = 7.20 Hz, 1 H), 3.66–3.88 (m, 1 H), 3.26 (m, 2 H), 3.16 (m, 1 H), 2.94 (br.s., 3 H), 2.86–2.92 (m, 1 H), 2.83 (d, J = 4.04 Hz, 1 H), 2.46–2.60 (m, 2 H), 2.20–2.36 (m, 2 H), 2.05–2.20 (m, 2 H). MS [M + 1] = 401.
N-[3-(Dimethylamino)propyl]-N-methyl-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (28)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N,N,N′-trimethyl-1,3-propanediamine (25.3 μL, 0.17 mmol) according to method A to give N-[3-(dimethylamino)propyl]-N-methyl-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide hydrochloride (27.7 mg, 0.071 mmol, 41.4% yield) as a yellow solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 9.12 (s, 1 H), 8.77 (d, J = 7.33 Hz, 1 H), 8.32 (d, J = 6.32 Hz, 2 H), 8.10–8.21 (m, 1 H), 7.54–7.75 (m, 3 H), 6.90–7.01 (m, 1 H), 3.41 (m, 2 H), 3.10 (m, 3 H), 2.87–2.93 (m, 8 H), 2.13 (m, 2 H). MS [M + 1] = 389.
N-(1-Methyl-4-piperidyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (29)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 4-amino-1-methylpiperidine (21.6 μL, 0.17 mmol) according to method A to give N-(1-methyl-4-piperidyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (32.4 mg, 0.084 mmol, 48.7% yield) as an orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.55 (d, J = 8.34 Hz, 2 H), 9.12 (s, 1 H), 8.97 (dd, J = 7.33, 1.77 Hz, 1 H), 8.75 (dd, J = 6.82, 1.77 Hz, 1 H), 8.22 (s, 1 H), 8.13 (d, J = 8.84 Hz, 1 H), 8.03 (d, J = 8.59 Hz, 1 H), 7.68–7.72 (m, 1 H), 7.56–7.61 (m, 1 H), 6.92 (t, J = 7.07 Hz, 1 H), 4.19 (br s, 1 H), 2.96 (br s, 2 H), 2.43 (s, 3 H), 2.33 (br s, 2 H), 2.21 (d, J = 9.60 Hz, 2 H), 1.85–1.95 (m, 2 H). MS [M + 1] = 387.
tert-Butyl 3-[(12-Oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carbonyl)amino]azetidine-1-carboxylate (30)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 1-Boc-3-(amino)azetidine (29.7 mg, 0.17 mmol) according to method A to give tert-butyl 3-[(12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carbonyl)amino]azetidine-1-carboxylate (76 mg, 0.17 mmol, 99% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.98 (br s, 1 H), 9.15 (s, 1 H), 9.00 (dd, J = 7.33, 1.77 Hz, 1H), 8.73 (dd, J = 7.07, 1.77 Hz, 1 H), 8.25 (s, 1 H), 8.13 (dd, J = 17.56, 7.71 Hz, 2 H), 7.70–7.76 (m, 1 H), 7.58–7.64 (m, 1 H), 6.91–6.95 (m, 1 H), 4.90 (d, J = 6.32 Hz, 1 H), 4.46–4.53 (m, 2 H), 4.11 (br s, 2 H), 1.52 (s, 9 H). MS [M + 1] = 445.
4-(4-Isopropylpiperazine-1-carbonyl)-12H-benzo[g]pyrido[2,1-b]quinazoline-12-one (31)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 1-isopropylpiperazine (26.4 μL, 0.17 mmol) according to method A to give 4-(4-isopropylpiperazine-1-carbonyl)-12H-benzo[g]pyrido[2,1-b]quinazoline-12-one (40.9 mg, 0.102 mmol, 59.4% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.11–9.14 (m, 1 H), 8.79–8.82 (m, 1 H), 8.30 (s, 1 H), 8.10–8.15 (m, 1 H), 7.99–8.03 (m, 1 H), 7.62–7.68 (m, 1 H), 7.53–7.59 (m, 1 H), 7.45 (dd, J = 6.57, 1.52 Hz, 1 H), 6.73–6.79 (m, 1 H), 4.10 (m, 2 H), 3.87 (m, 2 H), 3.48 (m, 1 H), 3.36 (m, 1 H), 2.74–2.92 (m, 1 H), 2.68 (m, 1 H), 2.40 (m, 1 H), 1.10 (d, J = 6.57 Hz, 6 H). MS [M + 1] = 401.
N-Methyl-N-(1-methylpyrrolidin-3-yl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (32)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N,N′-dimethyl-3-aminopyrrolidine (19.7 mg, 0.17 mmol) according to method A to give N-methyl-N-(1-methylpyrrolidin-3-yl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide trifluoroacetate (6 mg, 0.016 mmol, 9.0% yield) as a yellow-orange solid. 1H NMR (400 MHz, MeOD) δ ppm 9.11 (s, 1 H), 8.83–8.92 (m, 1 H), 8.24–8.33 (m, 1 H), 8.19 (d, J = 8.08 Hz, 1 H), 8.07 (d, J = 8.34 Hz, 1 H), 7.92 (s, 1 H), 7.55–7.74 (m, 3 H), 6.95 (q, J = 7.33 Hz, 1 H), 4.60 (d, J = 9.35 Hz, 1 H), 4.25 (d, J = 11.87 Hz, 1 H), 4.02–4.14 (m, 1 H), 3.47–3.62 (m, 1 H), 3.17–3.28 (m, 4 H), 3.07 (m, 3 H), 2.67–2.85 (m, 2 H). MS [M + 1] = 387.
4-[(3S)-3-(Dimethylamino)pyrrolidine-1-carbonyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-12-one (33)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and (3S)-(−)-3-(dimethylamino)pyrrolidine (19.7 mg, 0.17 mmol) according to method A to give 4-[(3S)-3-(dimethylamino)pyrrolidine-1-carbonyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-12-one hydrochloride (34.1 mg, 0.088 mmol, 51.2% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 1.96 (br s, 2 H), 9.12 (s, 1 H), 8.82 (td, J = 6.63, 1.64 Hz, 1 H), 8.31–8.35 (m, 1 H), 8.12 (d, J = 8.59 Hz, 1 H), 8.03 (d, J = 8.08 Hz, 1 H), 7.62–7.68 (m, 1 H), 7.53–7.58 (m, 1 H), 6.74–6.80 (m, 1 H), 3.47 (m, 2 H), 2.91 (m, 2 H), 2.43–2.49 (m, 1 H), 2.39 (s, 3 H), 2.24–2.33 (m, 1 H), 2.20 (s, 3 H), 2.11 (m, 1 H). MS [M + 1] = 387.
4-[3-(Diethylamino)pyrrolidine-1-carbonyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-12-one (34)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 3-(diethylamino)pyrrolidine (24.5 mg, 0.17 mmol) according to method A to give 4-[3-(diethylamino)pyrrolidine-1-carbonyl]-12H-benzo[g]pyrido[2,1-b]quinazoline-12-one (54.7 mg, 0.13 mmol, 76.6% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 9.12 (s, 1 H), 8.82 (dd, J = 7.58, 1.52 Hz, 1 H), 8.28–8.37 (m, 2 H), 8.12 (d, J = 8.59 Hz, 1 H), 7.65–7.74 (m, 2 H), 7.57–7.63 (m, 1 H), 6.97–7.02 (m, 1 H), 4.10 (m, 1 H), 3.97 (m, 1 H), 3.81–3.93 (m, 1 H), 3.63 (m 1 H), 3.38 (d, J = 7.33 Hz, 1 H), 3.14 (m, 1 H), 2.87 (m, 1 H), 2.33 (br s, 1 H), 2.26 (br s, 1 H), 1.33–1.38 (m, 1 H), 1.26–1.33 (m, 1 H), 1.14 (t, J = 7.07 Hz, 3 H), 1.01 (t, J = 7.07 Hz, 3 H). MS [M + 1] = 415.
N-[2-(Diisopropylamino)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (35)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N,N-diisopropylethylenediamine (29.9 μL, 0.17 mmol) according to method A to give N-[2-(diisopropylamino)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (33 mg, 0.079 mmol, 46 % yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.07 (br s, 1 H), 9.11 (s, 1 H), 8.92 (d, J = 7.33 Hz, 1 H), 8.58 (d, J = 6.32 Hz, 1 H), 8.42 (s, 1 H), 8.31 (d, J = 7.83 Hz, 1 H), 8.09 (d, J = 7.83 Hz, 1 H), 7.73 (t, J = 7.07 Hz, 1 H), 7.57–7.66 (m, 1 H), 7.05 (t, J = 7.33 Hz, 1 H), 3.50 (d, J = 5.56 Hz, 2 H), 3.13–3.23 (m, 2 H), 2.66–2.75 (m, 2 H), 1.07 (d, J = 6.57 Hz, 12 H). 1H NMR (400 MHz, CDCl3), δ ppm 11.33 (br s, 1 H), 9.12 (s, 1 H), 8.96 (d, J = 6.06 Hz, 1 H), 8.76 (d, J = 6.57 Hz, 1 H), 8.36 (s, 1 H), 8.13 (d, J = 8.08 Hz, 1 H), 8.01 (d, J = 9.09 Hz, 1 H), 7.63–7.72 (m, 1 H), 7.52–7.61 (m, 1 H), 6.90 (t, J = 7.07 Hz, 1 H), 3.66 (d, J = 5.56 Hz, 2 H), 3.22 (d, J = 6.57 Hz, 2 H), 2.81 (br s, 2 H), 1.14 (d, J = 6.32 Hz, 12 H). MS [M + 1] = 417.
N-(2-Morpholinoethyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (36)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N-(2-aminoethyl)morpholine (22.6 μL, 0.17 mmol) according to method A to give N-(2-morpholinoethyl)-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (28.7 mg, 0.071 mmol, 41.4 % yield) as an orange solid. 1H NMR (400 MHz, DMSO-d6) δ ppm 11.50 (br s, 1 H), 9.12 (s, 1 H), 8.96 (dd, J = 7.33, 1.77 Hz, 1 H), 8.75 (dd, J = 6.82, 1.77 Hz, 1 H), 8.38 (s, 1 H)8.13 (d, J = 8.59 Hz, 1 H), 8.00 (d, J = 8.34 Hz, 1 H), 7.64–7.73 (m, 1 H), 7.54–7.62 (m, 1 H), 6.90 (t, J = 7.07 Hz, 1 H), 3.88–3.97 (m, 4 H), 3.77 (q, J = 5.89 Hz, 2H), 2.78 (t, J = 6.19 Hz, 2 H), 2.68 (br s, 4 H). MS [M + 1] = 403.
N-[2-(4-Methylpiperazin-1-yl)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (37)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and 2-(4-methylpiperazin-1-yl)ethanamine (27.6 μL, 0.17 mmol) according to method A to give N-[2-(4-methylpiperazin-1-yl)ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (11.7 mg, 0.028 mmol, 16.3% yield) as a yellow-orange solid. 1H NMR (400 MHz, CDCl3) δ ppm 11.46 (br s, 1 H), 9.14 (s, 1 H), 8.98 (dd, J = 7.33, 1.77 Hz, 1 H), 8.76 (dd, J = 6.82, 1.77 Hz, 1 H), 8.43 (s, 1 H), 8.16 (d, J = 8.59 Hz, 1 H), 8.08 (d, J = 8.34 Hz, 1 H), 7.65–7.73 (m, 1 H), 7.60 (dd, J = 6.82, 1.26 Hz, 1 H), 6.92 (t, J = 7.07 Hz, 1 H), 3.74–3.83 (m, 2 H), 2.60–2.88 (m, 10 H), 2.38 (s, 3 H). MS [M + 1] = 416.
N-[2-[2-Methoxyethyl(methyl)amino]ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide (38)
This compound was synthesized from 6a (50 mg, 0.17 mmol) and N′-(2-methoxyethyl)-N′-methylethane-1,2-diamine (27.3 mg, 0.21 mmol) according to method A to give N-[2-[2-methoxyethyl(methyl)amino]ethyl]-12-oxo-12H-benzo[g]pyrido[2,1-b]quinazoline-4-carboxamide trifluoroacetate (15 mg, 0.037 mmol, 21.5 % yield) as an orange gum. 1H NMR (400 MHz, CDCl3) δ ppm 11.79 (br s, 1 H), 9.10 (s, 2 H), 9.00 (dd, J = 7.33, 1.52 Hz, 1 H), 8.68–8.73 (m, 1 H), 8.43 (s, 1 H), 8.10 (dd, J = 13.64, 8.34 Hz, 3 H), 7.70 (t, J = 7.58 Hz, 2 H), 7.56–7.62 (m, 2 H), 6.94 (t, J = 7.20 Hz, 1 H), 4.07 (br s, 2 H), 3.80 (t, J = 4.55 Hz, 2 H), 3.68 (br s, 1 H), 3.55 (br s, 2 H), 3.36–3.41 (m, 4 H), 3.07 (s, 3 H). 19F NMR (376 MHz, CDCl3) δ ppm −170.87 (br s, 3 F). MS [M + 1] = 405.
Metabolic Stability
Test compounds (1 μM) were incubated in duplicate 100 μL incubations for each time point (0, 5, 20, 30, and 60 min) in a 37 °C water bath. Reaction mixes contained 0.5 mg/mL pooled mixed gender human liver microsomes (Xenotech, LLC) in incubation buffer (10 mM MgCl2, 0.1 M potassium phosphate, pH 7.4). After a 5 min preincubation, reactions were initiated with addition of β-NADPH, nicotinamide adenine dinucleotide phosphate (final concentration 1 mM). Reactions were terminated by addition of 200 μL of 0.1% formic acid in acetonitrile (ACN) with internal standard (400 ng/mL warfarin). Reactions were vortexed then centrifuged for 15 min at 4000 rpm. Supernatants were transferred and diluted with equal volume of 0.1% formic acid in water. The half-life and the intrinsic clearance were determined using Michaelis–Menten nonlinear regression analysis as shown in the following equations: T1/2(min) = ln(2)/k and Clint ((μL/min)/mg protein) = 1000k/[HLM] (mg/mL), where k is the rate constant equal to the absolute value of the slope of the compound disappearance in the plot of the natural log of percent remaining vs time.
The samples were quantified using an Agilent 6540 QTOF with jet stream electrospray ionization source (ESI) and Agilent 1290 UHPLC front-end. Chromatographic separation was achieved over 1 min using a 2.1 mm × 50 mm, 1.8 μm, Zorbax Eclipse Plus RRHD column (Agilent Technologies) with a binary gradient starting at 0.1% formic acid in 5% ACN in water. The ACN concentration increased to 95% between 0.2 and 0.4 min and held until 0.7 min. The gradient returned to initial conditions at 0.75 min. The ESI source conditions were as follows: drying gas temp, 350 °C; sheath gas temp, 400 °C; drying and sheath gas flow, 12 L/min; nebulizer, 60 psig; voltage of cap, 3000 V; nozzle, 600 V; fragmentor, 150 V; skimmer, 100 V; OCT 1 RF Vpp, 750 V. The acquisition was performed by full scan MS from m/z 100 to m/z 500.
Cells and Viability Assay
The cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2. U2OS osteosarcoma cells were cultured in DMEM supplemented with 15% fetal bovine serum. Cells were plated in 96-well plates at a density of 10 000 cells/well in triplicate and incubated for 48 h with the compounds. Viability was determined using WST-1 cell proliferation reagent (Roche Diagnostics).
Immunofluorescence and Epifluorescence Microscopy
U2OS cells grown on coverslips were fixed in 3.5% paraformaldehyde, permeabilized with 0.5% NP-40, and blocked with 3% BSA as described in ref (13). Cells were stained for RPA194 (C-1, Santa Cruz Biotechnology) and NCL (4E2, Abcam). Alexa 488 and Alexa 594 conjugated anti-mouse or anti-rabbit antibodies were from Invitrogen. DNA was counterstained with DAPI (Invitrogen). Images were captured using Axioplan2 fluorescence microscope (Zeiss) equipped with AxioCam HRc CCD camera and AxioVision 4.5 software using EC Plan-Neofluar 20×/0.75 objective (Zeiss).
Determination of RPA194 and NCL IC50
U2OS cells grown on coverslips were treated with the compounds at 0.1, 0.5, 1, 5, and 10 μM or treated with vehicle (DMSO) for 3 h and fixed and stained as above. All compounds, except when indicated, were tested in duplicate independent biological experiments. Immunostaining for NCL and RPA194 was conducted separately, and cells were counterstained for DNA. Two to four fields of each treatment were captured using epifluorescence microscopy as above and contained an average of 200 cells/analysis. The images were quantified using FrIDA image analysis software as described in ref (14). Hue saturation and brightness range were defined individually for RPA194 and NCL, and all values were normalized to the DNA content. The fold change to control was determined. IC50 was determined by GraphPad Prism for Windows (version 6.01) using a three-parameter fit.
Immunoblotting
Lysis of cells was conducted in 0.5% NP-40 buffer (25 mM Tris-HCl, pH 8.0, 120 mM NaCl, 0.5% NP-40, 4 mM NaF, 100 μM Na3VO4, 100 KIU/mL aprotinin, 10 μg/mL leupeptin). Proteins were separated on SDS–PAGE gel and blotted as in ref (14). The following antibodies were used: NCL (4E2, Abcam), RPA194 (C-1, Santa Cruz Biotechnology), GAPDH (Europa Bioproducts). HRP-conjugated secondary antibodies were from DAKO.
Acknowledgments
This work has been supported by NIH Grants P30 CA006973 and 1R01CA172069 (M.L.) and Johns Hopkins University start-up funds.
Glossary
Abbreviations Used
- NCL
nucleolin
- Pol I
RNA polymerase I
- r
ribosomal
Author Present Address
# L.C.: Department of Chemistry, Yale University, New Haven, CT 06520, U.S.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
References
- Hernandez-Verdun D. Nucleolus: from structure to dynamics. Histochem. Cell. Biol. 2006, 125, 127–137. [DOI] [PubMed] [Google Scholar]
- Haag J. R.; Pikaard C. S. RNA polymerase I: a multifunctional molecular machine. Cell 2007, 131, 1224–1225. [DOI] [PubMed] [Google Scholar]
- Russell J.; Zomerdijk J. C. The RNA polymerase I transcription machinery. Biochem. Soc. Symp. 2006, 73, 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comai L. Mechanism of RNA polymerase I transcription. Adv. Protein Chem. 2004, 67, 123–155. [DOI] [PubMed] [Google Scholar]
- Boulon S.; Westman B. J.; Hutten S.; Boisvert F. M.; Lamond A. I. The nucleolus under stress. Mol. Cell 2010, 40, 216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montanaro L.; Treré D.; Derenzini M. Nucleolus, ribosomes, and cancer. Am. J. Pathol. 2008, 173, 301–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grummt I. Wisely chosen paths—regulation of rRNA synthesis. FEBS J. 2010, 277, 4626–4639. [DOI] [PubMed] [Google Scholar]
- Bywater M. J.; Pearson R. B.; McArthur G. A.; Hannan R. D. Dysregulation of the basal RNA polymerase transcription apparatus in cancer. Nat. Rev. Cancer 2013, 13, 299–314. [DOI] [PubMed] [Google Scholar]
- Whitesell L.; Lindquist S. Inhibiting the transcription factor HSF1 as an anticancer strategy. Expert Opin. Ther. Targets 2009, 13, 469–478. [DOI] [PubMed] [Google Scholar]
- Drygin D.; Lin A.; Bliesath J.; Ho C. B.; O’Brien S. E.; Proffitt C.; Omori M.; Haddach M.; Schwaebe M. K.; Siddiqui-Jain A.; Streiner N.; Quin J. E.; Sanij E.; Bywater M. J.; Hannan R. D.; Ryckman D.; Anderes K.; Rice W. G. Targeting RNA polymerase I with an oral small molecule CX-5461 inhibits ribosomal RNA synthesis and solid tumor growth. Cancer Res. 2011, 71, 1418–1430. [DOI] [PubMed] [Google Scholar]
- Haddach M.; Schwaebe M. K.; Michaux J.; Nagasawa J.; O’Brien S. E.; Whitten J. P.; Pierre F.; Kerdoncuff P.; Darjania L.; Stansfield R.; Drygin D.; Anderes K.; Proffitt C.; Bliesath J.; Siddiqui-Jain A.; Omori M.; Huser N.; Rice W. G.; Ryckman D. M. Discovery of CX-5461, the first direct and selective inhibitor of RNA polymerase I, for cancer therapeutics. ACS Med. Chem. Lett. 2012, 3, 602–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bywater M. J.; Poortinga G.; Sanij E.; Hein N.; Peck A.; Cullinane C.; Wall M.; Cluse L.; Drygin D.; Anderes K.; Huser N.; Proffitt C.; Bliesath J.; Haddach M.; Schwaebe M. K.; Ryckman D. M.; Rice W. G.; Schmitt C.; Lowe S. W.; Johnstone R. W.; Pearson R. B.; McArthur G. A.; Hannan R. D. Inhibition of RNA polymerase I as a therapeutic strategy to promote cancer-specific activation of p53. Cancer Cell 2012, 22, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen K.; Colis L.; Liu H.; Jäämaa S.; Moore H. M.; Enbäck J.; Laakkonen P.; Vaahtokari A.; Jones R. J.; af Hällström T. M.; Laiho M. Identification of novel p53 pathway activating small-molecule compounds reveals unexpected similarities with known therapeutic agents. PLoS One 2010, 5, e12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltonen K.; Colis L.; Liu H.; Trivedi R.; Moubarek M. S.; Moore H. M.; Bai B.; Rudek M. A.; Bieberich C. J.; Laiho M. A targeting modality for destruction of RNA polymerase I that possesses anticancer activity. Cancer Cell 2014, 25, 77–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A.; Sasikala W. D. Drug–DNA intercalation: from discovery to the molecular mechanism. Adv. Protein Chem. Struct. Biol. 2013, 92, 1–62. [DOI] [PubMed] [Google Scholar]