

Abstract
In Saccharomyces cerevisiae, Msh2-Msh3-mediated mismatch repair (MMR) recognizes and targets insertion/deletion loops for repair. Msh2-Msh3 is also required for 3′ non-homologous tail removal (3′NHTR) in double-strand break repair. In both pathways, Msh2-Msh3 binds double-strand/single-strand junctions and initiates repair in an ATP-dependent manner. However, we recently demonstrated that the two pathways have distinct requirements with respect to Msh2-Msh3 activities. We identified a set of aromatic residues in the nucleotide binding pocket (FLY motif) of Msh3 that, when mutated, disrupted MMR, but left 3′ NHTR largely intact. One of these mutations, msh3Y942A, was predicted to disrupt the nucleotide sandwich and allow altered positioning of ATP within the pocket. To develop a mechanistic understanding of the differential requirements for ATP binding and/or hydrolysis in the two pathways, we characterized Msh2-Msh3 and Msh2-msh3Y942A ATP binding and hydrolysis activities in the presence of MMR and 3′ NHTR DNA substrates. We observed distinct, substrate-dependent ATP hydrolysis and nucleotide turnover by Msh2-Msh3, indicating that the MMR and 3′ NHTR DNA substrates differentially modify the ATP binding/hydrolysis activities of Msh2-Msh3. Msh2-msh3Y942A retained the ability to bind DNA and ATP but exhibited altered ATP hydrolysis and nucleotide turnover. We propose that both ATP and structure-specific repair substrates cooperate to direct Msh2-Msh3-mediated repair and suggest an explanation for the msh3Y942A separation-of-function phenotype.
Keywords: Mismatch repair, Double-strand break repair, Msh2-Msh3, ATP binding/hydrolysis, 3′ non-homologous tail removal
1. Introduction
DNA mismatch repair (MMR) is a highly conserved DNA repair pathway that is critical for maintaining genome stability [1–3]. MMR is best known for recognizing and directing repair of nucleotide misincorporation or DNA slippage events that occur at the replication fork. MMR is initiated when replication errors are recognized and bound by MutS homologs, or Msh proteins. Prokaryotes encode a single MutS protein whereas most eukaryotes, including Saccharomyces cerevisiae, contain two distinct Msh complexes, Msh2-Msh3 and Msh2-Msh6, with separate but overlapping specificities. Msh2-Msh3 primarily binds and directs repair of both small (1 nucleotide) and larger (up to 17 nucleotide) insertion/deletion loops (IDLs) [4, 5]. Msh2-Msh6 primarily directs repair of misincorporation events and small (1–2 nucleotide) IDLs [4, 6]. Msh2-Msh3 also recognizes and binds some mispairs, particularly C-C mispairs [7]. Once bound to a mismatch (mispair or IDL), the Msh complex recruits the downstream MutL homolog (Mlh) complex, primarily Mlh1-Pms1 in yeast. The ternary complex formation is dependent on ATP-binding by the Msh complex and triggers subsequent steps in MMR, including helicase and exonuclease enzymes to remove the mismatch. Repair is completed by DNA resynthesis of the nascent strand and ligation of the DNA [1, 2, 6].
In addition to MMR of IDLs, S. cerevisiae Msh2-Msh3 is also required during genetic recombination [8–10]. It is required for the prevention of homeologous recombination, i.e. recombination between divergent sequences in which loop structures are formed [8, 9, 11]. Repair of large unpaired loops that can occur during meiotic recombination also requires Msh2-Msh3 as well as the structure-specific endonuclease Rad1-Rad10, which is part of the nucleotide excision repair (NER) pathway [12, 13]. Rad1-Rad10 cleaves at double-strand (ds)/single-strand (ss) DNA junctions with 3′ ssDNA tails [14–16]. Msh2-Msh3 and Rad1-Rad10 are also both required in a specialized pathway of double-strand DNA break repair (DSBR) that involves recombination intermediates with 3′ non-homologous tails (3′ NHTs), such as single strand annealing (SSA) and some gene conversion events [10, 17, 18]. DNA synthesis is required to complete repair, but DNA polymerases cannot prime from unannealed 3′ hydroxyl group. Therefore the 3′ NHTs must be removed to allow synthesis and subsequent ligation. Rad1-Rad10 is responsible for cleaving the tails, but requires partner proteins Msh2-Msh3 and Saw1 to be recruited to the 3′ NHTs [17, 19, 20]. Msh2-Msh3 has been proposed to stabilize the recombination intermediate to promote cleavage by Rad1-Rad10 [17, 20].
In MMR, DNA-binding and ATP-binding activities of bacterial MutS and yeast and human Msh2-Msh6 complexes have been well-studied and demonstrated to be coordinated; DNA-binding leads to conformational changes in MutS and Msh2-Msh6 that are transmitted to the ATP-binding domain via the transmitter region [21–28]. Analogous conformational changes likely occur in Msh2-Msh3; mutations in the putative transmitter region of Msh3 lead to defects in both MMR and 3′ NHTR in vivo [29]. Furthermore, it has been shown that the DNA-binding domains of MutS and Msh2-Msh6 modulate activity and conformational changes within the ATPase domain and vice versa [21, 30–37]. The presence of DNA stimulates steady-state ATP hydrolysis, an effect that is abrogated when there are no free DNA ends [30, 38, 39]. Therefore dissociation from DNA is thought to provoke hydrolysis. In turn, ATP-binding reduces MutS and Msh2-Msh6 complex binding to specific DNA substrates and is predicted to promote the formation of a sliding clamp conformation that allows the complex to move away from the mismatch.
Fewer studies have examined the relationship between Msh2-Msh3 DNA-binding and ATPase activities [40–43]. Nonetheless, the coordinated regulation of DNA-binding and ATP-binding and hydrolysis by Msh2-Msh3 is thought to be critical for proper MMR function [40, 42]. However, while there are similarities, the ATP binding and hydrolysis activities of human Msh2-Msh6 and human Msh2-Msh3 are distinct [41, 42]. Furthermore, the requirements for ATP binding and/or hydrolysis in the Msh3 subunit are distinct for MMR and 3′ NHTR [29]; mutations within the conserved FLY motif of Msh3 [44] predicted to alter the nucleotide binding pocket exhibited a strong defect in MMR but had much milder effects on 3′ NHTR in vivo [29]. These observations led us to hypothesize that the type of DNA substrate (MMR versus 3′ NHTR) might further regulate ATP-binding and/or hydrolysis by Msh2-Msh3.
To develop a mechanistic understanding of the differential requirements for ATP binding/hydrolysis in vivo in MMR and 3′ NHTR, we performed an in vitro analysis of Msh2-Msh3 ATP-binding and ATP hydrolysis activities in the presence of distinct DNA substrates and then compared them to the activities of Msh2-msh3Y942A under the same conditions. This mutation changes the Y (tyrosine) of the FLY motif, which is predicted to form half of a nucleotide (ATP/ADP) sandwich in the Msh3 nucleotide binding pocket, stacking with the adenine [44]. Replacing the Tyr with Ala is predicted to widen the adenine-binding portion of the pocket and lead to fewer constraints on the positioning of ATP or ADP in the pocket. We used a) a homoduplex DNA substrate to mimic non-specific DNA-binding, such as Msh2-Msh3 would encounter during a target search, b) an MMR-specific substrate (a +8 loop; (GT)4), c) a splayed or d) a 3′ flap substrate to mimic 3′ NHTR intermediates (Fig. 1a). We found that the kinetics of ATP hydrolysis by Msh2-Msh3 were substrate-dependent, supporting a model in which distinct DNA substrates promote signature Msh2-Msh3 ATPase activity. This regulation was disrupted in Msh2-msh3Y942A and therefore required an intact Msh3 nucleotide binding pocket. Based on these data, we suggest a possible mechanistic explanation for the msh3Y942A separation-of-function phenotype in vivo.
Figure 1. DNA substrates and purified Msh2-Msh3 and Msh2-msh3Y942A.
(a) The four different synthetic DNA substrates used in this study were homoduplex (non-specific), +8-loop (MMR), splayed Y and 3′flap (3′ NHTR) substrates. (b) Purified Msh2-Msh3 and Msh2-msh3Y942A (1.5 μg complex each) using the PBE94 purification (left) or the Q-Sepharose Fast Flow purification protocol (right). The protein complexes were analyzed by SDS-PAGE (8%) and stained with Coomassie Blue. Msh2 and Msh3 are indicated. The sizes of molecular weight markers (MW; Bio-Rad, broad range) are indicated alongside the gels in kDa.
2. Materials and methods
2.1 Reagents
Phosphoenol pyruvate (PEP), FTE nicotinamide adenine dinucleotide (NADH), pyruvate kinase, lactate dehydrogenase were obtained from Sigma. A 200 mM stock solution of PEP was made in 0.5 M Tris–acetate (pH 7.5). NADH was dissolved in 10 mM Tris–acetate (pH 7.5) and the concentration was determined spectrometrically, using an extinction coefficient of 6250 M–1 cm–1. ATP was obtained from Amersham Pharmacia Biotech and was dissolved in 0.5 M Tris–HCl (pH 7.5), with the concentration determined spectrophotometrically using an extinction coefficient of 1.54 × 105 M–1 cm–1. Oligonucleotides used to construct homoduplex, +8 loop, 3′ flap and splayed substrates were purchased from Integrated DNA Technologies, Coralville, IA and have been described previously [43].
Msh2-Msh3 was initially purified as described previously [45]. Over the course of this study, it became necessary to modify our purification protocol due to the fact that PBE94 is no longer available. In place of PBE94, we used Q-Sepharose Fast Flow (GE) as the first chromatography step. Induced cells were resuspended and frozen in 1 X MSH buffer (25 mM Tris-HCl pH 7.5, 1 mM EDTA) with 200 mM NaCl. The Q-Sepharose column was loaded at 200 mM NaCl and eluted with a linear gradient to 1M NaCl. Msh2-Msh3 eluted at approximately 250 mM NaCl. The final PBE94 column was similarly replaced with Q-Sepharose, loaded at 200 mM NaCl and eluted at 500 mM NaCl. Msh2-msh3Y942A purified exactly as the wild-type Msh2-Msh3 complex (Fig. 1b). The in vitro activities of both Msh2-Msh3 and Msh2-msh3Y942A were indistinguishable in side-by-side comparisons of the two purification protocols (data not shown). Protein concentrations were determined by Bradford assay.
Recently, it was demonstrated that the actual transcriptional start site for MSH3 was different than the previously annotated start site [7]. The construct that we have used for overexpression of MSH3 for purification contains both start sites [43, 45–47]. To ensure that the protein we were using is the correct version, we sent purified Msh3 for N-terminal sequencing (University of Pittsburgh) and determined that our purified protein has the correct start site (data not shown).
2.2 DNA substrates
The homoduplex (LS1/LS2), MMR (LS2/LS8), splayed (LS1/LS3) and 3′ flap (LS1/LS3/LS16) synthetic substrates have been described previously [43, 47]. DNA substrates were prepared at 20μM. Top strand oligonucleotides were mixed with excess of the corresponding bottom oligonucleotide (1:1.2) in 100 mM NaCl, 10 mM MgCl2 and 0.1 mM EDTA. The mixture was heated to 95 °C for 5 min and then allowed to cool slowly to room temperature, to allow the strands to anneal. The annealed oligonucleotide substrates were purified by gel filtration (HR S-300 spin columns; Amersham Biosciences) to remove any unannealed oligonucleotide. This step is important because Msh2-Msh3 binds well to single-stranded DNA [43], which interfered with the kinetic analysis (data not shown). All substrates were verified by gel electrophoresis. All substrates used in this study are shown schematically in Fig. 1a.
2.3 Gel mobility shift assay
The standard reaction (10 μl) contained 1 nM 32P 5′ end-labeled substrate in 20 mM HEPES (pH 7.5), 100 mM NaCl, 1 mM DTT, 40 μg/ml BSA, 2 mM MgCl2. The reactions were assembled on ice and then incubated at room temperature for 5 minutes. The samples were electrophoresed through 4% non-denaturing polyacrylamide gels in 45 mM Tris borate, 0.5 mM EDTA at 130V for 45 minutes in a water-cooled gel electrophoresis apparatus. The gels were dried and exposed to a PhosphorImager screen (Molecular Dynamics). ImageQuant was used to quantify bound and free DNA. The approximate Kd’s shown in Fig. 4b represent the protein concentration at 50% maximal binding. The values are an average of at least 3 independent experiments with 2 different protein preparations.
Figure 4. Msh2-msh3Y942A retains wild-type DNA-binding activity.
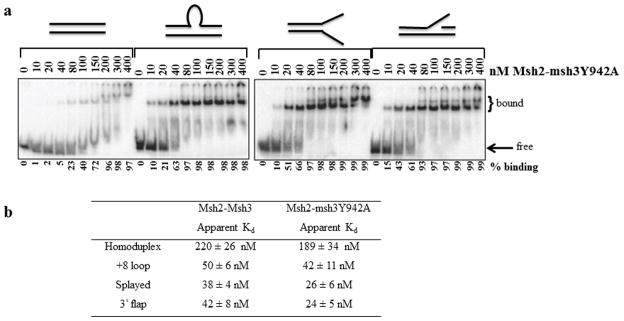
(a) Increasing concentrations (nM) of Msh2-msh3Y942A were incubated with labeled homoduplex, +8 loop, splayed Y and 3′ flap substrates and analyzed by electrophoretic mobility shift assay (see Material and Methods). Substrates are illustrated above the gels. (b) Apparent Kd’s of Msh2-Msh3 and Msh2-msh3Y942A based on titration experiments. Values for Msh2-Msh3 are from Surtees and Alani, 2006 [43]. The values for Msh2–msh3Y942A represent the protein concentration at 50% maximal binding for each substrate and are an average of at least three separate experiments (±S.E.M.).
2.4 Filter binding assays
The affinity of Msh2-Msh3 for nucleotide was measured using a double-filter method for nitrocellulose filter binding [48]. 20 μl reactions contained 1X reaction buffer (20mM Hepes pH 7.6, 40 μg/ml BSA, 2mM MgCl2, 1mM DTT, 100mM NaCl), 50nM protein, 250nM DNA (when present) and the indicated concentration of ATPγS spiked with 35S-ATPγS. Upon addition of protein, reactions were incubated for 10 minutes at 4°C. Nitrocellulose (0.45 μm, Biorad) was soaked in 1X reaction buffer overnight at 4°C. Hybond DEAE membrane (GE) was washed three times with 1M NaCl and in 0.1M NaOH for 10 seconds. The DEAE was placed on a dot blot apparatus and overlaid with nitrocellulose and a vacuum was applied. Reactions were applied to the nitrocellulose in duplicates, and washed three times with 1X reaction buffer. Membranes were dried and exposed to PhosphoImager screen (Molecular Dynamics), imaged using Typhoon scanner and quantified using ImageQuant. A standard curve of ATPγS was used to calculate the number of pmol ATPγS bound by Msh2-Msh3 or Msh2-msh3Y942A.
For the ADP competition experiments, Msh2-Msh3 or Msh2-msh3Y942A (50 nM) was pre-incubated with DNA substrate (250 nM), as described above. ADP (0.5 mM) was added and allowed to bind on ice for 10 minutes. ATPγS (8 μM) spiked with 35S-ATPγS was added to the reaction and incubated at room temperature for 30 or 60 seconds and then applied to nitrocellulose as described above. Binding of ATPγS in the presence of ADP was normalized to binding in the absence of ADP under equivalent conditions.
2.5 ATPase assays
Hydrolysis of ATP was monitored using a coupled spectrophotometric assay [45–46,55]. In this assay, the conversion of ATP to ADP and Pi is linked to the oxidation of NADH to NAD+, and is monitored as a decrease in absorbance at 340nm. Assays were performed at 30°C and monitored using a Varian, Cary-50 Bio UV-visible spectrophotometer. The reaction conditions include 20 mM Tris-acetate (pH 7.5), 0.3 mM NADH, 5 mM PEP, 20 U/mL pyruvate kinase, 20 U/mL lactate dehydrogenase, 2 mM magnesium acetate, DNA (at indicated concentrations) and Msh2-Msh3 (at indicated concentrations) and up to 1 mM ATP. Reactions were initiated with DNA, Msh2-Msh3 and ATP in that order. ATP titration was performed by addition of incremental amounts of ATP until maximal rates were achieved. Volume additions were taken into account for accurate calculation of ATP concentration. In a typical reaction, approximately 80 data points were fit to a linear curve. The rate of ATP hydrolysis at each ATP concentration was calculated by multiplying the slope of the line by 159 (the change in absorbance of NADH per unit time) [49]. In the optimization experiments (Fig. 3), 100 nM Msh2-Msh3 was used. In all subsequent experiments, 50 nM Msh2-Msh3 was used. The kinetic parameters of Msh2-Msh3 at 50 nM and 100 nM were similar at equivalent protein:DNA ratios (data not shown).
Figure 3. ATPase activity of Msh2-Msh3 is modulated by divalent cation and DNA substrate.
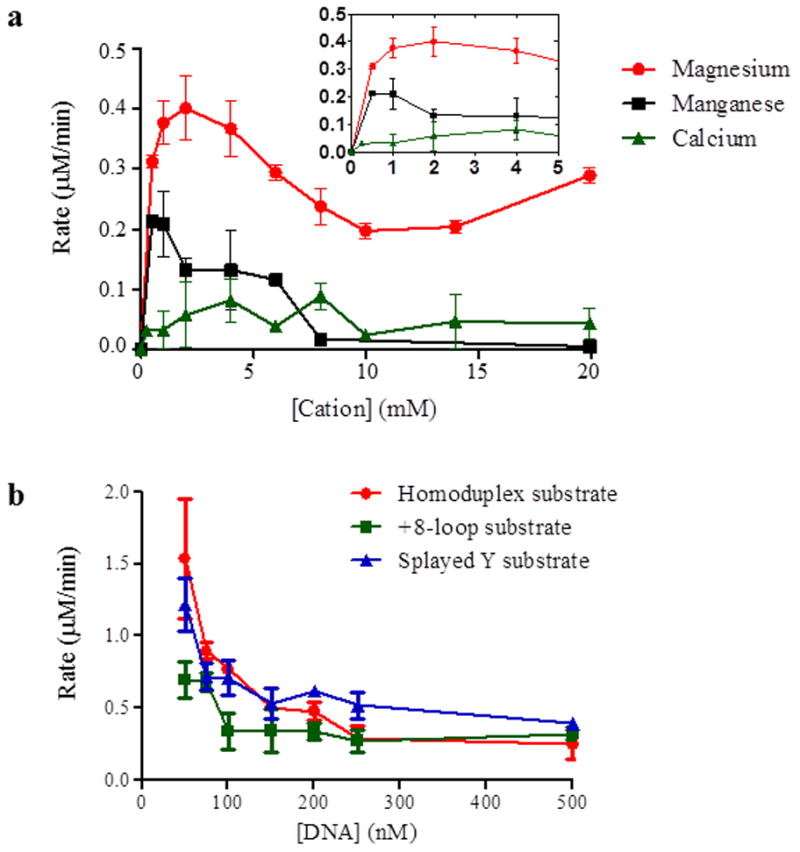
(a) ATPase activity of Msh2-Msh3 is dependent on magnesium. Cation titrations were performed with 100nM Msh2-Msh3 in the presence of 500 nM +8-loop substrate, 1 mM ATP and 100 mM NaCl. Magnesium acetate, manganese chloride and calcium chloride were titrated at the indicated concentrations. The inset shows low cation concentrations at higher resolution. (b) Homoduplex, +8-loop and splayed Y substrates were titrated into an ATPase reaction containing 50 nM Msh2-Msh3, 1 mM ATP and 100 mM NaCl. The DNA titration was started at 50 nM and continued to 500nM DNA substrate. The rate of ATP hydrolysis was plotted against the concentration of DNA. Red circles represent homoduplex DNA, green squares represent +8 loop DNA and blue triangles represent splayed DNA.
For ATPγS titration experiments, 50 nM Msh2-Msh3 was pre-incubated with 2 mM magnesium and 1 mM ATP in the absence of DNA or 250 nM DNA, 2 mM magnesium and 100 μM ATP, in the presence of DNA substrate. Different ATP concentrations were used in the absence or presence of DNA because of the different Km of Msh2-Msh3 for ATP under those conditions (Table 1). When steady state was achieved (constant slope), ATPγS was titrated into the reaction. Rate of ATP hydrolysis was determined after each ATPγS increment. At the end of the titration, rates were normalized to no ATPγS (100% activity). From the resulting curves, concentration required for inhibition of ATPase activity by 50% was calculated.
Table 1.
ATP titrations to determine time dependence of ATP hydrolysis
| ATPa (μM) | Time (min.)b (A&B) | Rate (μ | M/min)c | Time (min.) (A&B) | Rate (μM/min)d | |
|---|---|---|---|---|---|---|
| 1A | 1B | 2A | 2B | |||
| 0 | 15.2e | 0 | 0 | 16.1 | 0 | 0 |
| 5 | NAf | NA | NA | 26.1 | −0.005 | −0.005 |
| 10 | NA | NA | NA | 37.4 | 0.125 | 0.030 |
| 10 | NA | NA | NA | 46.9 | 0.058 | 0.069 |
| 10 | NA | NA | NA | 56.9 | 0.080 | 0.089 |
| 15 | NA | NA | NA | 67.3 | 0.150 | 0.153 |
| 30 | NA | NA | NA | 78.1 | 0.237 | 0.158 |
| 65 | 25.1 | 0.322 | 0.343 | 90.3 | 0.341 | 0.354 |
| 140 | 36.4 | 0.351 | 0.345 | 100.2 | 0.362 | 0.362 |
| 287.5 | 46.0 | 0.443 | 0.413 | 111.1 | 0.453 | 0.434 |
| 567.5 | 56.0 | 0.419 | 0.524 | 122.7 | 0.468 | 0.593 |
| 1125 | 66.4 | 0.645 | 0.610 | 132.0 | 0.633 | 0.631 |
Each titration was performed in duplicate (A and B). The reactions contained a fivefold excess of the +8 loop DNA substrate over Msh2-Msh3.
Total incubation time when rate was calculated.
Calculated rates for titration 1, starting at 65μM ATP.
Calculated rates for titration 2, starting at 5 μM ATP.
The time for replicate A is shown. The time for replicate B is less than 30 seconds later. It takes the reaction approximately 15 minutes to equilibrate.
Not applicable – measurements were not taken at these ATP concentrations.
2.6 Data Analysis
To obtain kinetic parameters, data obtained from coupled spectroscopy were analyzed using non-linear curve fitting in Prism v 5.04 (GraphPad Software, Inc.). Two kinetic models, Michaelis-Menten (MM) and Hill equations, were used to fit the data [50]. The MM equation is the simpler of the two and is
where v is the rate of ATP hydrolysis, Vmax is the maximal rate of ATP hydrolysis, [S] signifies substrate (ATP) concentration and Km is the concentration of substrate when the rate of the reaction is half Vmax indicating half saturation [50] (Fig. 2c). The data were also fit to the Hill equation, which takes into account multiple active sites present in the enzyme. The Msh2-Msh3 complex has two composite active sites, and therefore, Hill equation is an appropriate fit. The equation is defined as
Figure 2. ATP hydrolysis by Msh2-Msh3 was measured by coupled spectroscopy.
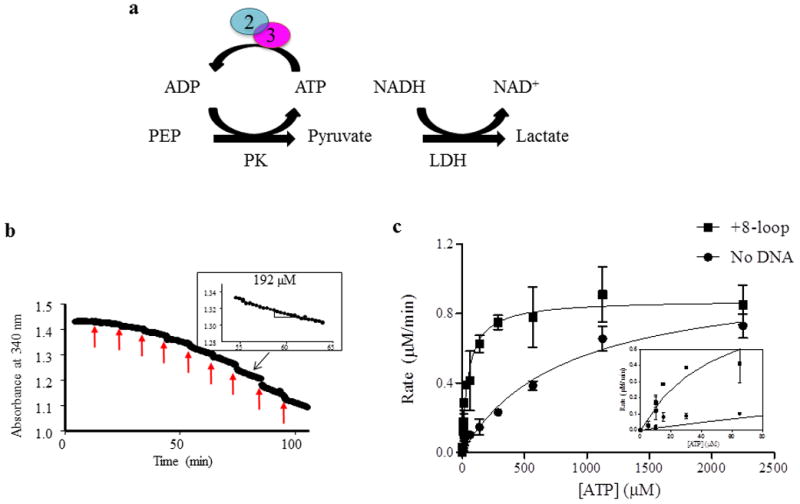
(a) Hydrolysis of ATP by Msh2-Msh3 was measured using a regeneration system and coupled spectroscopy. The hydrolysis reaction was coupled to a series of reactions that result in the oxidation of NADH to NAD+, which decreased the absorbance at 340 nm. The rate of decrease is a measure of rate of ATP hydrolysis (see Materials and Methods). (b) Coupled spectroscopy is a continuous experiment and a sample ATP titration is shown. The red arrows indicate the time of addition of additional ATP. The rate of hydrolysis is calculated as the slope of a line fit to the data points, as shown in the inset for 192 μM ATP. The black arrow indicates the part of the titration where the ATP concentration is 192 μM. (c)A representative experiment is shown to illustrate the type of curves that are generated with these data. The rate of hydrolysis, calculated as in (b), was plotted against the concentration of ATP and the data were approximated by the Michaelis-Menten equation. Vmax and Km were obtained from these equations (see Materials and Methods). In this particular experiment, the reactions include 50 nM Msh2-Msh3, 100 mM NaCl, 2 mM MgOAc and 250 nM DNA (+8 loop), when present.
where Y is the rate of ATP hydrolysis
[S] is the substrate concentration
Km is the concentration of substrate at half maximal rate of hydrolysis
h is the Hill constant which defines the cooperativity between active site.
If the Hill co-efficient is 1, the active sites are non-cooperative. If the co-efficient is less than 1, the active sites are negatively cooperative while co-efficient greater than 1 indicates positively cooperativity [50]. In the case of negative cooperativity, the binding and/or catalysis at one site inhibits catalysis and/or binding at the other. In the scenario of positive cooperativity, binding and/or catalysis at one active site promotes binding and/or catalysis at the other. When ATP binding appeared to exhibit negative cooperativity, a comparison was done in Prism to determine which model more accurately described the data.
From the kinetic parameters we calculated kcat and kcat/Km. kcat is the catalytic rate, the maximum number of substrate molecules converted to product per enzyme per minute and is calculated as the ratio of Vmax and enzyme concentration. kcat/Km is also known as catalytic efficiency. Each ATPase assay or ATPγS inhibition assay was performed at least two times in duplicate, with each of two different protein preparations.
3. Results
Msh2-Msh3 has two ATPase sites, both of which are required for its MMR and 3′ NHTR repair functions in vivo [29, 51]. We recently demonstrated that alterations to the ATP-binding pocket of S. cerevisiae Msh2-Msh3, including msh3Y942A, have different phenotypic outcomes with respect to MMR and 3′ NHTR and we proposed that this is due to distinct requirements in Msh3 with respect to ATP binding and/or hydrolysis [29]. To test this hypothesis, we first characterized the ATP binding and hydrolysis activity of Msh2-Msh3, in the presence of DNA substrates that mimic either MMR (+8 loop) or 3′ NHTR (splayed or 3′ flap) substrates, which has not been examined previously. We then compared these activities with those of Msh2-msh3Y942A. The msh3Y942A allele is defective for MMR but competent for 3′NHTR and heteroduplex rejection in vivo [29]. Based on the human Msh2-Msh3 crystal structure [44], we predicted that the Y942A change would open up the nucleotide binding site and alter the tight regulation of ATP-binding and/or hydrolysis required by MMR.
3.1 ATPase activity of Msh2-Msh3
We characterized the ATPase activity of Msh2-Msh3 using an enzyme-coupled spectroscopy assay in which conversion of ATP to ADP (and Pi) was linked to NADH oxidation to NAD+ (Fig. 2a and Materials and Methods) [52, 53]. Oxidation of NADH was monitored continuously as a decrease in absorbance at 340nm over time and the rate of ATP hydrolysis is derived from the slope of that line (Fig. 2b). The rates of ATP hydrolysis as a function of ATP concentration (derived from the slopes) were plotted and approximated by the Michaelis-Menten equation, from which Vmax and Km values were derived (Fig. 2c). In this example, the presence of a MMR-specific substrate (+8 loop) significantly decreased the Km for ATP (828.2 ± 151.4 μM in the absence of DNA; 49.3 ± 8.41 μM in the presence of +8 loop DNA substrate), but had little effect on the Vmax for ATP hydrolysis (1.0 μM/min without DNA versus 0.88 μM/min with the +8 loop DNA substrate).
The strength of this kinetic ATPase assay is that it allows continuous measurement of ATPase activity over time in the same reaction. However, by the time Msh2-Msh3 has reached Vmax at about 1.5 mM ATP, the protein will have spent approximately an hour at 30°C. We wanted to determine whether this long incubation affects the activity of the enzyme, i.e. are the rates measured at the later time points artificially reduced due to a loss of Msh2-Msh3 activity over time? To test this, we performed side-by-side ATP titrations, in duplicate (A and B) (Table 1). After equilibration in the absence of ATP, Titration 1 (1A and 1B) started at 65 μM ATP and continued up to 1125 μM ATP (left side of Table 1). Titration 2 (2A and 2B) started at a low ATP concentration (5 μM) and arrived at 65 μM after one hour (right side of Table 1). This titration was continued up to 1125 μM ATP. The rates measured at the same ATP concentrations were indistinguishable in the two titrations despite the fact that one reaction was incubated for an additional hour. Therefore Msh2-Msh3 does not appear to lose activity in the time frame of the assay and this is a powerful assay to characterize the kinetics of Msh2-Msh3 ATP hydrolytic activity.
3.2 Msh2-Msh3 exhibits distinct kinetic parameters in the presence of different DNA substrates
Msh2-Msh3 promotes MMR and 3′ NHTR and we previously isolated putative ATP binding mutants that behave differently in the two pathways [29]. To determine whether MMR and 3′ NHTR DNA substrates differentially modulate Msh2-Msh3 ATPase activity through altered nucleotide binding and/or hydrolysis, we performed a careful analysis of Msh2-Msh3 ATPase activity in the presence of homoduplex, MMR (+8 loop) and 3′ NHTR (splayed and 3′ flap) DNA substrates (Fig. 1a).
To identify optimal conditions for these experiments, we first performed titrations with divalent cations (Fig. 3a) and with the different DNA substrates (Fig. 3b) in the presence of Msh2-Msh3. ATP hydrolysis by Msh2-Msh3 required a divalent cation and magnesium (Mg(OAc)2) was preferred over manganese (MnCl2) or calcium (CaCl2) (Fig. 3a) with an optimal concentration of 2 mM, which we used in all subsequent experiments, consistent with previous work with Msh proteins [22, 23, 30, 40, 54–56]. Manganese was able to support ATP hydrolysis with an optimal concentration of 0.8 mM MnCl2, although the maximal rate was two-fold lower that that at the optimal magnesium concentration (0.4 μM/min. versus 0.2 μM/min.). CaCl2 did not support any ATP hydrolysis. Furthermore, Msh2-Msh3 did not hydrolyze GTP (data not shown).
In DNA titration experiments, Msh2-Msh3 aggregated when in excess over DNA (data not shown). Msh2-Msh3 also exhibited DNA substrate inhibition above stoichiometric concentrations (Fig. 3b), making it difficult to determine a KmDNA. However, we have previously measured apparent Kd’s of Msh2-Msh3 for different DNA substrates [43]. At 100 mM NaCl, the salt concentration used here, Msh2-Msh3 exhibited a similar affinity for the +8 and splayed substrates, with an approximate Kd of ~40–50 nM [43]. Msh2-Msh3 had an approximately four- to five-fold lower affinity for homoduplex DNA(~220 nM) [43]. Therefore, we performed ATP titrations at three different ratios of Msh2-Msh3:DNA (1:1, 1:5 and 1:10), corresponding to 50 nM DNA (1:1), close to the Kd for MMR and 3′NHTR substrates, 250 nM DNA (1:5), close to the Kd for homoduplex DNA and 500 nM DNA (1:10) (Table 2). We reasoned, based on Kd values, that the proportion of Msh2-Msh3 bound to MMR and 3′ NHTR substrates at 1:1 should be similar to the proportion bound to homoduplex DNA at 1:5. We included the 1:10 to determine any effect of increased Msh2-Msh3/DNA complex formation and excess DNA on the kinetic properties on Msh2-Msh3.
Table 2.
Kinetic Parameters of Msh2-Msh3 ATP hydrolysis in the presence of homoduplex, MMR and 3′ NHTR DNA substrates.
| 1:1 ratio | No DNA | Homoduplex DNA | + 8 loop substrate | Splayed Y substrate | 3′flap substrate |
|---|---|---|---|---|---|
| Vmax (nM/min) a | 1060 ± 56 | 923 ± 25 | 1465 ± 20 | 1400 ± 23 | 1305 ± 88 |
| Km (μM) b | 842 ± 132 | 95.4 ± 3.0 | 8.4 ± 2.1 | 7.9 ± 1.4 | 7.2 ± 2.5 |
| kcat (min−1) c | 21.2 | 18.5 | 29.3 | 28 | 26.1 |
| kcat/Km (min−1μM−1) | 0.025 | 0.19 | 3.5 | 3.5 | 3.6 |
| Relative to no DNA d | 1 | 7.4 | 134 | 136 | 144 |
| Hill co-efficient e | 1.1 ± 0.2 | 0.42 ± 0.07 | 1.1 ± 0.2 | 0.6 ± 0.1 | 0.6 ± 0.2 |
|
| |||||
| 1:5 ratio | |||||
| Vmax (nM/min) a | 1060 ± 56 | 500 ± 40 | 800 ± 60 | 700 ± 30 | 712 ± 32 |
| Km (μM) b | 842 ± 132 | 29.6 ± 4.1 | 23.9 ± 5.1 | 33.9 ± 6.4 | 45.1 ± 7.3 |
| kcat (min−1) c | 21.2 | 10.8 | 16.0 | 14.0 | 14.2 |
| kcat/Km (min−1μM−1) | 0.025 | 0.34 | 0.67 | 0.41 | 0.3 |
| Relative to no DNA d | 1 | 13.6 | 26.8 | 16.4 | 12.4 |
| Hill co-efficient e | 1.1 ± 0.2 | 0.3 ± 0.07 | 1.2 ± 0.2 | 0.9 ± 0.1 | 0.8 ± 0.2 |
|
| |||||
| 1:10 ratio | |||||
| Vmax (nM/min) a | 1060 ± 56 | 600 ± 40 | 900 ± 60 | 900 ± 30 | ND f |
| Km (μM) b | 842 ± 132 | 25.2 ± 2.1 | 71.3 ± 4.1 | 25.3 ± 6.4 | ND |
| kcat (min−1) c | 21.2 | 12 | 18 | 18 | |
| kcat/Km (min−1μM−1) | 0.025 | 0.5 | 0.25 | 0.71 | |
| Relative to no DNA d | 1 | 20 | 10 | 28.4 | |
| Hill co-efficient e | 1.1 ± 0.2 | 0.32 ± 0.06 | 1.0 ± 0.15 | 1.1 ± 0.11 | ND |
Maximum rate of ATP hydrolysis when data fit to Michaelis-Menten or Hill equation. Errors represent standard error of the mean.
Dissociation constant determined as the concentration of ATP at half maximal ATP hydrolysis rate data fit to Michaelis-Menten or Hill equation. Errors represent standard error of the mean.
Ratio of Vmax and concentration of protein
kcat/Km of Msh2-Msh3 in the presence of DNA substrate relative to the absence of DNA
Determined when data fit to Hill equation
ND = Not determined
Msh2-Msh3 ATPase activity was highest when the protein and DNA were at stoichiometric concentrations (Fig. 3b; Table 2, top panel). Relative to the absence of DNA, Vmax of Msh2-Msh3 was decreased ~ 10% in the presence of homoduplex and increased slightly (1.4-fold) in the presence of the specific MMR and 3′NHTR (both splayed and 3′ flap) DNA substrates, small differences that are reflected in the kcat values. In contrast, the differences in KmATP were much more significant. The KmATP decreased by 8.8-fold in the presence of homoduplex DNA compared to the absence of DNA, a striking effect given that the Kd for binding homoduplex DNA would suggest only a small fraction of Msh2-Msh3 would be in complex with this DNA substrate. In the presence of the specific substrates (+8, 3′ flap and splayed) the KmATP was decreased approximately 100-fold compared to the absence of DNA. As a result, the kcat/Km (catalytic efficiency) of Msh2-Msh3 is 19-fold higher in the presence of MMR and 3′ NHTR substrates than in the presence of homoduplex DNA.
When the stoichiometric ratio of protein:DNA was 1:5 (Table 2, middle panel), Vmax decreased about two-fold in the presence of DNA compared to the 1:1 ratio. Consequently, the kcat in the presence of all four DNA substrates decreased. The KmATP in the presence of homoduplex DNA decreased three-fold, possibly due to increased homoduplex DNA-binding at this ratio. In contrast, the KmATP in the presence of the three repair substrates increased 3- to 6-fold, indicating that as the equilibrium is shifted toward Msh2-Msh3 in complex with repair substrates, ATP binding and/or hydrolysis is altered (reduced). Nonetheless, the catalytic efficiency of Msh2-Msh3 in the presence of homoduplex at 1:5 (protein:DNA; ~ Kd for homoduplex) remained ten-fold lower than that of Msh2-Msh3 in the presence of repair substrates at stoichiometric (1:1; ~ Kd for MMR and 3′ NHTR substrates) concentrations of protein and DNA, at which complex formation is predicted to be similar. Finally, the kcat/Km in the presence of all DNA substrates was decreased five- to twelve-fold when DNA was in five-fold excess over protein. Notably, at this protein:DNA ratio, the kcat/Km values of Msh2-Msh3 in the presence of the 3′ NHTR substrates more closely resembled that in the presence of homoduplex DNA than the kcat/Km in the presence of the MMR substrate. Importantly, the kinetics of ATPase activity in the presence of two 3′ NHTR substrates (splayed and 3′ flap) were indistinguishable, consistent with the Msh2-Msh3 binding to these substrates similarly [43] (Fig. 4b).
To test the possibility that the increased complex formation and/or excess DNA alters Msh2-Msh3’s interaction with ATP, the ratio of Msh2-Msh3 to DNA was increased to 1:10 (Table 2, bottom panel). Vmax in the presence of homoduplex, +8-loop and the splayed Y substrates were similar to that observed at a 1:5 ratio. Notably, the KmATP was elevated in the presence of the +8 loop substrate, but not in the presence of either homoduplex or 3′ NHTR substrate. Consequently, the kcat/Km in the presence of the +8-loop substrate was ~ 3-fold lower at the 1:10 ratio. This is consistent with the idea that excess MMR substrate altered Msh2-Msh3 ATP binding. This may be due to higher order complex formation in the presence of a loop structure which slows hydrolysis. This does not appear to be the case in the presence of homoduplex DNA or the splayed (3′ NHTR) DNA substrate at the 1:10 ratio. Therefore, at all three ratios the kinetic data indicated that the homoduplex, MMR and 3′ NHTR DNA substrates modulated Msh2-Msh3 ATPase activity in distinct ways, indicating differential, context-specific regulation of Msh2-Msh3 ATPase activity.
Because the Msh2-Msh3 complex has two composite ATPase active sites (one in Msh2 and the other in Msh3) [44], the data were also approximated by the Hill equation to take into account the potential influence of each active site on the other [50] (see Materials and Methods). Notably, the Hill coefficient, which indicates cooperativity, was substrate-dependent. In the absence of any DNA or in the presence of the +8 substrate, the Hill coefficient was close to 1 at all three protein:DNA ratios, consistent with non-cooperative ATP binding/hydrolysis. However, in the presence of the homoduplex substrate the Hill coefficient was low (0.3–0.4) at all Msh2-Msh3:DNA ratios, indicating negative cooperativity in which ATP binding/hydrolysis at one site has an inhibitory effect on binding/hydrolysis at a second site [50] in the presence of homoduplex DNA. In the presence of the 3′ NHTR DNA substrates at stoichiometric concentrations, the Hill coefficient of 0.6 suggested weak negative cooperativity, but this was relieved as the DNA concentration was increased. These observations indicate that the regulation of the Msh2-Msh3 ATPase cycle is differentially modulated in a DNA substrate-dependent manner.
3.3 Msh2-msh3Y942A retains wild-type DNA- and ATPγS-binding activity
Msh2-Msh3 exhibited differential kinetics of ATP hydrolysis, depending on the DNA substrate present. But are these differences important in vivo for the distinct repair functions of Msh2-Msh3? To address this question, we took advantage of the msh3Y942A allele that is defective in MMR but competent for 3′ NHTR in vivo [29]. To determine whether the in vivo separation-of-function phenotype of msh3Y942A was a result of abrogating the substrate-dependent differences in the hydrolytic cycle, we wanted to examine Msh2-msh3Y942A ATPase activity in the presence of homoduplex, MMR and 3′ NHTR DNA substrates. But because DNA substrate (and presumably complex formation) has a significant impact on the kinetics of Msh2-Msh3 ATPase activity, we first assessed the ability of the mutant protein complex to bind and distinguish between homoduplex, MMR and 3′ NHTR DNA substrates. We performed gel mobility shift assays to determine whether Msh2-msh3Y942A retained wild-type DNA-binding activity (Fig. 4a). Based on the crystal structure of human Msh2-Msh3, Y942 was predicted to form part of the Msh3 nucleotide binding pocket, away from the DNA-binding domains [44] and therefore was not predicted to affect DNA interactions. In fact, the DNA-binding activity of Msh2-msh3Y942A was very similar to that of wild-type Msh2-Msh3 [43]. Its substrate specificity was intact and Msh2-msh3Y942A bound to the different substrates with affinities similar to those of Msh2-Msh3, although Msh2-msh3Y942A may have a slightly higher affinity for the 3′ NHTR substrates. Notably, neither Msh2-Msh3 nor Msh2-msh3Y942A DNA-binding distinguished between the splayed substrate or the 3′ flap substrate. Therefore the splayed substrate was the primary 3′ NHTR substrate used for subsequent analyses (see below).
We also compared the ability of Msh2-Msh3 and Msh2-msh3Y942A to bind ATPγS, a poorly hydrolysable analog of ATP in the absence or presence of DNA substrate (Fig. 5). Using a nitrocellulose filter binding assay, we observed largely substrate-independent ATPγS-binding by Msh2-Msh3 (Fig. 5). Binding by Msh2-Msh3 and Msh2-msh3Y942A was largely indistinguishable and was saturated at about 16 μM ATPγS with between 1.5 and 2 pmol of ATPγS bound per 1 pmol of Msh2-Msh3 complex (with two nucleotide binding sites per complex).
Figure 5. Msh2-Msh3 and Msh2-msh3Y942A bind ATPγS.

Filter binding assays were performed to assess ATPγS binding to Msh2-Msh3 or Msh2-msh3Y942A Comparisons between the two protein complexes are shown (a) in the absence of DNA, (b) in the presence of homoduplex DNA, (c) in the presence of the MMR (+8 loop) DNA and (d)in the presence of the 3′ NHTR (splayed) DNA substrate. The protein was incubated with increasing concentrations of ATPγS in the absence or presence of DNA substrates (1:5 protein to DNA ratio). There is 1 pmol Msh2-Msh3 (2 pmol ATP binding sites) in each reaction. The amount of ATPγS bound was determined using a standard curve. The curves represent the average of at least three separate experiments for Msh2-msh3Y942A or Msh2-Msh3 (± S.E.M.), respectively. HD is homoduplex DNA; loop is the +8 loop MMR DNA substrate and splayed is the 3′ NHTR splayed DNA substrate.
3.5 Msh2-msh3Y942A displays altered kinetics of ATP hydrolysis
We performed a kinetic analysis of the ATPase activity of Msh2-msh3Y942A, at a 1:1 (approximate Kd for MMR and 3′ NHTR substrates) and 1:5 (approximate Kd for homoduplex) protein to DNA ratio (Table 3). Because the effects of the splayed and 3′ flap substrates on Msh2-Msh3 kinetic properties were indistinguishable (Table 2) and because Msh2-msh3Y942A bound the splayed and 3′ flap substrates equivalently (Fig. 4), we only used the splayed substrate when analyzing Msh2-msh3Y942A. At the 1:1 ratio, the KmATP of Msh2-msh3Y942A was 2-fold lower than that of Msh2-Msh3 in the absence of DNA or in the presence of homoduplex DNA, but was unchanged in the presence of the MMR and 3′ NHTR DNA substrates (Table 3, upper panel). In contrast, at a 1:5 stoichiometry of protein to DNA (Table 3, lower panel), KmATP were ~2-fold lower for Msh2-msh3Y942A with all three DNA substrates tested. Unlike Msh2-Msh3, KmATP of Msh2-msh3Y942A in the presence of the MMR and 3′NHTR substrates was not affected by DNA concentration, indicating that increased Msh2-msh3Y942A ATP binding was not altered by complex formation with either the MMR or 3′ NHTR substrate. The lower KmATP under all DNA conditions for Msh2-msh3Y942A could be a result of more efficient ATP binding or faster nucleotide exchange. Alternatively, both subunits could be more fully occupied by ATP, which is not thought to be the case for Msh2-Msh3 [42].
Table 3.
Kinetic parameters of Msh2-msh3942A ATP hydrolysis in the presence of homoduplex, MMR and 3′ NHTR DNA substrates
| 1:1 ratio | No DNA | Homoduplex DNA | + 8 loop substrate | Splayed Y substrate |
|---|---|---|---|---|
| Vmax (nM/min) a | 498 ± 58 | 450 ± 30 | 750 ± 50 | 730 ± 40 |
| Km (μM) b | 522 ± 44 | 42.1 ± 12.4 | 12.8 ± 2.0 | 8.3 ± 3.5 |
| kcat (min−1) c | 10.0 | 9 | 15 | 14.6 |
| kcat/Km (min−1μM−1) | 0.02 | 0.21 | 1.2 | 1.8 |
| Relative to no DNA d | 1 | 10.5 | 60 | 90 |
| Hill co-efficient e | 0.9 ± 0.1 | 0.8 ± 0.09 | 1.1 ± 0.1 | 0.6 ± 0.2 |
|
| ||||
| 1:5 ratio | ||||
| Vmax (nM/min) a | 498 ± 58 | 320 ± 30 | 560 ± 60 | 390 ± 38 |
| Km (μM) b | 522 ± 44 | 10.2 ± 3.1 | 11.3 ± 3.1 | 14.3 ± 5.4 |
| kcat (min−1) c | 10.0 | 6.4 | 11.2 | 7.8 |
| kcat/Km (min−1μM−1) | 0.02 | 0.6 | 1.0 | 0.5 |
| Relative to no DNA d | 1 | 30 | 50 | 25 |
| Hill co-efficient e | 0.9 ± 0.1 | 0.82 ± 0.07 | 1.0 ± 0.2 | 0.9 ± 0.1 |
Maximum rate of ATP hydrolysis when data fit to Michaelis-Menten or Hill equation
Dissociation constant determined as the concentration of ATP at half maximal ATP hydrolysis rate data fit to Michaelis-Menten or Hill equation
Ratio of Vmax and concentration of protein
kcat/Km of Msh2-msh3Y942A in the presence of DNA substrate relative to the absence of DNA
Determined when data fit to Hill equation
Overall, despite the lower KmATP, the mutant complex had reduced hydrolytic activity compared to Msh2-Msh3. Under all conditions, the maximal rate of ATP hydrolysis was approximately 50% that of Msh2-Msh3 (compare Tables 2 and 3). One possibility is the altered msh3 nucleotide binding pocket does not bind ATP in the correct conformation and/or allows ATP to dissociate prior to hydrolysis. Alternatively, the communication between the ATPase sites of Msh2 and Msh3 may be disrupted leading to sub-optimal hydrolytic activity.
Notably, when the data were approximated by the Hill equation, the Hill coefficient for Msh2-msh3Y942A in the presence of homoduplex DNA was close to 1, whereas for Msh2-Msh3 is was 0.3. Therefore negative cooperativity in the presence of the homoduplex substrate is lost with Msh2-msh3Y942A (Table 3), indicating that an intact nucleotide binding pocket in Msh3 is required for this level of regulation. In contrast, the weak negative cooperativity in the presence of the 3′ NHTR substrate at stoichiometric concentrations was unchanged at 0.6.
3.5 ATPγS inhibition of Msh2-Msh3 ATPase activity is substrate-dependent
The ATP hydrolysis data indicate that: 1) the Msh2-Msh3 ATPase cycle is differentially affected by homoduplex, MMR and 3′ NHTR DNA substrates and 2) the Msh2-msh3Y942A complex has an altered hydrolytic cycle relative to Msh2-Msh3. But these data do not indicate what step (or steps) in the cycle are differentially regulated by DNA substrate and by the msh3Y942A mutation. To address the first question, i.e. the step(s) in the Msh2-Msh3 hydrolytic cycle modulated by DNA substrate, we characterized the ability of ATPγS to inhibit ATPase activity of Msh2-Msh3 (Fig. 6). Because ATPγS is a very weakly hydrolysable analog of ATP, when bound to Msh2-Msh3 it will effectively block hydrolysis. Furthermore, ATPγS can only bind if a nucleotide binding site has been vacated by ADP (or ATP). Therefore ATPγS inhibition is an indirect measure of nucleotide turnover within the complex.
Figure 6. Hydrolysis of ATP by Msh2-Msh3 was inhibited by ATPγS.

Increasing concentrations of ATPγS were titrated into a steady-state ATPase reaction in the presence of 50 nM protein and 250 nM DNA substrate, when present. The rates of hydrolysis were normalized to the rate in the absence of ATPγS (set at 100%). The dotted line in each panel represents the concentration of ATPγS required for 50% inhibition of ATP hydrolysis. The ATPγS titration was performed in the absence of DNA (a) or in the presence of homoduplex DNA (b), +8 loop DNA (c) or splayed Y DNA (d). (e) The concentrations of ATPγS required for 50% inhibition of Msh2-Msh3 ATPase activity in the presence of each DNA substrate.
Msh2-Msh3 ATPase reactions in the absence (Fig. 6a)or the presence of homoduplex DNA (Fig. 6b), MMR DNA substrate (Fig. 6c) or 3′ NHTR substrate (Fig. 6d) were allowed to reach steady-state, at which point ATPγS was titrated into the reactions. We determined the concentration of ATPγS required for 50% inhibition of ATPase activity (Fig. 6e). In the absence of DNA, a high concentration of ATPγS (6.3 mM) was required to achieve 50% inhibition of Msh2-Msh3 ATPase activity (Fig. 6a), consistent with the high KmATP (Table 2) and with slow ADP→ATP exchange in the absence of DNA [40]. In contrast, ATPγS efficiently inhibited Msh2-Msh3 ATPase activity in the presence of DNA, consistent with the more rapid ADP→ATP exchange observed by Wilson et al, 1999 [40]. Importantly, we observed differences depending on the DNA substrate present in the reaction. In the presence of homoduplex DNA, 50% inhibition of Msh2-Msh3 ATPase activity was achieved at 5.2 μM ATPγS (Fig. 6b), suggesting that Msh2-Msh3 binds ATPγS efficiently when bound to the non-specific substrate. In contrast, in the presence of the MMR substrate (Fig. 6c), 50% inhibition required 52.5 μM ATPγS, a 16-fold increase relative to the homoduplex substrate, indicating less efficient ATPγS binding in the presence of the MMR substrate. Notably, the 3′ NHTR substrate did not have the same effect on ATPγS binding in this assay as the MMR substrate. 50% inhibition occurred at 8.5 μM, more closely resembling inhibition in the presence of homoduplex DNA and demonstrating a clear difference in the effect of the MMR and 3′ NHTR substrates in this assay. This is in contrast to the relatively subtle differences observed in the ATPase assays and suggests that specific steps in the hydrolytic cycle are differentially regulated by DNA substrate.
We considered the possibility that ADP dissociation (following hydrolysis) was substrate-dependent, thereby differentially affecting the ability of ATPγS to bind Msh2-Msh3in the ATPγS inhibition experiments. To examine the effect of ADP on ATPγS binding, we pre-bound Msh2-Msh3 to ADP in the absence or presence of DNA substrates and then measured the amount of ATPγS bound after a 30 or 60 second incubation, by nitrocellulose filter binding (Fig. 7a). The amount of ATPγS binding in the presence of ADP was normalized to the amount of ATPγS binding in the absence of ADP, in the absence or presence of the three DNA substrates. ADP was able to inhibit ATPγS binding by Msh2-Msh3 with and without DNA. At 30 seconds, there was slightly more ATPγS binding in the presence of homoduplex DNA (0.77 ± 0.05) than in the absence of DNA (0.54 ± 0.15) or in the presence of either repair substrate (0.51± 0.16 for MMR; 0.53 ± 0.1) but binding was indistinguishable at 60 seconds. These data indicate that there is not a substantial substrate-dependent effect on ATP (ATPγS) binding in the presence of ADP and suggest that ADP turnover is not the substrate-dependent step in the Msh2-Msh3 ATPase cycle.
Figure 7. ADP alters ATPγS binding by Msh2-Msh3 and Msh2-msh3Y942A.

Filter binding assays were performed to assess ATPγS binding to (a) Msh2-Msh3 or (b) Msh2-msh3Y942A pre-bound to ADP in the absence of DNA (pink), in the presence of homoduplex DNA (light blue), in the presence of MMR DNA (+8 loop) substrate (purple) or in the presence of 3′NHTR (splayed) substrates (green) after 30 seconds or 60 seconds. The 30 second (left; solid) and 60 second (right; checkerboard) time points are shown side-by-side. The amount of ATPγS bound in the presence of ADP was normalized to the equivalent condition in the absence of ADP (dark blue) and expressed as a fraction. The mean of at least three independent experiments is plotted (± S.E.M.) for each condition.
3.6 Msh2-msh3Y942A ATPase activity is more sensitive to ATPγS inhibition than Msh2-Msh3
The two-fold reduction in the maximal ATPase activity of Msh2-msh3Y942A indicated that the FLY motif mutation alters the hydrolytic cycle of Msh2-Msh3. The wild-type DNA-binding and ATPγS binding by Msh2-msh3Y942A that we observed indicated that these are not the affected steps (Fig. 5, Tables 2 and 3). However, in contrast to Msh2-Msh3, Msh2-msh3Y942A ATPase activity was very sensitive to inhibition by ATPγS under all conditions (Fig. 6), indicating that the mutant complex is more accessible to ATPγS binding. In the absence of DNA, 50% inhibition occurred at 0.8 mM ATPγS, an 8-fold reduction. Similarly, in the presence of the DNA substrates, the concentrations of ATPγS required for 50% inhibition of Msh2-msh3Y942A ATPase activity were 4- to 11-fold lower than in the presence of wild-type Msh2-Msh3, indicating more efficient ATPγS binding. These observations are consistent with an altered nucleotide binding pocket in Msh2-msh3Y942A. One possibility is that the difference is due to increased ADP (or ATP) dissociation from the mutant complex, regardless of substrate. Importantly, the relative sensitivity to ATPγS remains dependent on DNA substrate. In the presence of homoduplex and 3′ NHTR DNA substrates, 0.6 μM and 0.75 μM ATPγS were required for 50% inhibition, respectively, while 12.5 μM ATPγS was required in the presence of the MMR substrate. This indicates that Msh2-msh3Y942 retains the ability to discriminate between DNA substrates, consistent with the gel mobility shift assays (Fig. 4).
Notably, the ability of ADP to inhibit Msh2-msh3Y942A binding to ATPγS was more substrate- and time-dependent than that of Msh2-Msh3 (Fig. 7b). At 30 and 60 seconds, ADP had little or no effect on ATPγS binding in the absence of DNA (0.88 ± 0.14 and 0.96 ± 0.16, respectively). In contrast, at 30 seconds ADP inhibited ATPγS binding to levels that were very similar to binding by Msh2-Msh3 in the presence of either repair substrates (0.57 ± 0.1 for MMR; 0.72 ± 0.1 for 3′ NHTR). However, unlike Msh2-Msh3, the inhibitory effect of ADP was lost at the 60 second time point in the presence of the MMR substrate (1.12 ± 0.17), and to a lesser extent in the presence of the 3′ NHTR substrate (0.88 ± 0.13). Finally, Msh2-msh3Y942A binding to ATPγS was enhanced by the presence of ADP in the presence of homoduplex DNA at both the 30 and 60 second time points, which may explain the loss of negative cooperativity observed in Table 3. Therefore the msh3Y942A mutation within the putative nucleotide binding pocket substantially altered ATPγS binding relative to Msh2-Msh3 binding when ADP was present.
4. Discussion
This is the first characterization of the ATPase activity of yeast Msh2-Msh3. Overall, the kinetic parameters for yeast Msh2-Msh3 in the presence of homoduplex and +8-loop DNA substrates are consistent with those for the human Msh2-Msh3 complex [40, 44, 54], although Gupta et al observed <2-fold difference in the kcat between homoduplex and +6-loop DNA substrates [44]. The effect of 3′ NHTR substrates (splayed or 3′ flaps) on Msh2-Msh3 ATPase activity has not been characterized previously, nor has the effect of different DNA concentrations. The KmATP we calculated for Msh2-Msh3 in the absence of DNA is substantially higher than that observed in studies with human Msh2-Msh3 [40, 44, 54]. This may be due to differences in the yeast and human protein complexes. However, we note that we continued the ATP titration to significantly higher concentrations than those studies, which may also account for the difference. Negative cooperativity has not been noted previously, although Owen et al (2009) did note that nucleotide binding in one subunit of human Msh2-Msh3 inhibited nucleotide binding in the other subunit in the absence of DNA [42]. Our preliminary UV cross-linking data with yeast Msh2-Msh3 similarly indicate that ATP preferentially interacts with Msh2 in the absence of DNA (R.E. and J.A.S., unpublished data).
4.1 Regulation of the Msh2-Msh3 ATPase cycle is substrate-dependent
Msh2-Msh3 binds to several different DNA structures in vitro [43] and acts in distinct pathways of DNA repair, including MMR and 3′ NHTR. Here, we demonstrated that Msh2-Msh3 distinguishes between the non-specific and the specific substrates to which it binds, as indicated by negative cooperativity in the presence of homoduplex DNA (Table 2). Furthermore, our data show that Msh2-Msh3 also distinguishes between specific MMR and 3′ NHTR substrates, most notably in the ATPγS inhibition experiments (Fig. 6). The differential sensitivity of Msh2-Msh3 ATPase activity to inhibition by ATPγS is consistent with substrate-dependent regulation of the hydrolytic cycle. Msh2-Msh3 was significantly more resistant to ATPγS inhibition in the presence of the MMR substrate compared to either homoduplex or 3′ NHTR DNA (Fig. 6). We suggest that this is not simply a result of decreased ADP→ATP exchange; ADP effectively competed for ATPγS binding similarly with all three substrates (Fig. 7), although this assay did not look directly at ADP release. With human Msh2-Msh3, Wilson et al (1999) demonstrated a 2-fold increase in ATP-dependent ADP release in the presence of a loop substrate versus a homoduplex substrate [40]. In contrast, Owen et al (2009) noted a two-fold decrease in ADP release in the presence of a loop substrate versus homoduplex, also with human Msh2-Msh3 [42]. The two groups used different assays and therefore it is difficult to compare their results; neither group tested the effect of a 3′ NHTR substrate. Nonetheless, a two-fold change in either direction would not fully account for the 6- to 10-fold differences we observed in the ATPγS inhibition assays in the presence of MMR versus homoduplex (and 3′ NHTR) substrates (Fig. 6). One possibility is that the MMR (IDL) structure inhibits or slows ATP hydrolysis by Msh2-Msh3 more than alternative DNA substrates, as has been observed in pre-steady-state experiments with MutS and Msh2-Msh6 [57, 58]. Nevertheless, these differences lead us to propose that the substrate-dependent regulation of the ATP hydrolytic cycle is critical in differentiating between repair pathways.
How might the different substrates modulate the ATPase cycle? Our previous foot printing data indicated subtle distinctions in the way that Msh2-Msh3 interacts with the +8-loop versus splayed and 3′ flap DNA structures [43]. The human Msh2-Msh3 crystal structure also indicated differences in the Msh2-Msh3-DNA interactions, depending on the size of the IDL [44]. We propose that the different DNA substrates induce signature conformational changes in the DNA-binding domains of Msh2-Msh3 that are relayed to the ATPase domains of Msh2-Msh3. These, in turn, induce specific conformational changes in the ATPase domains of Msh2 and/or Msh3 resulting in the distinct regulation of Msh2-Msh3 function in the presence of homoduplex, MMR and 3′ NHTR DNA substrates. Consistent with this hypothesis, yeast Msh2-Msh3 is more resistant to ATP-induced dissociation from a 3′ NHTR substrate than from an MMR substrate [43]. It is worth noting that Owen et al (2009) similarly noted differences in human Msh2-Msh3 ATP binding and hydrolysis in the presence of homoduplex versus a +8 loop versus a trinucleotide repeat structure [42].
4.2 Msh2-msh3Y942A alters DNA substrate-dependent regulation of ATP binding and hydrolysis
In vivo, msh3Y942A, a mutation within the putative nucleotide binding pocket of Msh3, disrupted MMR while leaving 3′ NHTR largely intact, suggesting different requirements for ATP binding and/or hydrolysis in the two pathways [29]. The distinct substrate-dependent enzyme kinetics of Msh2-Msh3 suggested that these differences might underlie the different molecular requirements observed in vivo. To assess the extent to which the FLY motif within the nucleotide binding pocket regulates these differences, we characterized the ATP binding and hydrolysis activities of Msh2-msh3Y942A. Based on structural information, the msh3Y942A change was predicted to disrupt the putative nucleotide sandwich in the Msh3 nucleotide binding pocket [29, 44]. If this is the case, our data strongly indicate that the sandwich is not required for ATP-binding, but rather regulates the properties of nucleotide binding and “nucleotide turnover”, which we define as the combined effects on nucleotide hydrolysis and/or exchange.
We observed two major differences between Msh2-Msh3 and Msh2-msh3Y942A in our analysis. First, Msh2-msh3Y942A did not exhibit negative cooperativity with respect to ATP binding/hydrolysis in the presence of homoduplex DNA, illustrating that a functional Msh3 nucleotide binding pocket is required for this regulation of ATP binding and catalysis. Second, the ATPase activity of Msh2-msh3Y942A was significantly more sensitive to ATPγS inhibition than Msh2-Msh3 (Fig. 5) although it retained its ability to distinguish between DNA substrates in this assay. Thus msh3Y942A does not block ATP binding, but rather appears to alter the mode of ATP binding. Increased ATPγS binding by Msh2-msh3Y942A in the presence of ADP is consistent with this interpretation. One possibility is that reduced conformational constraints within the nucleotide binding pocket allow faster nucleotide dissociation (prior to hydrolysis) and/or alternative positioning of nucleotide within the nucleotide binding pocket. This would, in turn, inhibit hydrolysis (Table 3)by reducing the probability of finding the nucleotide in the pocket and correctly placed to allow cleavage.
It is remarkable that, despite its altered ATP hydrolysis and nucleotide exchange activities, msh3Y942A retained ~84% activity in 3′ NHTR [29]. These activities are clearly not essential in that pathway, but are critical for MMR, which was largely abrogated in the presence of this mutation. It may simply be that the reduced catalytic activity of Msh2-msh3Y942A is sufficient for 3′ NHTR, but not MMR. However, the increased sensitivity of ATPase activity to inhibition by ATPγS and enhanced ATPγS binding in the presence of ADP (altered “nucleotide turnover”) suggests that the highly ordered sequence of conformational changes within the ATPase domain required for MMR has been disrupted. Notably, mutation of the highly conserved glycine in the Msh3 Walker A motif (msh3G796A or msh3G796D), predicted to block ATP binding, had a much more substantial effect on 3′ NHTR than msh3Y942A [29]. Therefore, while Msh3 ATP binding is essential for 3′ NHTR, efficient ATP hydrolysis may not be required.
4.3 ATP and DNA repair substrates cooperate to regulate Msh2-Msh3 repair pathway selection
Based on our data, we suggest that Msh2-Msh3 shuttles between free, target search and distinct repair states (Fig. 8). The repair state is determined by the specific DNA substrate, information that is then transmitted to the ATPase domain to regulate nucleotide binding, hydrolysis and exchange. In the absence of DNA, Msh2-Msh3 exhibits basal ATPase activity, characterized by a low affinity for ATP (high Km). Previous work with human Msh2-Msh3 indicates that DNA-binding is inhibited in the presence of ATP and that human Msh2-Msh3 likely binds DNA in an ADP-bound form in vivo [42]. Upon binding homoduplex DNA, the affinity of Msh2-Msh3 for ATP increases relative to free Msh2-Msh3 (see decreased Km in Tables 2 and 3), resulting in an increase in hydrolysis and subsequent nucleotide exchange (ADP for ATP). Furthermore, the negative cooperativity observed in the presence of homoduplex DNA indicates altered interactions with ATP that we suggest prevents the initiation of repair (Fig. 8). This homoduplex-dependent negative cooperativity requires correct nucleotide binding in the Msh3 pocket; it is lost in Msh2-msh3Y942A. Therefore, the MMR defect in msh3Y942A could be, in part, a result of unregulated initiation of MMR, i.e. repair in the absence of a mismatch.
Figure 8. Distinct DNA repair substrates differentially regulate Msh2-Msh3 ATP hydrolytic cycle.
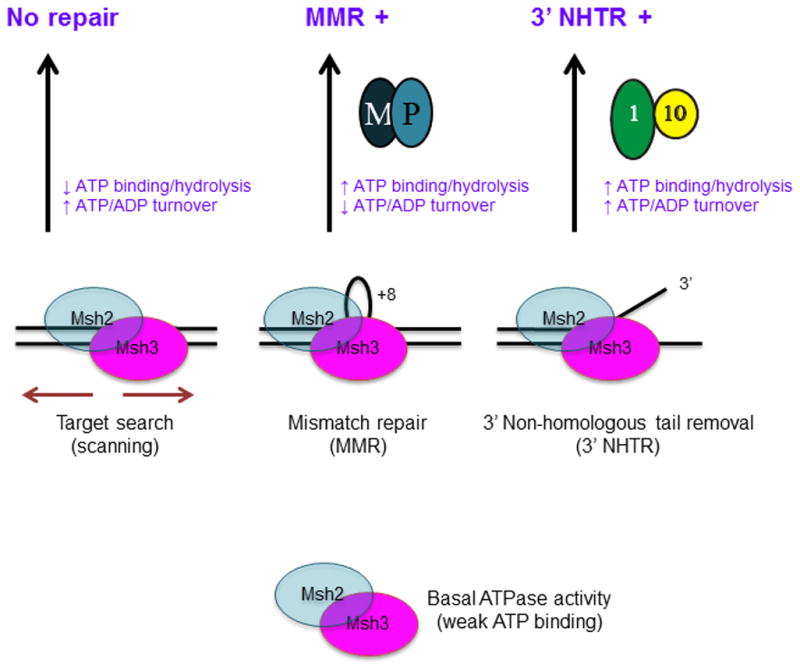
We propose that Msh2-Msh3 shuttles between different free and DNA bound states. The repair mechanism activated depends on the specific conformational changes induced by the different DNA substrates. These conformational changes then induce distinct changes within the ATPase domain, modulating its activities. Increased ATP binding and higher catalytic efficiency by Msh2-Msh3 in the presence of MMR and 3′ NHTR substrates result in repair. Increased ATP hydrolysis (relative to homoduplex) and slowed “nucleotide turnover” (indicated by reduced ATPγS inhibition of ATPase activity relative to homoduplex and 3′ NHTR) in response to the MMR substrate are predicted to be critical for the proper initiation of Msh2-Msh3-mediated MMR, perhaps allowing interactions with Mlh1-Pms1 (M-P). In contrast, 3′ NHTR does not require slowed “nucleotide turnover”, indicating intact interactions between Msh2-Msh3 and Rad1-Rad10 (1–10). Altered ATP binding/hydrolysis by Msh2-msh3Y942A in the presence of homoduplex DNA, indicated by loss of negative cooperativity (Table 3), could promote inappropriate initiation of MMR. Decreased ATP hydrolysis and/or increased nucleotide turnover by Msh2-msh3Y942A in the presence of MMR substrates inactivate MMR, but do not significantly impair 3′ NHTR in vivo [29]. See text for additional details.
Once Msh2-Msh3 locates an IDL, negative cooperativity is relieved, allowing the efficient and appropriate initiation of MMR (Fig. 8). The ATPγS inhibition experiments (Fig. 6) indicate that nucleotide hydrolysis and/or exchange, which together regulate the timing of the ATP hydrolytic cycle, is slower in the presence of an IDL than in the presence of homoduplex (or 3′ NHTs), which may be necessary to interact with Mlh complexes (M-P; Fig. 8) [59] and/or to generate a “sliding clamp” conformation [25, 30]. When Msh2-Msh3 is bound to the 3′NHT, the requirements for the hydrolytic cycle are relaxed (Fig. 8); ATPγS inhibition is more efficient than in the presence of the MMR substrate (Fig. 6) but is nonetheless compatible with 3′ NHTR in vivo .
While our data do not distinguish between the relative contributions of Msh2 and Msh3 to nucleotide binding, hydrolysis and exchange, they are consistent with the model proposed by Owen et al (2009) [42]. In this model, Msh2-Msh3 binds DNA with Msh2 in an ADP-bound form and Msh3 in a nucleotide-free state. IDL binding stimulates ATP binding by Msh3. ATP hydrolysis in Msh3 then stimulates ADP→ATP exchange in Msh2, which triggers sliding or dissociation away from the IDL. If no hydrolysis occurs in Msh3 (as would be the case when ATPγS is bound), then ADP→ATP exchange in Msh2 would be suppressed, as indicated by reduced ATPγS binding in our ADP competition experiment (Fig. 7). The substrate-dependence of ATPγS inhibition of ATP hydrolysis (Fig. 6) allows us to propose that the rate of hydrolysis following ATP binding (in Msh3 and/or Msh2) is slowed in the presence of the MMR but not in the presence of the 3′ NHTR substrate.
In Msh2-msh3Y942A, inhibition of ATPγS binding is relieved (to varying degrees) with all DNA substrates, consistent with the prediction that msh3Y942A interferes with the coordination between the Msh2 and Msh3 nucleotide binding sites, facilitating inappropriate nucleotide binding in one or both sites. MMR could be disrupted by the altered timing and/or reduced efficiency of the hydrolytic cycle and/or affect interactions with Mlh1-Pms1. The conformational changes induced by ATP binding are essential for Msh2-Msh6 to interact with Mlh1-Pms1 [59]. Similar changes may be required for Msh2-Msh3 to functionally interact with Mlh1-Pms1 and could be inhibited by increased “nucleotide turnover” in Msh2-msh3Y942A. In contrast, interactions between Msh2-msh3Y942A and Rad1-Rad10 in 3′ NHTR are apparently unaffected or not sufficiently impaired to block 3′ NHTR (Fig. 8). Msh2-Msh3 thus appears to use different modes of nucleotide binding and hydrolysis in the presence of distinct DNA substrates to authorize and regulate the initiation of repair and possibly repair pathway selection.
Highlights.
Msh2-Msh3 requires ATP binding for its function in MMR and 3′NHTR
The kinetics of ATP hydrolysis and turnover by Msh2-Msh3 are DNA substrate-specific
Msh2-Msh3 ATP hydrolysis displays negative cooperativity with homoduplex DNA
A mutated Msh3 ATP binding pocket relieves negative cooperativity and alters turnover
Repair substrates and ATP coordinate to differentially modulate Msh2-Msh3 activity
Acknowledgments
We thank Dr. Mark Sutton and Dr. Manju Hingorani for critical discussions and for reviewing the manuscript. We gratefully acknowledge the technical support of Dr. Andrew Bukata and Evan Myers. Work in the Surtees lab is supported by NIH GM087459. Work in the Bianco lab is supported by NIH GM100156. The sponsor was not involved in conducting this research or preparing this manuscript.
Footnotes
Conflict of Interest Statement
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hsieh P, Yamane K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mechanisms of Ageing and Development. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li GM. Mechanisms and functions of DNA mismatch repair. Cell Res. 2008;18:85–98. doi: 10.1038/cr.2007.115. [DOI] [PubMed] [Google Scholar]
- 3.Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
- 4.Sia E, Kokoska R, Dominska M, Greenwell P, Petes T. Microsatellite instability in yeast: dependence on repeat unit size and DNA mismatch repair genes. Mol Cell Biol. 1997;17:2851–2858. doi: 10.1128/mcb.17.5.2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jensen LE, Jauert PA, Kirkpatrick DT. The Large Loop Repair and Mismatch Repair Pathways of Saccharomyces cerevisiae Act on Distinct Substrates During Meiosis. Genetics. 2005;170:1033–1043. doi: 10.1534/genetics.104.033670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kunkel TA, Erie DA. DNA MISMATCH REPAIR. Annual Review of Biochemistry. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 7.Harrington JM, Kolodner RD. Saccharomyces cerevisiae Msh2-Msh3 Acts in Repair of Base-Base Mispairs. Mol Cell Biol. 2007;27:6546–6554. doi: 10.1128/MCB.00855-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans E, Alani E. Roles for Mismatch Repair Factors in Regulating Genetic Recombination. Mol Cell Biol. 2000;20:7839–7844. doi: 10.1128/mcb.20.21.7839-7844.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Surtees JA, Argueso JL, Alani E. Mismatch repair proteins: key regulators of genetic recombination. Cytogen Genome Res. 2004;107:146–159. doi: 10.1159/000080593. [DOI] [PubMed] [Google Scholar]
- 10.Paques F, Haber JE. Multiple Pathways of Recombination Induced by Double-Strand Breaks in Saccharomyces cerevisiae. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nicholson A, Hendrix M, Jinks-Robertson S, Crouse GF. Regulation of Mitotic Homeologous Recombination in Yeast: Functions of Mismatch Repair and Nucleotide Excision Repair Genes. Genetics. 2000;154:133–146. doi: 10.1093/genetics/154.1.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kirkpatrick DT, Petes TD. Repair of DNA loops involves DNA-mismatch and nucleotide-excision repair proteins. Nature. 1997;387:929–931. doi: 10.1038/43225. [DOI] [PubMed] [Google Scholar]
- 13.Kearney HM, Kirkpatrick DT, Gerton JL, Petes TD. Meiotic Recombination Involving Heterozygous Large Insertions in Saccharomyces cerevisiae: Formation and Repair of Large, Unpaired DNA Loops. Genetics. 2001;158:1457–1476. doi: 10.1093/genetics/158.4.1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bardwell AJ, Bardwell L, Johnson DK, Friedberg EC. Yeast DNA recombination and repair proteins Rad1 and Rad10 consititute a complex in vivo mediated by localized hydrophobic domains. Mol Microbiol. 1993;8:1177–1188. doi: 10.1111/j.1365-2958.1993.tb01662.x. [DOI] [PubMed] [Google Scholar]
- 15.Bardwell AJ, Bardwell L, Tomkinson AE, Friedberg EC. Specific cleavage of model recombination and repair intermediates by the yeast Rad1-RAD10 DNA endonuclease. Science. 1994;265:2082–2085. doi: 10.1126/science.8091230. [DOI] [PubMed] [Google Scholar]
- 16.Davies AA, Friedberg EC, Tomkinson AE, Wood RD, West SC. Role of the Rad1 and Rad10 Proteins in Nucleotide Excision Repair and Recombination. J Biol Chem. 1995;270:24638–24641. doi: 10.1074/jbc.270.42.24638. [DOI] [PubMed] [Google Scholar]
- 17.Sugawara N, Paques F, Colaiacovo M, Haber JE. Role of Saccharomyces cerevisiae Msh2 and Msh3 repair proteins in double-strand break-induced recombination. Proceedings of the National Academy of Sciences. 1997;94:9214–9219. doi: 10.1073/pnas.94.17.9214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lyndaker AM, Alani E. A tale of tails: insights into the coordination of 3′ end processing during homologous recombination. BioEssays. 2009;31:315–321. doi: 10.1002/bies.200800195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li F, Dong J, Pan X, Oum JH, Boeke JD, Lee SE. Microarray-Based Genetic Screen Defines SAW1, a Gene Required for Rad1/Rad10-Dependent Processing of Recombination Intermediates. Molecular Cell. 2008;30:325–335. doi: 10.1016/j.molcel.2008.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li F, Dong J, Eichmiller R, Holland C, Minca E, Prakash R, Sung P, Yong Shim E, Surtees JA, Eun Lee S. Role of Saw1 in Rad1/Rad10 complex assembly at recombination intermediates in budding yeast. EMBO J. 2013;32:461–72. doi: 10.1038/emboj.2012.345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Junop MS, Oblomova G, Rausch K, Hsieh P, Yang W. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol Cell. 2001;7:1–12. doi: 10.1016/s1097-2765(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 22.Obmolova G, Ban C, Hsieh P, Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 23.Lamers MH, Perrakis A, Enzlin JH, Winterwerp HHK, de Wind N, Sixma TK. The crystal structure of DNA mismatch repair protein MutS binding to a G[middot]T mismatch. Nature. 2000;407:711–717. doi: 10.1038/35037523. [DOI] [PubMed] [Google Scholar]
- 24.Warren JJ, Pohlhaus TJ, Changela A, Iyer RR, Modrich PL, Beese Lorena S. Structure of the Human MutS[alpha] DNA Lesion Recognition Complex. Molecular Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Qiu R, DeRocco VC, Harris C, Sharma A, Hingorani MM, Erie DA, Weninger KR. Large conformational changes in MutS during DNA scanning, mismatch recognition and repair signalling. EMBO J. 2012;31:2528–2540. doi: 10.1038/emboj.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sharma A, Doucette C, Biro FN, Hingorani MM. Slow Conformational changes in Muts and Dna direct Ordered Transitions Between Mismatch Search, Recognition and Signaling of Dna Repair. Journal of Molecular Biology. doi: 10.1016/j.jmb.2013.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pieniazek Susan N, Hingorani Manju M, Beveridge DL. Dynamical Allosterism in the Mechanism of Action of DNA Mismatch Repair Protein MutS. Biophysical Journal. 2011;101:1730–1739. doi: 10.1016/j.bpj.2011.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mukherjee S, Feig M. Conformational Change in MSH2-MSH6 upon Binding DNA Coupled to ATPase Activity. Biophysical Journal. 2009;96:L63–L65. doi: 10.1016/j.bpj.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kumar C, Williams GM, Havens B, Dinicola MK, Surtees JA. Distinct Requirements within the Msh3 Nucleotide Binding Pocket for Mismatch and Double-Strand Break Repair. Journal of Molecular Biology. 2013;425:1881–1898. doi: 10.1016/j.jmb.2013.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gradia S, Acharya S, Fishel R. The Human Mismatch Recognition Complex hMSH2-hMSH6 Functions as a Novel Molecular Switch. Cell. 1997;91:995–1005. doi: 10.1016/s0092-8674(00)80490-0. [DOI] [PubMed] [Google Scholar]
- 31.Gradia S, Acharya S, Fishel R. The Role of Mismatched Nucleotides in Activating the hMSH2-hMSH6 Molecular Switch. J Biol Chem. 2000;275:3922–3930. doi: 10.1074/jbc.275.6.3922. [DOI] [PubMed] [Google Scholar]
- 32.Mendillo ML, Putnam CD, Mo AO, Jamison JW, Li S, Woods VL, Kolodner RD. Probing DNA- and ATP-mediated Conformational Changes in the MutS Family of Mispair Recognition Proteins Using Deuterium Exchange Mass Spectrometry. Journal of Biological Chemistry. 2010;285:13170–13182. doi: 10.1074/jbc.M110.108894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hess MT, Das Gupta R, Kolodner RD. Dominant Saccharomyces cerevisiae msh6 mutations cause increased mispair binding and decreased dissociation from mispairs by Msh2-Msh6 in the presence of ATP. J Biol Chem. 2002;277:25545–25553. doi: 10.1074/jbc.M202282200. [DOI] [PubMed] [Google Scholar]
- 34.Kijas AW, Studamire B, Alani E. msh2 Separation of Function Mutations Confer Defects in the Initiation Steps of Mismatch Repair. Journal of Molecular Biology. 2003;331:123–138. doi: 10.1016/s0022-2836(03)00694-6. [DOI] [PubMed] [Google Scholar]
- 35.Alani E, Lee JY, Schofield MJ, Kijas AW, Hsieh P, Yang W. Crystal Structure and Biochemical Analysis of the MutS{middle dot}ADP{middle dot}Beryllium Fluoride Complex Suggests a Conserved Mechanism for ATP Interactions in Mismatch Repair. J Biol Chem. 2003;278:16088–16094. doi: 10.1074/jbc.M213193200. [DOI] [PubMed] [Google Scholar]
- 36.Hess MT, Mendillo ML, Mazur DJ, Kolodner RD. Biochemical basis for dominant mutations in the Saccharomyces cerevisiae MSH6 gene. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:558–563. doi: 10.1073/pnas.0510078103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Acharya S, Foster PI, Brooks R, Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 38.Gradia S, Subramanian, Wilson T, Acharya S, Makhov A, Griffith J, Fishel R. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp in mismatched DNA. Mol Cell. 1999;3:255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 39.Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P. Interaction of Escherichia coli MutS and MutL at a DNA mismatch. J Biol Chem. 2001;276:28291–28299. doi: 10.1074/jbc.M103148200. [DOI] [PubMed] [Google Scholar]
- 40.Wilson T, Guerrette S, Fishel R. Dissociation of Mismatch Recognition and ATPase Activity by hMSH2-hMSH3. J Biol Chem. 1999;274:21659–21664. doi: 10.1074/jbc.274.31.21659. [DOI] [PubMed] [Google Scholar]
- 41.Tian L, Gu L, Li G-M. Distinct Nucleotide Binding/Hydrolysis Properties and Molar Ratio of MutS{alpha} and MutS{beta} Determine Their Differential Mismatch Binding Activities. J Biol Chem. 2009;284:11557–11562. doi: 10.1074/jbc.M900908200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Owen BAL, Lang HW, McMurray CT. The nucleotide binding dynamics of human MSH2-MSH3 are lesion dependent. Nat Struct Mol Biol. 2009;16:550–557. doi: 10.1038/nsmb.1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Surtees JA, Alani E. Mismatch Repair Factor MSH2-MSH3 Binds and Alters the Conformation of Branched DNA Structures Predicted to form During Genetic Recombination. Journal of Molecular Biology. 2006;360:523–536. doi: 10.1016/j.jmb.2006.05.032. [DOI] [PubMed] [Google Scholar]
- 44.Gupta S, Gellert M, Yang W. Mechanism of mismatch recognition revealed by human MutSβ bound to unpaired DNA loops. Nat Struct Mol Biol. 2012;19:72–78. doi: 10.1038/nsmb.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kantartzis A, Williams Gregory M, Balakrishnan L, Roberts Rick L, Surtees Jennifer A, Bambara Robert A. Msh2-Msh3 Interferes with Okazaki Fragment Processing to Promote Trinucleotide Repeat Expansions. Cell Reports. 2012;2:216–222. doi: 10.1016/j.celrep.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Habraken Y, Sung P, Prakash L, Prakash S. Binding of insertion/deletion DNA mismatches by the heterodimer of yeast mismatch repair proteins MSH2 and MSH3. Current Biology. 1996;6:1185–1187. doi: 10.1016/s0960-9822(02)70686-6. [DOI] [PubMed] [Google Scholar]
- 47.Lee SD, Surtees JA, Alani E. Saccharomyces cerevisiae MSH2-MSH3 and MSH2-MSH6 Complexes Display Distinct Requirements for DNA Binding Domain I in Mismatch Recognition. Journal of Molecular Biology. 2007;366:53–66. doi: 10.1016/j.jmb.2006.10.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wong I, Lohman TM. A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proceedings of the National Academy of Sciences. 1993;90:5428–5432. doi: 10.1073/pnas.90.12.5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bianco PR, Xu C, Chi M. Type I restriction endonucleases are true catalytic enzymes. Nucleic Acids Research. 2009;37:3377–3390. doi: 10.1093/nar/gkp195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dixon M. In: Enzymes. Webb EC, editor. New York: Academic Press; 1958. [Google Scholar]
- 51.Studamire B, Price G, Sugawara N, Haber JE, Alani E. Separation-of-Function Mutations in Saccharomyces cerevisiae MSH2 That Confer Mismatch Repair Defects but Do Not Affect Nonhomologous-Tail Removal during Recombination. Mol Cell Biol. 1999;19:7558–7567. doi: 10.1128/mcb.19.11.7558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bianco PR, Hurley EM. The Type I Restriction Endonuclease EcoR124I, Couples ATP Hydrolysis to Bidirectional DNA Translocation. Journal of Molecular Biology. 2005;352:837–859. doi: 10.1016/j.jmb.2005.07.055. [DOI] [PubMed] [Google Scholar]
- 53.Slocum SL, Buss JA, Kimura Y, Bianco PR. Characterization of the ATPase Activity of the Escherichia coli RecG Protein Reveals that the Preferred Cofactor is Negatively Supercoiled DNA. Journal of Molecular Biology. 2007;367:647–664. doi: 10.1016/j.jmb.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Owen BAL, Yang Z, Lai M, Gajec M, Badger Jd, Hayes JJ, Edelmann W, Kucherlapati R, Wilson TM, McMurray CT. (CAG)n-hairpin DNA binds to Msh2-Msh3 and changes properties of mismatch recognition. Nat Struct Mol Biol. 2005;12:663–670. doi: 10.1038/nsmb965. [DOI] [PubMed] [Google Scholar]
- 55.Modrich P, Lahue R. Mismatch Repair in Replication Fidelity, Genetic Recombination, and Cancer Biology. Annual Review of Biochemistry. 1996;65:101–133. doi: 10.1146/annurev.bi.65.070196.000533. [DOI] [PubMed] [Google Scholar]
- 56.Bjornson KP, Allen DJ, Modrich P. Modulation of MutS ATP hydrolysis by DNA cofactors. Biochemistry. 2000;39:3176–3183. doi: 10.1021/bi992286u. [DOI] [PubMed] [Google Scholar]
- 57.Antony E, Hingorani MM. Mismatch Recognition-Coupled Stabilization of Msh2-Msh6 in an ATP-Bound State at the Initiation of DNA Repair†. Biochemistry. 2003;42:7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Antony E, Hingorani MM. Asymmetric ATP Binding and Hydrolysis Activity of the Thermus aquaticus MutS Dimer Is Key to Modulation of Its Interactions with Mismatched DNA†. Biochemistry. 2004;43:13115–13128. doi: 10.1021/bi049010t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hargreaves VV, Putnam CD, Kolodner RD. Engineered Disulfide-forming Amino Acid Substitutions Interfere with a Conformational Change in the Mismatch Recognition Complex Msh2-Msh6 Required for Mismatch Repair. Journal of Biological Chemistry. 2012;287:41232–41244. doi: 10.1074/jbc.M112.402495. [DOI] [PMC free article] [PubMed] [Google Scholar]