

Abstract
Recent evidence suggests that dystonia, a movement disorder characterized by sustained involuntary muscle contractions, can be associated with cerebellar abnormalities. The basis for how functional changes in the cerebellum can cause dystonia is poorly understood. Here we identify alterations in physiology in Atcayji-hes mice which in addition to ataxia, have an abnormal gait with hind limb extension and toe walking, reminiscent of human dystonic gait. No morphological abnormalities in the brain accompany the dystonia, but partial cerebellectomy causes resolution of the stiff-legged gait, suggesting that cerebellar dysfunction contributes to the dystonic gait of Atcayji-hes mice. Recordings from Purkinje and deep cerebellar nuclear (DCN) neurons in acute brain slices were used to determine the physiological correlates of dystonia in the Atcayji-hes mice. Approximately 50% of cerebellar Purkinje neurons fail to display the normal repetitive firing characteristic of these cells. In addition, DCN neurons exhibit increased intrinsic firing frequencies with a subset of neurons displaying bursts of action potentials. This increased intrinsic excitability of DCN neurons is accompanied by a reduction in after-hyperpolarization currents mediated by small-conductance calcium-activated potassium (SK) channels. An activator of SK channels reduces DCN neuron firing frequency in acute cerebellar slices and improves the dystonic gait of Atcayji-hes mice. These results suggest that a combination of reduced Purkinje neuron activity and increased DCN intrinsic excitability can result in a combination of ataxia and a dystonia-like gait in mice.
Keywords: dystonia, ataxia, cerebellum, Purkinje cells, deep cerebellar nuclei, patch-clamp, electrophysiology, mutant mice
Introduction
Dystonia is a disorder of movement characterized by sustained involuntary muscle contractions, which forcefully distort the body into typical postures (Berardelli et al., 1998; Fahn, 1984; Frucht, 2013). Voluntary movement exacerbates the abnormal movements. Only in severe cases is muscle activity recorded in subjects at complete rest (Berardelli et al., 1998). A role for the striatum in dystonia is evident from numerous studies (Marsden et al., 1985)(reviewed in (Lehericy et al., 2013)). Recent evidence also suggests a role for the cerebellum in the pathophysiology of some forms of dystonia (Lehericy et al., 2013). For example, a reduction in cerebellar gray matter has been described in patients with focal dystonia (Delmaire et al., 2007), and cerebellar dysfunction appears to be the primary cause for dystonia in patients who display marked dystonia affecting the neck, vocal cords, face, and upper and lower limbs, and face (Le Ber et al., 2006). Moreover, patients with primary dystonia have increased metabolic activity in the cerebellum (Eidelberg et al., 1998) and imaging studies of patients with the most common inherited form of dystonia, DYT1, have revealed abnormalities in cerebellar outflow (Argyelan et al., 2009). These new data suggest that dystonia is a network disorder involving multiple brain regions, including the cerebellum (Neychev et al., 2011; Niethammer et al., 2011).
Rodent models also provide evidence supporting a role for the cerebellum in dystonia. These models include the tottering mouse (Campbell and Hess, 1999), the genetically dystonic rat (Lorden et al., 1988), and a mouse model of rapid onset dystonia Parkinsonism (Calderon et al., 2011). The movement disorder in these models can be attributed to defects in the cerebellum since the dystonia resolves following cerebellar lesioning (Calderon et al., 2011; Campbell and Hess, 1999; LeDoux et al., 1993). Both primary generalized dystonia in humans and the dystonia seen in these animal models are not accompanied by degenerative neuropathology (den Dunnen, 2013) suggesting that physiologic dysfunction underlies the motor abnormalities.
A cerebellar link to dystonia is further suggested by the manifestations of mutations in the human ATCAY gene and its homologs in the mouse and rat. A mutation in human ATCAY causes a rare form of ataxia isolated to the Grand Cayman Islands (Bomar et al., 2003). Mutations in the homologous mouse gene Atcay, causes a variable motor phenotype. In sidewinder mice, there is complete absence of expression of caytaxin, the protein encoded by Atcay and results in a marked ataxic phenotype (Sikora et al., 2012). An Intracisternal A-particle (IAP) element insertion in the mouse Atcay gene causes caytaxin to be expressed at a very low, but detectable level and results in dystonia in addition to ataxia in the Hesitant mouse, Atcayji-hes (Bomar et al., 2003; Kapfhamer et al., 1996; LeDoux, 2011; Sikora et al., 2012). A similar insertion mutation in the rat homolog is responsible for the movement disorder in the genetically dystonic rat (Xiao and Ledoux, 2005), which exhibits generalized dystonia resulting in premature death. Physiologic abnormalities in the cerebellum have been documented in the genetically dystonic rat. In anesthetized dystonic rats, Purkinje cell simple spike firing rates are reduced (Stratton et al., 1988), whereas recordings from awake dystonic rats indicate no difference in Purkinje cell simple spike firing (LeDoux and Lorden, 2002), but do show abnormal burst firing patterns in neurons from the deep cerebellar nuclei (DCN) (LeDoux et al., 1998). The mechanism for these alterations in firing has not been established. Here, using the Atcayji-hes mice mentioned above, we describe a dystonic phenotype that primarily affects gait. We suggest that the phenotype is of cerebellar origin. Using recordings from brain slices, we show that both reduced Purkinje neuron activity and increased DCN intrinsic excitability are associated with cerebellar dystonia. Importantly, pharmacological activation of small-conductance calcium-activated potassium channels improves both the DCN firing abnormalities and the dystonic phenotype in these mice. Our findings demonstrate that correcting electrophysiological defects in cerebellar dystonia is a potential therapeutic strategy.
Materials and methods
Mice and genotyping
All animal procedures were approved by the University of Michigan Committee on the Use and Care of Animals. Hesitant mice (C3H-Atcayji-hes/J, stock number 001904) were obtained from the Jackson Laboratory and bred in-house. Genotyping was done as described previously (Bomar et al., 2003). Briefly, mouse tail biopsies were performed between post-natal day 6 and 8 and genomic DNA was extracted using the PUREGENE® DNA Purification Kit according to the manufacturer’s protocol (Gentra, Cat. No. 158222 or 158267). Genomic PCR for all strains was performed by amplifying a target region from 25 ng of genomic DNA with the following buffer conditions: PCR Buffer set (Invitrogen, MgCl2 Cat. No. Y02016 and 10× PCR buffer Cat. No. Y02028). The primers used were: 5′-cctccctgcacagacacaatag-3′, 5′-gggatgttagggtttaccacca-3′, 5′-tacaacagaattccagggtcca-3′.
Phenotype characterization
Video sequences of the mice at 12, 21, 35 days and 5 months were analyzed to characterize the phenotype. Mice were examined both in their home cage as well as in an open-field situation. Mice were examined for at least 10 min with video recordings of between 2 and 5 min. Video recordings were taken of mice that were subjected to partial cerebellectomy 1hr, 2hr and 24hr after surgery. Gait analysis was performed from the footprint pattern of mice. The footpads of mice were dipped in non-toxic paint and mice were allowed to walk on a strip of paper. Gait analysis was performed on three consecutive steps, where there were no pauses in the gait. Hind forefoot discordance was measured as the distance between the hind foot and forefoot pads in each step for three sequential steps. Stride length was measured between the central pads of the two consecutive prints on each side. Base width was measured between as the perpendicular distance between lines connecting consecutive paw prints on each side. Rotarod analysis was performed as previously described (Shakkottai et al., 2004).
Partial cerebellectomies and histology
Mice were anesthetized with vaporized isoflurane (4% for induction, 1.5% for maintenance). An incision was made over the skull along the midline and hydrogen peroxide was used to expose the skull and remove connective tissue. Anterior-posterior and medial-lateral coordinates were calculated from Bregma and the dorsal-ventral coordinates were calculated from the dorsal surface. Measurements were made on an experimentally flat skull. The area over DCN was removed by drilling and removing the skull flap. Using a 26 gauge syringe needle, the area of the DCN was damaged, with partial tissue removal. Carprofen was administered prophylactically for analgesia. 24–48 hours following surgery, brains were collected and fresh frozen using 2-methylbutane. 40 micron sections were obtained using a cryostat and mounted on superfrost slides. Nissl staining was performed to confirm the site of the lesion.
Cerebellar slice recordings
Preparation of brain slices for electrophysiological recordings were done as described previously (Shakkottai et al., 2004; Shakkottai et al., 2011). Mice were anesthetized by isoflurane inhalation, the brains were extracted and chilled in an ice-cold solution containing the following (in mM): 87 NaCl, 2.5 KCl, 25 NaHCO3, 1 NaH2PO4, 0.5 CaCl2, 7 MgCl2, 75 sucrose, and 10 glucose, bubbled with 5% CO2/95% O2. Parasagittal cerebellar slices (300 μm thick) were cut using a vibratome for Purkinje cell recordings. For DCN recordings coronal sections were produced in the same manner (300 μm thick). Slices were incubated at 33°C in artificial CSF (ACSF) containing the following (in mM): 125 NaCl, 3.5 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2, and 10 glucose, bubbled with 5% CO2 + 95% O2 (carbogen) for 45 min. Slices were then placed in an recording chamber and perfused continuously with warmed (33°C) carbogen-bubbled ACSF at 2–3 ml/min. Recordings were performed from visually identified Purkinje neurons or DCN neurons for cell attached and whole-cell recordings in parasagittal or coronal cerebellar slices respectively. For whole cell recordings from Purkinje neurons, borosilicate glass patch pipettes (with resistances of 3–5 MΩ) were filled with internal recording solution containing the following (in mM): 119 K gluconate, 2 Na gluconate, 6 NaCl, 2 MgCl2, 10 EGTA, 10 HEPES, 14 Tris-phosphocreatine, 4 MgATP, and 0.3 Tris-GTP. Whole-cell recordings were made in ACSF 1–5 h after slice preparation using an Axopatch 200B amplifier, Digidata 1440A interface, and pClamp-10 software (MDS Analytical Technologies). Voltage data were acquired in the fast current-clamp mode of the amplifier and filtered at 2 kHz. The fast current-clamp mode is necessary to reduce distortion of action potentials observed when patch-clamp amplifiers are used in current-clamp mode (Magistretti et al., 1996; Swensen and Bean, 2003). Series resistance was monitored but not compensated; cells were rejected if the series resistance exceeded 15 MΩ. Data were digitized at 100 kHz. Voltage traces were corrected for a ~10 mV liquid junction potential. A synaptic inhibitor cocktail containing 50 μM picrotoxin to block GABAA receptors in addition to 10 μM DNQX and 5 μM CPP to block glutamatergic transmission was used when indicated.
Immunohistochemistry
Mice were transcardially perfused with 0.01M Phosphate Buffered Saline (PBS) followed by 4% paraformaldehyde in phosphate buffered saline. Brains were removed, post-fixed for 24 hours, and transferred to 20% sucrose. 40 μm sagittal sections through the cerebellum were generated using a cryostat (Leica 3050S, −19°C) and placed in PBS. Free-floating sections were washed with PBS containing 0.1% Triton-X-100 (PBS-Tx), blocked in 0.3% H2O2 followed by 5% normal donkey serum, and incubated overnight in primary antibody (1:1000 mouse anti-calbindin, Swant #300; 1:2000 rabbit anti-glial fibrillary acidic protein, Dako Z0334). Sections were rinsed with PBS-Tx, incubated in secondary antibody for one hour (1:500 biotin-conjugated donkey anti-rabbit or donkey anti-mouse, Jackson Immunoresearch 715-065-152, 715-065-150, respectively), followed by 2 hours in ABC solution (Vector Laboratories PK-6100). Sections were exposed to 3,3′-diaminobenzidine (DAB – Sigma D4293), rinsed with PBS, mounted onto gelatin-coated slides, dehydrated, and cover slipped with Permount (Fisher Scientific). Staining was visualized under bright field microscopy using 2.5X and 10X objectives. Molecular layer thickness and Purkinje cell counts were performed as described previously (Shakkottai et al., 2011).
Western blotting
Whole brain membranes were purified from brain lysates prepared from a pool of 5 adult mouse brains per sample. Briefly, whole brains were homogenized in TrisEGTA containing 1x Protease Inhibitor Cocktail (Thermo Scientific). After centrifugation for 10 minutes at 2,500xg, membranes were pelleted from the supernatant by ultracentrifugation for 30 minutes at 37,000rpm. Membrane pellets were resuspended in homogenization buffer. For each sample, 50 μg of membrane protein was run on a 4–15% gradient BioRad Criterion gel. Western blotting was carried out with polyclonal antibody to Nav1.6 at 1:100 (#ASC-009, Alomone).
Trial of SKA-31 in vivo
Four Atcayji-hes mice between 20–35 days old were used for the trial. SKA-31 was administered at a dose of 2.5 mg/kg dissolved in peanut oil as a vehicle, reconstituted on the morning of the trial. Video recordings were done prior to the treatment for an initial baseline assessment, and later at 10 min, 20 min, 30 min and 60 min, and 24 hours after treatment. Observations were made in both the home-cage environment and in an open-field situation. The scoring of the hind limb extension was made by choosing three consecutive steps and noting the average of the maximum hind-limb extension in the three steps. Video clips were extracted, where three consecutive steps were seen. Frames were advanced in each gait cycle, in order to identify the frame with maximum hind limb extension in each gait cycle. Maximum hind limb extension was calculated as the ratio of the distance of the hind limb to the distance of the forelimb from the ground during the phase in the gait cycle when the rump to nose angle was steepest. Once the video clips of three consecutive steps were identified, the clips were anonymized so that the investigator scoring the maximum hind-limb extension was blinded to the treatment condition.
Data analyses and statistics
Statistical significance was assessed by either an unpaired Student’s t test for parametric data and or a Rank Sum Test for non-parametric data. Data were analyzed as non-parametric if it failed a Normality Test (Shapiro-Wilk). A Fisher’s exact test was used for categorical data. A paired Student’s t test was used to determine the effect of pharmacologic agents on firing properties. Values of p < 0.05 were considered significant. Data are expressed as mean ± SEM unless specified otherwise. Data were analyzed using SigmaPlot 11(Systat Software), and Excel (Microsoft).
Results
Atcayji-hes mice exhibit a stiff-legged gait that is reminiscent of human dystonic gait (for example, see videos accompanying (Arif et al., 2011)). The gait is associated with prolonged hind-limb and tail extension during the stance and swing phases of gait accompanied by toe-walking, a pattern that differs markedly from the normal gait seen in wild-type littermate controls (Figure 1A, Movies 1 and 2). The abnormal gait is first evident at post-natal day 11, when the animal starts to develop a stride, and is fully developed and completely penetrant by post-natal day 16. Dystonia in the Atcayji-hes mice is characterized by stiffness only during movement, with no limb stiffness evident at rest (Movie 2). It should be noted that Atcay mutations in mice have variable phenotype severity depending on the genetic background, with a combination of ataxia (broad-based gait with postural instability) and dystonia (stiff legged gait) (Bomar et al., 2003; Kapfhamer et al., 1996; Sikora et al., 2012). In order to determine whether both ataxic and dystonic components to the gait are present in Atcayji-hes mice, foot imprints on paper were analyzed. In normal mice the stride length is relatively uniform from one step to the next, and the hind foot and forefoot placements nearly superimpose (figure 1A). Atcayji-hes mice had a shorter stride length and discordance in the placement of hind and forefeet (figure 1A–C). There was more variability in the stride length in Atcayji-hes mice compared to in wild-type littermates but this was not statistically significant (figure 1C). There was no difference in the base width between wild-type and Atcayji-hes mice (figure 1B). There was a marked impairment of Atcayji-hes mice to perform on the rotarod (figure 1C). There was no postural instability during casual gait (Movie 2). The discordant foot placement would suggest that a dysmetria of foot placement coexists with the stiff-legged gait. This would suggest that ataxia likely coexists with dystonia in the Atcayji-hes mice.
Figure 1. The cerebellum contributes to the stiff-legged dystonic gait of Atcayji-hes mice.

A. Footprint pattern of Atcayji-hes mice and littermate controls. B. Atcayji-hes mice exhibit reduced stride length with a normal base width. C. Atcayji-hes mice exhibit significantly abnormal foot placement and have impaired performance on the rotarod (D). E.. Atcayji-hes mice have a stiff legged gait with abnormal hind limb extension F. Following a partial cerebellectomy, the same mouse has a broad-based ataxic gait without hind limb extension during gait (n = 4 mice). G. The site of the cerebellar lesion involving the DCN and overlying cerebellar cortex is shown (n = 4 mice). Arrow: DCN, Arrowheads: Cerebellar cortical destruction. Data in each graph are presented as mean ± SEM.
A mutation in the Atcay gene in the genetically dystonic rat results in a severe generalized dystonic phenotype resulting in premature death prior to post-natal day 40 (LeDoux et al., 1993). This phenotype, although cerebellar in origin (LeDoux et al., 1993), differs from the focal gait-induced dystonic phenotype in the Atcayji-hes mice. We therefore sought to confirm that the gait dystonia in the Atcayji-hes mice is cerebellar in origin. A partial cerebellectomy with damage to the DCN resulted in conversion of the stiff-legged dystonic gait into a broad-based ataxic gait (Figures 1A–C, Movies 2 and 3). These results suggest that the cerebellum contributes to the stiff-legged gait in the Atcayji-hes mice.
In other rodent models of dystonia, histopathology shows no apparent apoptosis, neurodegeneration, or necrosis. To confirm that abnormalities in cerebellar development or degeneration are not associated with the motor impairment in Atcayji-hes mice, Nissl staining was used to examine the layers of the cerebellum. Cerebellar folia are normally formed in the Atcayji-hes mice, with a normal mature location of cerebellar neurons, suggesting no gross neurodevelopmental deficits (Figure 2, top row). Calbindin staining, specifically for Purkinje neurons, showed normal location of Purkinje neurons, with similar dendritic arborization in Atcayji-hes mice and wild-type littermate controls (figure 2A–C). Purkinje cell counts were similar in Atcayji-hes mice and wild-type littermate controls. The molecular layer thickness, a measure of Purkinje neuron dendritic arborization was also similar in in Atcayji-hes mice and wild-type littermate controls (2A,B,D). Additionally, no astrogliosis was present in Atcayji-hes mice confirming a lack of neurodegeneration (Figure 2A, bottom row). These observations suggest that the cause of the dystonic gait in Atcayji-hes mice is related to a specific neuronal functional deficit, rather than being due to a structural or degenerative change.
Figure 2. Dystonia in the Atcayji-hes mice develops in the absence of changes in cerebellar architecture or neurodegeneration.

A. Top row. Nissl staining of brain sections from wild-type and Atcayji-hes mice show no evidence of neurodevelopmental abnormalities. Middle row: Calbindin staining for Purkinje neurons shows intact Purkinje neurons with normal molecular layer thickness. Purkinje neuron terminals in the DCN are also intact (middle panel in each row for each genotype). Bottom row: GFAP staining reveals no evidence for gliosis in the cerebellar cortex or DCN suggesting an absence of neurodegeneration. N=3 mice of each genotype. Scale bars = 250 μM. B. Higher magnification images of calbindin staining the Purkinje cell layer shows no evidence of Purkinje cell loss or reduction in dendritic arborization. The lack of changes in Purkinje neuron morphology is summarized in C. and D. Data in the graphs are presented as mean ± SEM.
We next determined whether abnormalities in firing were present in Purkinje neurons, the sole projection neurons from the cerebellar cortex. In cell attached recordings, > 95% of Purkinje neurons from wild-type mice exhibited tonic, repetitive firing (Figure 3A, summarized in Figure 3C). Approximately 50% of Purkinje neurons from Atcayji-hes mice failed to demonstrate repetitive firing (Figure 3B,C). Whole-cell recordings demonstrate that cells not displaying repetitive spiking in the cell-attached configuration had a membrane potential of −55 mV (Figure 3B), which is close to the normal resting membrane potential of Purkinje neurons (Shakkottai et al., 2011; Shakkottai et al., 2009). This result suggests that the lack of spiking was not due to non-specific loss of membrane integrity. Purkinje neurons from Atcayji-hes mice that displayed no spontaneous spiking only fired one or a few action potentials in response to injection of depolarizing current, unlike wild-type neurons which continue to display increasing frequency of spikes with progressively increasing injected current (Figure 3D,E). Atcayji-hes Purkinje neurons that exhibited normal spiking had a mean firing frequency similar to wild-type littermate controls (Figure 3F). In other mouse models of ataxia that exhibit dystonia, irregular spiking of Purkinje neurons has been postulated to cause motor symptoms (Walter et al., 2006). Atcayji-hes Purkinje neurons that exhibited repetitive spiking, however, had increased precision of spiking (Figure 3G) compared to littermate controls. A reduction in firing frequency with reduced Purkinje neuron excitability has been described in multiple mouse of models of ataxia associated with reduced expression of the voltage-gated sodium channel Nav1.6 (Levin et al., 2006; Raman et al., 1997; Shakkottai et al., 2009). There was, however, no reduction in expression of Nav1.6 in the Atcayji-hes mice (Figure 3H). These results indicate that a significant subset of Purkinje neurons from Atcayji-hes display reduced Purkinje neuron excitability.
Figure 3. Purkinje neurons from Atcayji-hes mice display an absence of repetitive spiking.

A. Cell attached recording from a Purkinje neuron from a wild-type mouse showing normal repetitive spiking. B. A whole-cell recording from a Atcayji-hes mouse Purkinje neuron that displayed no repetitive spiking in the cell-attached configuration, showing a membrane potential close to the resting membrane potential of wild-type Purkinje neurons. C. Summary of patterns of firing in Atcayji-hes mice and littermate controls. Numbers within the bars represent numbers of cells from which recordings were made. D. Wild-type Purkinje neurons maintain repetitive spiking in response to injection of depolarizing current. E. Atcayji-hes Purkinje neurons that displayed no repetitive spiking cannot sustain repetitive spiking even with injection of depolarizing current. F. The average firing frequency of the ~ 50% Atcayji-hes Purkinje neurons that do display repetitive spiking is similar to wild-type littermate controls. G. The regularity of spiking is preserved in Atcayji-hes Purkinje neurons that display repetitive spiking. CV: Coefficient of variation. H. Expression of the voltage-gated sodium channel, Nav1.6, is not significantly different between Atcayji-hes and wild-type mice (n = 5 animals from each genotype). Data in the graphs are presented as mean ± SEM.
In several other mouse models of movement disorders, a reduction in Purkinje neuron firing frequency is associated with ataxia and not dystonia (Hansen et al., 2013; Levin et al., 2006; Sausbier et al., 2004; Shakkottai et al., 2009). We therefore determined whether abnormalities in cerebellar output at the level of the DCN were associated with dystonia in the Atcayji-hes mice. Recordings were performed in cerebellar slices from Atcayji-hes mice during the dystonic period. Similar to cerebellar Purkinje neurons, DCN neurons normally exhibit autonomous pacemaking (Aizenman and Linden, 1999; Raman et al., 2000; Shakkottai et al., 2004). Wild-type DCN neurons uniformly exhibited regular spiking (Figure 4A,D). The majority of DCN neurons from Atcayji-hes mice also exhibited tonic, repetitive spiking, but the firing frequency was significantly higher than in wild-type littermate controls (Figure 4B,D,E). A small but significant subset of DCN neurons from Atcayji-hes mice exhibited a burst pattern of firing not seen in wild-type littermate controls (Figure 4C,D). Although the spiking of Atcayji-hes DCN neurons appeared to be more irregular than wild-type littermate controls, this did not reach statistical significance (Figure 4F). Because the majority of Purkinje neuron connections are severed in a slice preparation (Telgkamp and Raman, 2002), the increased firing frequency of Atcayji-hes DCN neurons cannot be attributed to reduced Purkinje neuron excitability. Additionally, the firing frequency of Atcayji-hes DCN neurons remained unchanged in the presence of agents that inhibit synaptic transmission (50 μM picrotoxin to block GABAA receptors in addition to 10 μM DNQX and 5 μM CPP to block glutamatergic transmission) (Figure 4G). These data suggest that increased intrinsic DCN neuron excitability is the cause of the increased firing frequency in Atcayji-hes mice. Increased DCN neuron firing and bursting have previously been attributed to a loss of small conductance calcium-activated potassium (SK) channel currents (Aizenman and Linden, 1999; Shakkottai et al., 2004). We therefore determined whether the after-hyperpolarization (AHP) current, a measure of the SK channel current (Shakkottai et al., 2004) was altered in DCN neurons from Atcayji-hes mice. The AHP current was significantly reduced in DCN neurons from Atcayji-hes mice, as compared to wild-type littermate controls (Figure 4H–J). These results suggest that an increase in intrinsic excitability of DCN neurons due to reduced SK channel-mediated AHP currents, results in increased firing frequency and burst firing of Atcayji-hes DCN neurons.
Figure 4. DCN neurons from Atcayji-hes mice exhibit increased intrinsic excitability due to reduced SK channel afterhyperpolarization (AHP) current.

A. Cell attached recording from a DCN neuron from a wild-type mouse showing repetitive spiking. B. Cell attached recording from a DCN neuron from a Atcayji-hes mouse showing repetitive spiking at an increased firing frequency C. A subset of Atcayji-hes DCN neurons exhibit a burst pattern of firing. D. Summary of patterns of firing in DCN neurons in Atcayji-hes mice and wild-type littermate controls. E. DCN neurons from Atcayji-hes mice exhibit repetitive spiking at a significantly increased firing frequency. F. Although the spiking of Atcayji-hes mice appeared less regular, this was not statistically significant. G. The increase in firing frequency of Atcayji-hes DCN neurons persisted in the presence of inhibitors of synaptic transmission. H. An SK channel AHP could be elicited in wild-type DCN neurons. I. The AHP was significantly smaller in Atcayji-hes DCN neurons, and summarized in J. Data in the graphs are presented as mean ± SEM.
Since the SK channel-dependent AHP was reduced in Atcayji-hes DCN neurons, we decided to test whether an activator of SK channels, SKA-31(Sankaranarayanan et al., 2009), could reduce DCN excitability. In cerebellar slices, application of SKA-31 significantly reduced DCN firing frequency of Atcayji-hes DCN neurons (Figure 5A–C). In order to determine whether increased DCN excitability underlies the dystonic phenotype in Atcayji-hes mice, SKA-31 was administered by intraperitoneal injection to dystonic Atcayji-hes mice. At a dose of 2.5 mg/kg, 20 minutes following intraperitoneal injection, there was a significant reduction in the stiff-legged gait (Figures 5D–F), with maximal improvement between 20 and 30 minutes. In other models of dystonia, a visual score has been used to assess the severity of dystonia (Raike et al., 2012). Since the Atcayji-hes mice have dystonia-like hind-limb extension during gait, the maximum hind-limb extension during the gait cycle was scored in lieu of the visual score, so as to have a more quantitative assessment of dystonia. Following SKA-31 administration, an ataxic component to the gait was revealed, with a broader base (Movies 4 and 5). Home cage activity was unchanged following SKA-31 administration. The effect of SKA-31 was completely reversible. The improvement in the dystonic gait was not as prominent at 1 hour and completely disappeared 2 hours following administration, consistent with the known pharmacokinetics of SKA-31 in the brain (Sankaranarayanan et al., 2009). These results indicate that an SK channel activator, which reduces DCN excitability, improves the dystonic gait in the Atcayji-hes mice.
Figure 5. An activator of SK channels reduces DCN neuron excitability and improves the dystonic gait in Atcayji-hes mice.
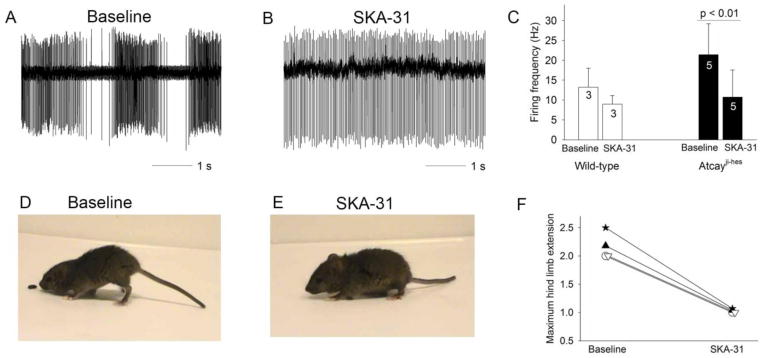
A. An Atcayji-hes DCN neuron that exhibited burst firing in a cerebellar slice was converted by perfusion of 10 μM SKA-31 into a tonic firing neuron with a reduced firing frequency (B.). C. SKA-31 reduces the firing frequency of both wild-type and Atcayji-hes DCN neurons. D,E. The dystonic phenotype in an Atcayji-hes mouse (D) is maximally improved 20 minutes following the intraperitoneal administration of 2.5 mg/kg SKA-31(E). F. The severity of dystonia was assessed by the degree of hind limb extension during gait. 20 minutes following administration of SKA-31, the degree of hind limb extension was significantly reduced (n = 4 animals). Data in the graphs are presented as mean ± SEM.
Discussion
Rodent models of cerebellar dystonia
Most of the existing rodent models of dystonia, where the cerebellum is suggested to contribute to the motor symptoms, exhibit generalized twisting and abnormal movements(Wilson and Hess, 2013). In genetically dystonic rats, which exhibit generalized dystonic movements, cerebellectomy improves the motor phenotype and allows the rats to survive to adulthood (LeDoux et al., 1995). In tottering mice, which exhibit episodic generalized dystonic movements caused by a mutation in a voltage-gated calcium channel gene (Campbell and Hess, 1999; Doyle et al., 1997), during dystonic episodes, electromyographic activity becomes significantly coherent with activity in the cerebellar cortex (Chen et al., 2009). In a mouse model of rapid onset dystonia-parkinsonism, when mice developed intermittent dystonic postures, there was a tight correlation between dystonic postures and an increase in amplitude of cerebellar EEG activity (Calderon et al., 2011). Finally, delivery of the excitatory glutamate agonist kainic acid directly into the cerebellum elicits generalized dystonia in both mice and rats (Alvarez-Fischer et al., 2012; Pizoli et al., 2002). Although stress appears to increase the likelihood of these movements (Raike et al., 2013), specific voluntary activity does not provoke the abnormal movements as can be seen in human dystonias (Berardelli et al., 1998; Fahn, 1984). Regional abnormalities in the cerebellar cortex can produce abnormal movements in focal body parts in rodents, but these too are not linked to specific activities (Raike et al., 2012). In the current study we describe a specific walking-induced gait abnormality in the Atcayji-hes mice that is reminiscent of human gait dystonia. The abnormal hind limb extension in the Atcayji-hes mice is not present at rest and only evoked during walking. Lesioning of the cerebellum improves the dystonic component to the gait, suggesting that the stiff-legged component to the gait is cerebellar in origin. The dystonic gait is accompanied by reduced Purkinje neuron excitability and increased DCN excitability. Our results suggest that the cerebellum can contribute to a specific task-associated dystonia.
Relationship between SK channels and Caytaxin
Caytaxin is a neuron-restricted protein encoded by the gene Atcay/ATCAY (Bomar et al., 2003). The function of Caytaxin remains unknown. Studies that have looked at proteins that interact with caytaxin have determined that the peptidyl-prolyl cis/trans-isomerase, Pin1, and kidney-type glutaminase (KGA) directly bind Caytaxin (Buschdorf et al., 2008; Buschdorf et al., 2006). Pin1 catalytically regulates the conformation of substrates after their phosphorylation to further control protein function and regulates a broad range of cellular processes including the cell cycle, cell signaling, transcription and splicing, DNA damage response pathways, germ cell development, and neuronal survival (Lu and Zhou, 2007). It is possible that Caytaxin, through its interaction with Pin1, regulates SK channel conformation or expression.
SK channels as regulators of neuronal excitability
SK channels are postulated to play an important role in setting the intrinsic excitability of cerebellar pacemaker neurons (Aizenman and Linden, 1999; Cingolani et al., 2002; Shakkottai et al., 2004; Womack and Khodakhah, 2003). Purkinje neuron excitability is altered in mouse models of ataxia, and improving Purkinje neuron dysfunction by administering activators of SK channels has therapeutic potential (Kasumu et al., 2012; Shakkottai et al., 2011; Walter et al., 2006). In a mouse model of ataxia that also exhibits paroxysmal dystonia, cerebellar perfusion of an SK channel activator not only reduced the frequency of the episodes of dystonia, but also decreased the severity of attacks (Walter et al., 2006). The improvement in the motor phenotype was associated with improved regularity of pacemaking of cerebellar Purkinje neurons (Walter et al., 2006).
In this study, we demonstrate that DCN neurons in Atcayji-hes mice exhibit increased intrinsic excitability that is associated with a reduction in SK currents. Systemic administration of an SK channel activator, SKA-31, reduces the gait-associated abnormal hind limb extension in these mice. Since the motor phenotype of Atcayji-hes mice is modified by a partial cerebellectomy, it suggests that the cerebellum contributes to the stiff-legged gait in these mice. The site of action of systemically delivered SK channel activators in the Atcayji-hes is also suggested to be the cerebellar output DCN neurons. SK channels are however widely expressed in the nervous system, including other motor areas such as cerebellar Purkinje neurons (Cingolani et al., 2002), and neurons of the basal ganglia (Deister et al., 2009; Nazzaro et al., 2012). A potential site of action of SKA-31 in Purkinje neurons and the basal ganglia cannot therefore be entirely ruled out.
DCN neuron excitability and relationship to motor function
How may the aberrant activity of DCN neurons in the Atcayji-hes mice translate into increased muscle activity? Normally, DCN neurons demonstrate characteristic patterns of activity, as seen in recordings from non-human primates performing simple movements. Recordings in monkeys at rest show that most DCN neurons fire at high maintained frequencies of ~ 40 Hz. During wrist flexion-extension movements, for a cell related to these movements the discharge frequency alternates between rates up to 400–500 Hz and lower than the resting rate consistently in time with the movement (Thach, 1968). Since the majority of cerebellar nuclear neurons are excitatory, an increase in firing of cerebellar nuclear neurons would be expected to facilitate muscle activity and a decrease in firing frequency would reduce facilitation of muscle groups (Wetts et al., 1985). Although only a subset of cerebellar output neurons are presumed to directly regulate motor activity of the limb, with others discharging in relation to joint position and the direction of the intended next movement (Thach, 1978), at least a subset of cerebellar output neurons can directly activate muscle groups (Rispal-Padel et al., 1982). Additionally, an interaction between the cerebellar and basal ganglia motor control systems may allow the cerebellar output neurons to influence muscle activity (Calderon et al., 2011; Hoshi et al., 2005). Aberrant activity of cerebellar output neurons can therefore directly modulate activity of downstream motor pathways to cause dystonia.
In a previously described mouse model of ataxia, the SK3-1B transgenic mice, an increase in DCN excitability due to a reduction in SK channels produced profound ataxia (Shakkottai et al., 2004). In the Atcayji-hes mice, we demonstrate in this study that DCN neuron intrinsic excitability is increased in a manner similar to that previously described in the SK3-1B transgenic mice. In spite of similar physiologic abnormalities in the DCN, SK3-1B transgenic mice fail to show features of dystonia (Shakkottai et al., 2004). This is likely explained by the fact that in SK3-1B transgenic mice, Purkinje neuron excitability is unaltered (Shakkottai et al., 2004), unlike Purkinje neurons in the Atcayji-hes that display a reduction in excitability. Purkinje neurons, which are autonomously active inhibitory GABAergic neurons, subject the DCN to a barrage of inhibitory postsynaptic potentials (Chen and Hillman, 1993; Gauck and Jaeger, 2000; Monaghan et al., 1986; Uusisaari et al., 2007). The combination of increased intrinsic DCN neuron excitability and a reduction in the inhibitory influence of Purkinje neurons in the Atcayji-hes mice likely exaggerates the hyperexcitability of DCN neurons above a threshold required for DCN neurons to directly activate muscle groups, and explains why these mice have both ataxia and dystonia.
Cerebellar ataxia can result from both the loss of cerebellar output (Carrea and Mettler, 1947; Dow and Moruzzi, 1958; Flament and Hore, 1986) as well as an increase in excitability of cerebellar output neurons (Shakkottai et al., 2004). Cerebellectomy involving DCN neurons in the Atcayji-hes mice likely converts the mild ataxic and more prominent dystonic gait due to DCN neuron hyperexcitability (a pathologic gain-of function of the cerebellum) into profound ataxia resulting from a loss of cerebellar function. This would suggest that the degree of DCN hyperexcitability may influence the manifestation of motor symptoms, with ataxia that transitions into both dystonia and ataxia with increasing DCN excitability.
Supplementary Material
Gait of a wild-type mouse.
Stiff-legged dystonic gait of an Atcayji-hes mouse prior to a partial cerebellectomy.
Conversion of the stiff-legged dystonic gait in an Atcayji-hes mouse to a broad-based ataxic gait, 24 hours following a partial cerebellectomy.
Stiff-legged dystonic gait of an Atcayji-hes mouse prior to administration of SKA-31
Attenuation of the dystonic gait of the same Atcayji-hes mouse as in Movie 4, 30 minutes following intraperitoneal administration of SKA-31
Research Highlights.
We suggest that the cerebellum contributes to both ataxia and the dystonia-like, stiff-legged gait in Atcayji-hes mice
Cerebellar output (DCN) neurons exhibit increased intrinsic excitability
DCN neurons display reduced calcium-activated potassium (SK) currents
Activating SK channels improves DCN neuron hyperexcitability
Activating SK channels in vivo improves the dystonic gait in Atcayji-hes mice
Acknowledgments
Funding was provided through the Dystonia Medical Research Foundation (VGS), the NIH 1K08NS072158 (VGS) and NIH T32 training grant NS007222 (KL-C) from the Department of the Neurology at the University of Michigan. We would like to thank Dr. Janelle O’Brien for assistance with the Nav1.6 immunoblot. We are grateful to James Dell’Orco, Natasha Kennedy, Samantha Ferraro, and Alexi Vasbinder for technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aizenman CD, Linden DJ. Regulation of the rebound depolarization and spontaneous firing patterns of deep nuclear neurons in slices of rat cerebellum. J Neurophysiol. 1999;82:1697–709. doi: 10.1152/jn.1999.82.4.1697. [DOI] [PubMed] [Google Scholar]
- Alvarez-Fischer D, et al. Prolonged generalized dystonia after chronic cerebellar application of kainic acid. Brain Res. 2012;1464:82–8. doi: 10.1016/j.brainres.2012.05.007. [DOI] [PubMed] [Google Scholar]
- Argyelan M, et al. Cerebellothalamocortical connectivity regulates penetrance in dystonia. J Neurosci. 2009;29:9740–7. doi: 10.1523/JNEUROSCI.2300-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arif B, et al. An unusual neurological syndrome of crawling gait, dystonia, pyramidal signs, and limited speech. Mov Disord. 2011;26:2279–83. doi: 10.1002/mds.23860. [DOI] [PubMed] [Google Scholar]
- Berardelli A, et al. The pathophysiology of primary dystonia. Brain. 1998;121(Pt 7):1195–212. doi: 10.1093/brain/121.7.1195. [DOI] [PubMed] [Google Scholar]
- Bomar JM, et al. Mutations in a novel gene encoding a CRAL-TRIO domain cause human Cayman ataxia and ataxia/dystonia in the jittery mouse. Nat Genet. 2003;35:264–9. doi: 10.1038/ng1255. [DOI] [PubMed] [Google Scholar]
- Buschdorf JP, et al. Nerve growth factor stimulates interaction of Cayman ataxia protein BNIP-H/Caytaxin with peptidyl-prolyl isomerase Pin1 in differentiating neurons. PLoS One. 2008;3:e2686. doi: 10.1371/journal.pone.0002686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buschdorf JP, et al. Brain-specific BNIP-2-homology protein Caytaxin relocalises glutaminase to neurite terminals and reduces glutamate levels. J Cell Sci. 2006;119:3337–50. doi: 10.1242/jcs.03061. [DOI] [PubMed] [Google Scholar]
- Calderon DP, et al. The neural substrates of rapid-onset Dystonia-Parkinsonism. Nat Neurosci. 2011;14:357–65. doi: 10.1038/nn.2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell DB, Hess EJ. L-type calcium channels contribute to the tottering mouse dystonic episodes. Mol Pharmacol. 1999;55:23–31. doi: 10.1124/mol.55.1.23. [DOI] [PubMed] [Google Scholar]
- Carrea RM, Mettler FA. Physiologic consequences following extensive removals of the cerebellar cortex and deep cerebellar nuclei and effect of secondary cerebral ablations in the primate. J Comp Neurol. 1947;87:169–288. doi: 10.1002/cne.900870302. [DOI] [PubMed] [Google Scholar]
- Chen G, et al. Low-frequency oscillations in the cerebellar cortex of the tottering mouse. J Neurophysiol. 2009;101:234–45. doi: 10.1152/jn.90829.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Hillman DE. Colocalization of neurotransmitters in the deep cerebellar nuclei. J Neurocytol. 1993;22:81–91. doi: 10.1007/BF01181572. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, et al. Developmental regulation of small-conductance Ca2+-activated K+ channel expression and function in rat Purkinje neurons. J Neurosci. 2002;22:4456–67. doi: 10.1523/JNEUROSCI.22-11-04456.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deister CA, et al. Calcium-activated SK channels influence voltage-gated ion channels to determine the precision of firing in globus pallidus neurons. J Neurosci. 2009;29:8452–61. doi: 10.1523/JNEUROSCI.0576-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmaire C, et al. Structural abnormalities in the cerebellum and sensorimotor circuit in writer’s cramp. Neurology. 2007;69:376–80. doi: 10.1212/01.wnl.0000266591.49624.1a. [DOI] [PubMed] [Google Scholar]
- den Dunnen WF. Neuropathological diagnostic considerations in hyperkinetic movement disorders. Front Neurol. 2013;4:7. doi: 10.3389/fneur.2013.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dow RS, Moruzzi G. The physiology and pathology of the cerebellum. University of Minnesota Press; Minneapolis: 1958. [Google Scholar]
- Doyle J, et al. Mutations in the Cacnl1a4 calcium channel gene are associated with seizures, cerebellar degeneration, and ataxia in tottering and leaner mutant mice. Mamm Genome. 1997;8:113–20. doi: 10.1007/s003359900369. [DOI] [PubMed] [Google Scholar]
- Eidelberg D, et al. Functional brain networks in DYT1 dystonia. Ann Neurol. 1998;44:303–12. doi: 10.1002/ana.410440304. [DOI] [PubMed] [Google Scholar]
- Fahn S. The varied clinical expressions of dystonia. Neurol Clin. 1984;2:541–54. [PubMed] [Google Scholar]
- Flament D, Hore J. Movement and electromyographic disorders associated with cerebellar dysmetria. J Neurophysiol. 1986;55:1221–33. doi: 10.1152/jn.1986.55.6.1221. [DOI] [PubMed] [Google Scholar]
- Frucht SJ. The definition of dystonia: current concepts and controversies. Mov Disord. 2013;28:884–8. doi: 10.1002/mds.25529. [DOI] [PubMed] [Google Scholar]
- Gauck V, Jaeger D. The control of rate and timing of spikes in the deep cerebellar nuclei by inhibition. J Neurosci. 2000;20:3006–16. doi: 10.1523/JNEUROSCI.20-08-03006.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen ST, et al. Changes in Purkinje cell firing and gene expression precede behavioral pathology in a mouse model of SCA2. Hum Mol Genet. 2013;22:271–83. doi: 10.1093/hmg/dds427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi E, et al. The cerebellum communicates with the basal ganglia. Nat Neurosci. 2005;8:1491–3. doi: 10.1038/nn1544. [DOI] [PubMed] [Google Scholar]
- Kapfhamer D, et al. The neurological mouse mutations jittery and hesitant are allelic and map to the region of mouse chromosome 10 homologous to 19p13.3. Genomics. 1996;35:533–8. doi: 10.1006/geno.1996.0394. [DOI] [PubMed] [Google Scholar]
- Kasumu AW, et al. Selective positive modulator of calcium-activated potassium channels exerts beneficial effects in a mouse model of spinocerebellar ataxia type 2. Chem Biol. 2012;19:1340–53. doi: 10.1016/j.chembiol.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Ber I, et al. Predominant dystonia with marked cerebellar atrophy: a rare phenotype in familial dystonia. Neurology. 2006;67:1769–73. doi: 10.1212/01.wnl.0000244484.60489.50. [DOI] [PubMed] [Google Scholar]
- LeDoux MS. Animal models of dystonia: Lessons from a mutant rat. Neurobiol Dis. 2011;42:152–61. doi: 10.1016/j.nbd.2010.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeDoux MS, et al. Single-unit activity of cerebellar nuclear cells in the awake genetically dystonic rat. Neuroscience. 1998;86:533–45. doi: 10.1016/s0306-4522(98)00007-4. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, Lorden JF. Abnormal spontaneous and harmaline-stimulated Purkinje cell activity in the awake genetically dystonic rat. Exp Brain Res. 2002;145:457–67. doi: 10.1007/s00221-002-1127-4. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, et al. Cerebellectomy eliminates the motor syndrome of the genetically dystonic rat. Exp Neurol. 1993;120:302–10. doi: 10.1006/exnr.1993.1064. [DOI] [PubMed] [Google Scholar]
- LeDoux MS, et al. Selective elimination of cerebellar output in the genetically dystonic rat. Brain Res. 1995;697:91–103. doi: 10.1016/0006-8993(95)00792-o. [DOI] [PubMed] [Google Scholar]
- Lehericy S, et al. The anatomical basis of dystonia: current view using neuroimaging. Mov Disord. 2013;28:944–57. doi: 10.1002/mds.25527. [DOI] [PubMed] [Google Scholar]
- Levin SI, et al. Impaired motor function in mice with cell-specific knockout of sodium channel Scn8a (NaV1.6) in cerebellar purkinje neurons and granule cells. J Neurophysiol. 2006;96:785–93. doi: 10.1152/jn.01193.2005. [DOI] [PubMed] [Google Scholar]
- Lorden JF, et al. Neuropharmacological correlates of the motor syndrome of the genetically dystonic (dt) rat. Adv Neurol. 1988;50:277–97. [PubMed] [Google Scholar]
- Lu KP, Zhou XZ. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat Rev Mol Cell Biol. 2007;8:904–16. doi: 10.1038/nrm2261. [DOI] [PubMed] [Google Scholar]
- Magistretti J, et al. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends Neurosci. 1996;19:530–4. doi: 10.1016/s0166-2236(96)40004-2. [DOI] [PubMed] [Google Scholar]
- Marsden CD, et al. The anatomical basis of symptomatic hemidystonia. Brain. 1985;108(Pt 2):463–83. doi: 10.1093/brain/108.2.463. [DOI] [PubMed] [Google Scholar]
- Monaghan PL, et al. Immunocytochemical localization of glutamate-, glutaminase- and aspartate aminotransferase-like immunoreactivity in the rat deep cerebellar nuclei. Brain Res. 1986;363:364–70. doi: 10.1016/0006-8993(86)91024-3. [DOI] [PubMed] [Google Scholar]
- Nazzaro C, et al. SK channel modulation rescues striatal plasticity and control over habit in cannabinoid tolerance. Nat Neurosci. 2012;15:284–93. doi: 10.1038/nn.3022. [DOI] [PubMed] [Google Scholar]
- Neychev VK, et al. The functional neuroanatomy of dystonia. Neurobiol Dis. 2011;42:185–201. doi: 10.1016/j.nbd.2011.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niethammer M, et al. Hereditary dystonia as a neurodevelopmental circuit disorder: Evidence from neuroimaging. Neurobiol Dis. 2011;42:202–9. doi: 10.1016/j.nbd.2010.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizoli CE, et al. Abnormal cerebellar signaling induces dystonia in mice. J Neurosci. 2002;22:7825–33. doi: 10.1523/JNEUROSCI.22-17-07825.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raike RS, et al. Limited regional cerebellar dysfunction induces focal dystonia in mice. Neurobiol Dis. 2012;49C:200–210. doi: 10.1016/j.nbd.2012.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raike RS, et al. Stress, caffeine and ethanol trigger transient neurological dysfunction through shared mechanisms in a mouse calcium channelopathy. Neurobiol Dis. 2013;50:151–9. doi: 10.1016/j.nbd.2012.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, et al. Ionic currents and spontaneous firing in neurons isolated from the cerebellar nuclei. J Neurosci. 2000;20:9004–16. doi: 10.1523/JNEUROSCI.20-24-09004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, et al. Altered subthreshold sodium currents and disrupted firing patterns in Purkinje neurons of Scn8a mutant mice. Neuron. 1997;19:881–91. doi: 10.1016/s0896-6273(00)80969-1. [DOI] [PubMed] [Google Scholar]
- Rispal-Padel L, et al. Cerebellar nuclear topography of simple and synergistic movements in the alert baboon (Papio papio) Exp Brain Res. 1982;47:365–80. doi: 10.1007/BF00239355. [DOI] [PubMed] [Google Scholar]
- Sankaranarayanan A, et al. Naphtho[1,2-d]thiazol-2-ylamine (SKA-31), a new activator of KCa2 and KCa3.1 potassium channels, potentiates the endothelium-derived hyperpolarizing factor response and lowers blood pressure. Mol Pharmacol. 2009;75:281–95. doi: 10.1124/mol.108.051425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sausbier M, et al. Cerebellar ataxia and Purkinje cell dysfunction caused by Ca2+-activated K+ channel deficiency. Proc Natl Acad Sci U S A. 2004;101:9474–8. doi: 10.1073/pnas.0401702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, et al. Enhanced neuronal excitability in the absence of neurodegeneration induces cerebellar ataxia. J Clin Invest. 2004;113:582–90. doi: 10.1172/JCI20216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, et al. Early changes in cerebellar physiology accompany motor dysfunction in the polyglutamine disease spinocerebellar ataxia type 3. J Neurosci. 2011;31:13002–14. doi: 10.1523/JNEUROSCI.2789-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakkottai VG, et al. FGF14 regulates the intrinsic excitability of cerebellar Purkinje neurons. Neurobiol Dis. 2009;33:81–8. doi: 10.1016/j.nbd.2008.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sikora KM, et al. Expression of Caytaxin protein in Cayman Ataxia mouse models correlates with phenotype severity. PLoS One. 2012;7:e50570. doi: 10.1371/journal.pone.0050570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratton SE, et al. Spontaneous and harmaline-stimulated Purkinje cell activity in rats with a genetic movement disorder. J Neurosci. 1988;8:3327–36. doi: 10.1523/JNEUROSCI.08-09-03327.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–63. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telgkamp P, Raman IM. Depression of inhibitory synaptic transmission between Purkinje cells and neurons of the cerebellar nuclei. J Neurosci. 2002;22:8447–57. doi: 10.1523/JNEUROSCI.22-19-08447.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thach WT. Discharge of Purkinje and cerebellar nuclear neurons during rapidly alternating arm movements in the monkey. J Neurophysiol. 1968;31:785–97. doi: 10.1152/jn.1968.31.5.785. [DOI] [PubMed] [Google Scholar]
- Thach WT. Correlation of neural discharge with pattern and force of muscular activity, joint position, and direction of intended next movement in motor cortex and cerebellum. J Neurophysiol. 1978;41:654–76. doi: 10.1152/jn.1978.41.3.654. [DOI] [PubMed] [Google Scholar]
- Uusisaari M, et al. Morphological and electrophysiological properties of GABAergic and non-GABAergic cells in the deep cerebellar nuclei. J Neurophysiol. 2007;97:901–11. doi: 10.1152/jn.00974.2006. [DOI] [PubMed] [Google Scholar]
- Walter JT, et al. Decreases in the precision of Purkinje cell pacemaking cause cerebellar dysfunction and ataxia. Nat Neurosci. 2006;9:389–97. doi: 10.1038/nn1648. [DOI] [PubMed] [Google Scholar]
- Wetts R, et al. Cerebellar nuclear cell activity during antagonist cocontraction and reciprocal inhibition of forearm muscles. J Neurophysiol. 1985;54:231–44. doi: 10.1152/jn.1985.54.2.231. [DOI] [PubMed] [Google Scholar]
- Wilson BK, Hess EJ. Animal models for dystonia. Mov Disord. 2013;28:982–9. doi: 10.1002/mds.25526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Womack MD, Khodakhah K. Somatic and dendritic small-conductance calcium-activated potassium channels regulate the output of cerebellar purkinje neurons. J Neurosci. 2003;23:2600–7. doi: 10.1523/JNEUROSCI.23-07-02600.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J, Ledoux MS. Caytaxin deficiency causes generalized dystonia in rats. Brain Res Mol Brain Res. 2005;141:181–92. doi: 10.1016/j.molbrainres.2005.09.009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Gait of a wild-type mouse.
Stiff-legged dystonic gait of an Atcayji-hes mouse prior to a partial cerebellectomy.
Conversion of the stiff-legged dystonic gait in an Atcayji-hes mouse to a broad-based ataxic gait, 24 hours following a partial cerebellectomy.
Stiff-legged dystonic gait of an Atcayji-hes mouse prior to administration of SKA-31
Attenuation of the dystonic gait of the same Atcayji-hes mouse as in Movie 4, 30 minutes following intraperitoneal administration of SKA-31