

Abstract
Voltage-gated Ca2+ (CaV) channels mediate Ca2+ ions influx into cells in response to depolarization of the plasma membrane. They are responsible for initiation of excitation-contraction and excitation-secretion coupling, and the Ca2+ that enters cells through this pathway is also important in the regulation of protein phosphorylation, gene transcription, and many other intracellular events. Initial electrophysiological studies divided CaV channels into low-voltage-activated (LVA) and high-voltage-activated (HVA) channels. The HVA CaV channels were further subdivided into L, N, P/Q, and R-types which are oligomeric protein complexes composed of an ion-conducting CaVα1 subunit and auxiliary CaVα2δ, CaVβ, and CaVγ subunits. The functional consequences of the auxiliary subunits include altered functional and pharmacological properties of the channels as well as increased current densities. The latter observation suggests an important role of the auxiliary subunits in membrane trafficking of the CaVα1 subunit. This includes the mechanisms by which CaV channels are targeted to the plasma membrane and to appropriate regions within a given cell. Likewise, the auxiliary subunits seem to participate in the mechanisms that remove CaV channels from the plasma membrane for recycling and/or degradation. Diverse studies have provided important clues to the molecular mechanisms involved in the regulation of CaV channels by the auxiliary subunits, and the roles that these proteins could possibly play in channel targeting and membrane Stabilization.
INTRODUCTION
Voltage-gated Ca2+ (CaV) channels are a family of transmembrane proteins widely distributed in excitable cells and also found in many nonexcitable cells. These channels open when the plasma membrane becomes depolarized mediating Ca2+ entry in response to action potentials and subthreshold depolarizing signals. Ca2+ entering the cell through CaV channels serves as the second messenger of electrical signaling, initiating a variety of cellular events including neurotransmitter and hormone release, muscle contraction, enzyme activation, and gene expression, among many others. Signal transduction in different cell types involves diverse molecular subtypes of CaV channels, which mediate currents with distinct physiological and pharmacological properties.1
CaV channels have been grouped into two distinct classes depending on their voltage sensitivity (Table 1). Channels that are activated at relatively hyperpolarized voltage ranges (less than or equal to −40 mV) have been named low-voltage-activated (LVA) or T-type, whereas channels that are activated at potentials more positive to −40 mV have been termed high-voltage-activated (HVA). HVA CaV channels can be further subdivided into L-, N-, P-, Q-, and R-types (Table 1), by virtue of their distinct functional and pharmacological profiles.1
TABLE 1.
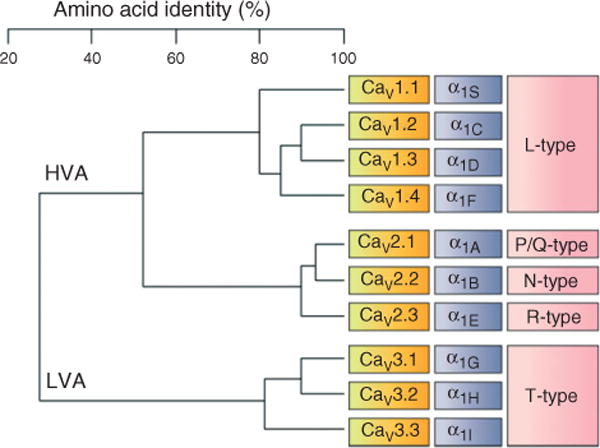
CaVα1 subunits can be divided into three classes according to amino acid sequence identity, as shown in the dendrogram. The CaV1 and CaV2 classes are termed High-Voltage-Activated (HVA) channels. The CaV3α1 subunits form the Low-Voltage-Activated (LVA) channels. The original names, molecular nomenclature, and type of currents are given in purple, yellow, and pink, respectively.
|
Members of the HVA class are heteromultimers of a pore-forming CaVα1 subunit that coassembles into a functional channel complex with auxiliary CaVβ, CaVα2δ, and in some cases with CaVγ subunits (Figure 1(a)). In contrast, members of the LVA channel CaV class are thought to be CaVα1 subunit monomers.2 The CaVα1 subunit defines the channel subtype, whereas the auxiliary subunits regulate the CaVα1 subunit function and plasma membrane expression.1,3 Molecular studies have shown that the CaVα1 subunit is comprised of four homologous repeats each containing six transmembrane helices (S1–S6) and a pore lining P-loop domain (Figure 1(b)). These homologous membrane repeats are connected via cytoplasmic loops and flanked by intracellular N-and C-termini.1
FIGURE 1.
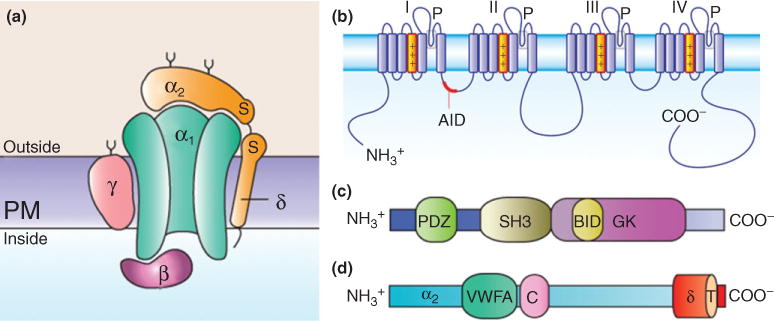
Subunit composition of high-voltage-activated Ca2+ channels and structural domains of the auxiliary subunits. (a) Channel complex is composed of the pore-forming CaVα1 and the auxiliary CaVα2δ, CaVβ, and CaVγ subunits. The CaVα2δ and CaVγ subunits contain transmembrane domains, whereas CaVβs are intracellular. (b) The CaVα1 subunits consist of four transmembrane domains (I–IV) and the linker joining I and II encompasses the α-interaction domain (AID). (c)The CaVβ subunits are formed by three conserved domains: PSD95/Dlg1/ZO-1 (PDZ), Scr homology 3 (SH3), and guanylate kinase (GK). The CaVβ subunit interaction domain (BID) is one of the regions involved in the interaction of the protein with the CaVα1 subunit. (d) The CaVα2δ subunits consist of α2 (blue), which is an extracellular subunit, disulphide-bonded to the δ subunit (orange), which is membrane-associated. The approximate positions of the vWA domain and the two bacterial chemosensory domains (Cache; C) are also indicated. The sites of interaction between the CaVα1 subunit and the CaVα2δ subunit are unknown.
Ten different CaVα1 subunits grouped into three subfamilies are expressed in mammals1 (Table 1). The CaV1 subfamily encodes L-type Ca2+ channels and includes four members (CaV1.1–CaV 1.4). The CaV2 subfamily consist of three members (CaV2.1–CaV2.3) that encode the neuronal P/Q-type, N-type, and R-type channels, respectively. The P- and Q-type channels appear to arise from alternate splicing of CaV2.1 and/or coassembly with distinct CaVβ subunits.4,5 Last, the CaV3 subfamily includes the LVA (T-type) channels with three members (CaV3.1–CaV3.3).
EFFECTS OF THE AUXILIARY SUBUNITS ON HVA CaV CHANNEL FUNCTION, TRAFFICKING, AND MEMBRANE STABILITY
CaV channel trafficking and regulation mechanisms involve many different aspects including intrinsic channel protein structural determinants, interactions with regulatory elements such as second messengers and/or with extrinsic proteins to the channel complex. However, as we shall discuss later, associations with auxiliary subunits CaVβ and CaVα2δ are thought to play crucial roles in fine-tuning channel gating, regulating channel modulation by other proteins and signaling molecules, as well as in forward trafficking and removal of CaV1 and CaV2 channels from the plasma membrane.6,7
THE CaVβ SUBUNITS
The CaVβ subunits are cytoplasmic proteins that bind to the proximal region of the intracellular loop between domains I and II of the CaV1α1 and CaV2α1 subunits termed the α-interaction domain (AID)8 (Figure 1(b)). There are four subfamilies of CaVβ subunits (CaVβ1–CaVβ4), each with splice variants, encoded by four distinct genes.9 All CaVβ subunits contain conserved Src homology 3 (SH3) and a guanylate kinase-like (GK) domains9,10 (Figure 1(c)), placing them into the membrane-associated guanylate kinase (MAGUK) protein family, scaffold proteins that function at neuronal synapses, tight junctions, immunological synapses, and neutrophil membranes.11 The general structural features of guanylate kinases are preserved in the GK domain of the CaVβ subunit (Cavβ-GK) though many key amino acid residues are absent rendering it catalytically inactive.12 Instead, this domain has evolved into a protein–protein interaction module, binding tightly to the CaVα1 subunit through the AID.13–15 Likewise, a large portion of the CaVβ-GK domain remains free to interact with other proteins, such as RGK GTPases16–18 and the largeconductance Ca2+-dependent K+ channels.19 The SH3 and Cavβ-GK conserved domains are flanked by regions of variable sequence and length between isoforms and splice variants of each isoform.9,12,20
When coexpressed in heterologous systems with CaV1α1 or CaV2α1, CaVβ auxiliary subunits significantly enhance Ca2+ currents and affect the voltage dependence and kinetics of current activation and inactivation.21–27 Furthermore, CaVβ subunit presence may be important for CaV1 and CaV2 channel modulation by different protein kinases,28–32 G proteins,33–35 Ras-related monomeric small GTP-binding proteins36–39 as well as by proteins of the vesicle release machinery including Rab3-interacting molecule 1.40–43 Results from CaVβ subunit knockout mice have confirmed the physiological relevance of these auxiliary subunits. CaVβ1 and CaVβ2 subunit knockouts are lethal,44,45 while CaVβ3 and CaVβ4 knockouts result in severe malfunction of the central nervous system.46,47
Likewise, diverse studies have suggested that the CaVβ subunits may participate in physiological events independent of their association with CaV channels. It has been suggested that a truncated splice variant of CaVβ4 lacking the GK and the C-terminal domains interacts directly with the heterochromatin protein 1 (HP1), attenuating its function as gene silencing factor.48 More recently, Zhang et al.49 confirmed that full-length CaVβ subunits may function as transcription regulators. In particular, CaVβ3 can interact and suppress the transcriptional activity of Pax6(S), a critical factor for the development of the eye and the nervous system. In addition, upon neuronal differentiation, CaVβ4 can interact with B56δ, a nuclear regulatory subunit of the phosphatase 2A (PP2A), and the heterochromatin protein, HP1γ. This complex relocates to the nucleus, where it regulates the dephosphorylation of histones, a key mechanism in transcriptional regulation, and associates to the tyrosine hydroxylase (TH) promoter through the nuclear transcription factor thyroid hormone receptor α. This signaling cascade evidences the role of CaVβ4 as a repressor recruiting platform to control neuronal gene expression.50,51
Given their essential role in the functional expression and modulation of HVA CaV channels, it is not surprising that CaVβ auxiliary subunit mutations have been implicated in disease. Cavβ knockout animals and spontaneous occurring mutants have severe phenotypes that are in some cases lethal.9,12,52 For instance, the CaVβ1a subunit isoform is selectively expressed in skeletal muscle where it partners with CaV 1.1α1 subunit and is necessary for excitation-contraction coupling. Consequently, the Cavβ1a subunit knockout mice, similar to CaV1.1α1 knockouts, are motionless and die after birth from asphyxiation.44 Similarly, CaVβ2b expression is predominant in the heart and knockouts of this subunit die prenatally due to lack of cardiac contractions.45 CaVβ3 subunit knockouts show altered perception of inflammatory pain as a result of reduced N-type CaV channel expression in dorsal root ganglia47 and have compromised sympathetic control, likely due to reduced N- and L-type channel activity.53 Last, lethargic (lh) mice are naturally occurring CaVβ4 subunit knockouts. The lethargic phenotype includes ataxia, seizures, absence epilepsy, and paroxysmal dyskinesia,46,52 associated with reduced excitatory neurotransmission in thalamic neurons.54
In humans, two point mutations in the CaVβ2b subunit have been implicated in cardiovascular diseases. The first mutation contributes to a type of sudden death syndrome characterized by a short QT interval and an elevated ST-segment,55 and the other is linked to the Brugada syndrome.56 Likewise, mutations in the CACNB4 gene have been linked to epilepsy and episodic ataxia.57
As mentioned earlier, CaV1α1 and CaV2α1 channel subunits show poor surface expression by themselves, however, upon coexpression of the auxiliary subunits currents are significantly increased reflecting either enhanced channel open probability or cell surface expression.58,59 The increase in surface expression can be observed both in native and recombinant channels with any of the CaVβs and seems to be dependent on binding to the AID, as point mutations that disrupt the AID/CaVβ interaction reduce or abolish CaVβ-mediated current stimulation.8 Interestingly, in heterologous systems the GK domain can recapitulate the function of the full-length CaVβ subunits increasing channel surface expression.60,61
An initial explanation of how the CaVβ subunits enhance channel surface expression was that they antagonize an endoplasmic reticulum (ER) retention signal located in the I-II linker that severely restricts the plasma membrane incorporation of the CaVα1 subunit58 (Figure 2(a)). More recently, this idea has been enriched based on data obtained after transferring the intracellular linkers of the CaV1.2α1 subunit into a T-type channel (CaV3.1). The results of these experiments suggest that the I-II linker of CaV1.2α1 has an ER export signal of 9 amino acids downstream the AID.62 On the other hand, all other intracellular linkers, including the N- and C-termini, were found to contain ER retention signals. Consequently, it was proposed that the intracellular regions in the CaVα1 subunit form a complex that yields a prevailing ER retention signal, and when the CaVβ subunit binds to the I-II linker, it triggers a switch in the channel complex such that the ER export signal becomes dominant, enhancing CaVα1 surface expression (Figure 2(b)). In this process, the CaVα1 C-terminus may play also an important role for Cavβ-dependent channel upregulation.12,62,63
FIGURE 2.
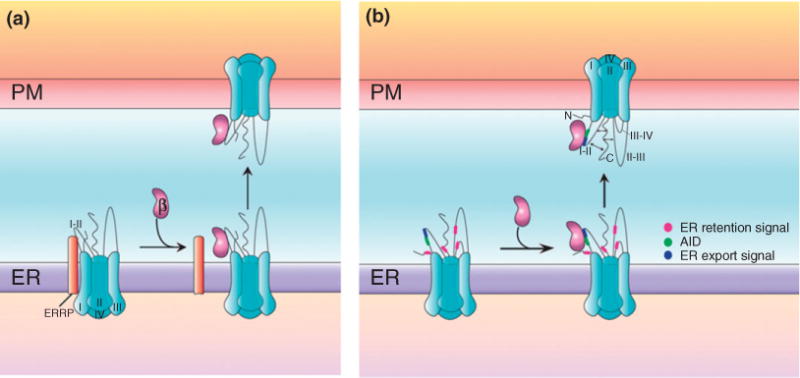
Two distinct models for CaVβ subunit-induced increase in CaV channel expression at the plasma membrane. (a) In the absence of CaVβ, the CaVα1 subunit remains trapped within the endoplasmic reticulum (ER) by binding via the I-II loop onto an ER retention protein (ERRP) of unknown identity. Expression and subsequent binding of the CaVβ subunit to the I-II loop relieves the trafficking clamp imposed by the ERRP and allows CaV channel complexes to be targeted to the cell surface. (b) In the absence of CaVβ, an ER export signal (blue) present on the CaVα1 subunit I-II loop is functionally overcome by ER retention signals (pink) present in different regions of the protein, leading to channels being retained in the ER. Upon CaVβ binding to the CaVα1 subunit, a C-terminus-dependent conformational change of the intracellular domains occurs that diminishes the strength of ER retention signals leading to channel transport to the plasma membrane.
There are also studies that point to the importance of intramolecular domains in the physiology of the CaVβ subunits. In particular, it has been reported that CaVβ3 contains two PEST-like sequences, potential signals for rapid protein degradation,64 sensitive to the Ca2+-dependent protease calpain. CaVβ3 mutants lacking the PEST sequences induce increased CaV2.2 current densities,64 suggesting that CaVβ subunit proteolytic cleavage may be important for Ca2+ channel surface expression.
Likewise, the role of CaVβs as critical determinants of channel cell surface expression has been also evidenced by protein–protein interaction assays. Using a yeast two-hybrid screening approach, Béguin et al.36 initially uncovered a link between regulation of CaV channel trafficking and signaling by small guanosine triphosphatases (GTPases). Using the CaVβ3 subunit as bait to screen a cDNA library these authors identified the Ras-like GTPase kir/Gem as an interacting partner. Interestingly, the binding of kir/Gem to CaVβ3 inhibited their assembly with the CaVα1 subunit, such that kir/Gem cotransfection drastically inhibited CaV channel functional expression.36
In line with this, using an extracellular epitope to probe for surface expression of the channels, subsequent studies showed a drastic reduction of fluorescent signal when channels and their subunits were coexpressed with the small GTPase RGK.37,38 This led to the initial conclusion that the RGK proteins disrupt the CaVα1/CaVβ subunit association, thus retaining newly synthesized channels in the ER and reducing the number of functional channels in the plasma membrane. However, it has been shown more recently that the reduction in surface expression of CaV 1.2 channels by small GTPases (e.g. Rem), seems to occur by enhancing the dynamin-dependent endocytosis pathway rather than by a sequestration of the CaVβ subunits by the small GTPase.16 Interestingly, restoring CaV1.2 surface expression by coexpressing a dominant negative dynamin mutant was not sufficient to restore current densities, suggesting that Rem has at least one other mode of action to inhibit currents, most likely an effect on channel open probability.65,66
Recent work from Colecraft’s group offers new insight into the complex mode of RGK-mediated CaV channel inhibition. According to their proposal which is based on a customized mechanism at both the channel and GTPase level, distinct RGKs differentially use CaVβ-binding-dependent and CaVα1-binding-dependent mechanisms to inhibit CaV1 and CaV2 channels.17 In this scenario, Rem may inhibit CaV 1.2 channel using both CaVβ-binding-dependent and independent mechanisms and binding to CaVβ is required for Rem-mediated decrease in CaV1.2 channel surface density and open probability. On the other hand, Rem associates directly with CaV1.2α1 to initiate β-binding-independent inhibition while inhibits CaV2.2 channels using a solely CaVβ-binding-dependent mechanism.17
The CaVβ subunits have been also implicated in trafficking of CaV channels to specific subcellular regions. Vendel et al.67 found that CaVβ4a, one of the two alternative splicing variants of the N-terminal A domain of the CaVβ4 auxiliary subunit,68 may exert functions other than modulation of channel gating and trafficking by interacting with the microtubule-associated protein 1A (MAP1A) as well as synaptotagmin I, an important protein for presynaptic vesicle release. These interactions suggest that the CaVβ4a subunit may act as a scaffolding element to facilitate coupling of Ca2+ signaling with neurotransmitter release. Since the Cavβ4a-synaptotagmin I interaction is disrupted by Ca2+,67 it has been speculated that at basal Ca2+ levels CaVβ4a may interact both with the CaV2.1α1 subunit and the vesicle protein to organize the machinery necessary for vesicle release, and that Ca2+ entry via CaV channels disrupts this interaction, thus releasing the vesicle to allow fusion with the plasma membrane.43
More recently, it has been found that different members of the RIM family, putative effectors of Rab3, may functionally link CaV channels to proteins in the active zone at the presynapsis which are related to the machinery for exocytosis69 (Figure 3). Moreover, it has been reported that several proteins in the active zone including RIM1 modulate neuronal CaV2.1 channels through interactions with the CaVβ subunits. These interactions not only modify the inactivation properties of the channels and cause a sustained Ca2+ influx, but also anchor the neurotransmitter-containing vesicles to the channels in the vicinity.40,41 Similar results have been reported for recombinant L-type channels (of the CaV1.2 and CaV1.3 class) in insulin-secreting cells.42
FIGURE 3.
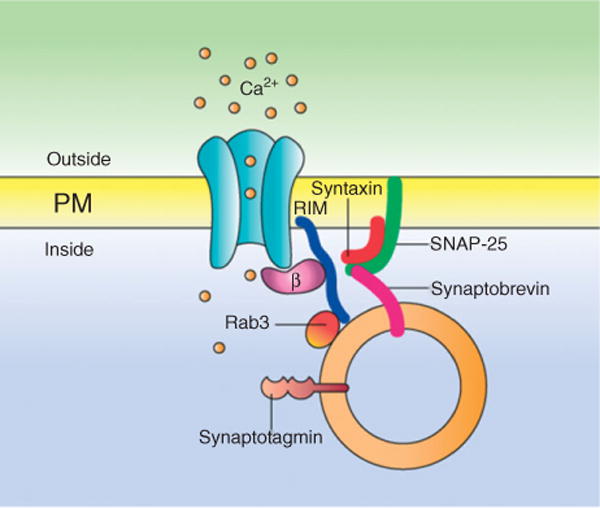
Functional coupling between CaV channels and secretory vesicles via the CaVβ subunit. (a) RIMs anchor synaptic vesicles next to channels through its interaction with zone-specific proteins (Rab3) and the CaVβ subunit. After depolarization, RIMs regulate the time course of channel inactivation resulting in a sustained Ca2+ influx. This molecular organization favors hormone and neurotransmitter release.
THE CaVα2δ SUBUNITS
As in the case of CaVβs, the ability of the CaVα2δ auxiliary subunits to promote membrane expression of HVA CaV channels is well established.6,70 Four subtypes of CaVα2δ proteins encoded by four separate genes with several known splice variants have been described.71,72 In general, the Cacna2d genes (coding the CaVα2δ subunits) are translated as precursor proteins that are proteolytically cleaved into a transmembrane δ region and an extracellular α2 domain, that remain linked by a disulphide bond.3,6,70 The extracellular α2 region is heavily glycosylated and contains several functional domains, including a 178 amino acid von Willebrand factor-A (vWFA) domain similar to extracellular matrix-binding regions of integrins, a 5 amino acid metal ion-dependent adhesion site (MIDAS) motif and a poorly understood 92 amino acid Cache domain6 (Figure 1(d)). The transmembrane domain of δ and some extracellular regions of α2 are thought to associate with the CaVα1 subunit of HVA channels.73
Although the functional relevance of the CaVα2δ subunits is not fully understood, there is compelling evidence indicating a role in trafficking and also in the modulation of channel biophysical properties. Heterologous expression of CaVα2δ with various CaVα1 and CaVβ subunits results in increased current densities, as well as altered current kinetics and current–voltage relationships.23,25,74–79 The increase in current density is generally explained by improved targeting of CaV channels to the plasma membrane74,78 and significant reduction in the rate of entry of surface resident channels into degradation pathways.80 Additionally, the CaVα2δ subunits have been involved in cell surface organization of CaV channels. It has been shown that CaVα2δ-1 (and likely the other CaVα2δ subunits) not only partitions into lipid rafts itself but also mediates raft-partitioning of neuronal CaV2.2 channel complexes.81–83
Recently, a mechanism has been proposed for CaV channel localization to lipid rafts which challenges the conventional structural model of the CaVα2δ subunits. This suggests that CaVα2δ associates with the plasma membrane via a glycosylphosphatidylinositol (GPI) anchor in the δ domain.84 In order to examine the role of membrane anchoring of the CaVα2δ-1 subunit on its biochemical and functional properties, Dolphin and her colleagues generated a CaVα2δ-1 truncated at the putative GPI-anchor ω-site, which removes the C-terminal hydrophobic domain (α2δ-1 ΔC-term85). Unexpectedly, the α2δ-1 ΔC-term protein was able to produce a significant increase of Ca2+ currents in cells expressing CaV2.1/CaVβ1 channels. Likewise, these authors did not observe any effect upon external application of secreted α2δ-1Δ C-term, which suggested that an intracellular interaction with other subunits may be needed for the complete functionality of the CaVα2δ-1 subunit in the channel complex. Last, a large proportion of α2δ-1 Δ C-term was secreted into the medium, but despite the lack of a C-terminal membrane anchor, the α2δ-1Δ C-term proteins remained partially associated with the plasma membrane. It has been speculated that this interaction may occur via a noncovalent linkage formed during the maturation and trafficking of the protein.85 Other studies, have also shown that the raft localization of CaVα2δ is preserved after replacement of the GPI anchoring motif,83 which is consistent with the idea that the CaVα2δ subunits may reside in lipid rafts via protein–protein and/or specialized lipid–protein interactions.
As mentioned earlier, during biosynthesis the CaVα2δ subunits are generated as precursor proteins which undergo post-translational cleavage, oxidation, and glycosylation to yield mature proteins comprised of disulphide-linked α2 and δ glycopolypeptides.86–88 Although the precise functional role of the post-translational modifications of the CaVα2δ subunits has not yet been fully defined, it has been suggested that they could play a role in channel trafficking. Initial experiments by Gurnett et al.75 showed that N-linked glycosylation play a critical role for surface expression of CaV2.2 channels since the Ca2+ current stimulation normally observed after CaVα2δ coexpression was virtually abolished by deglycosylation. Moreover, by combining electrophysiology with site-directed mutagenesis two N-glycosylation sites were found in the α2 domain of the protein that are required for the auxiliary subunit-induced current stimulation. Given that the mutation of these sites prevented current stimulation without altering its kinetic properties, a regulation on the number of functional channels at the plasma membrane was suggested.86
In addition, alterations in the proteolytic cleavage that typically separates the extracellular α2 from the transmembrane δ domain89 may interfere with proper assembly and trafficking of the CaV2.2 channel complex. After mutation of the amino acids in the proteolytic site, CaVα2δ becomes insensitive to proteolytic cleavage and loses its ability to increase currents. These changes are not accompanied with alterations in the voltage dependence or kinetics of the channels, suggesting a reduction in the number of channels targeted to the plasma membrane.87
Disulfide bond formation seems to be also important for channel cell surface expression. Recent studies have shown that a pair of conserved cysteine residues at positions 404 and 1047, located in the vWFA region of α2 and the extracellular domain of δ, respectively, form a single intermolecular disulfide bridge required for normal α2δ−1 subunit function.88 In these studies, it was shown that whereas there was a significant increase in current density through CaV2.2α1/β3 channels upon cotransfection with wild-type CaVα2δ−1, Cys404Ser, and Cys1047Ser mutations had little influence in current density, implying that formation of an intra-subunit disulfide bond between these residues is essential for CaVα2δ-1 functional enhancement of currents, and perhaps for CaV channel trafficking to the plasma membrane.
Likewise, some insights into the role of the CaVα2δ auxiliary subunits in vivo come from a series of spontaneously occurring mutations in mice. These include two mutations in CaVα2δ-2 (ducky and entla) that result in similar phenotypes characterized by ataxia, paroxysmal dyskinesia, and spike wave seizures.52,90,91 In humans, a mutation in CaVα2δ-4 that introduces a premature stop codon that truncates one-third of the corresponding open reading frame presumably underlies a channelopathy leading to cone-rod dysfunction in the visual system.92 More recently, a novel gene defect in CACNA2D2 encoding the CaVα2δ-2 subunit has been associated with early infantile epileptic encephalopathy a disease that usually manifest as severely impaired cognitive and motor development.93 Last, mutations in the CACNA2D1 gene might be associated with some forms of cardiac dysfunction, including the Brugada and long QT syndromes.94,95
Interestingly, it has been reported that experimental peripheral nerve injury results in increased levels of Caα2δ-1 mRNA in damaged dorsal root ganglion (DRG) sensory neurons, as well as a corresponding increase in protein levels in the neuronal cell bodies and at their presynaptic terminals in the spinal cord.96–98 Furthermore, mice overexpressing the CaVα2δ-1 subunit show a neuropathic phenotype of hyperalgesia and tactile allodynia in the absence of nerve injury,99 suggesting that CaVα2δ-1 is instrumental to the excitability of DRG neurons and the expression of neuropathic pain.
Evidence supporting the role of the CaVα2δ subunits in the promotion of surface expression of channels comes, in part, from the fact that CaVα2δ is the principal target for a group of small molecules with analgesic effects called gabapentinoids.100 Although the mechanisms by which these drugs alleviate pain is not well understood, they seem to act intracellularly after chronic but not acute treatment.79,101–105 Therefore, Vega-Hernandez and Felix102 initially formulated the hypothesis that gabapentinoids could be impairing the ability of CaVα2δ to enhance the number of channels in the plasma membrane, via an effect on trafficking. Subsequent studies showed that indeed GBP acting chronically by displacing an endogenous ligand that is normally a positive modulator of CaVα2δ, could be interfering with the function of the vWFA domain impairing CaV channel trafficking to the membrane78,104 (Figure 4).
FIGURE 4.
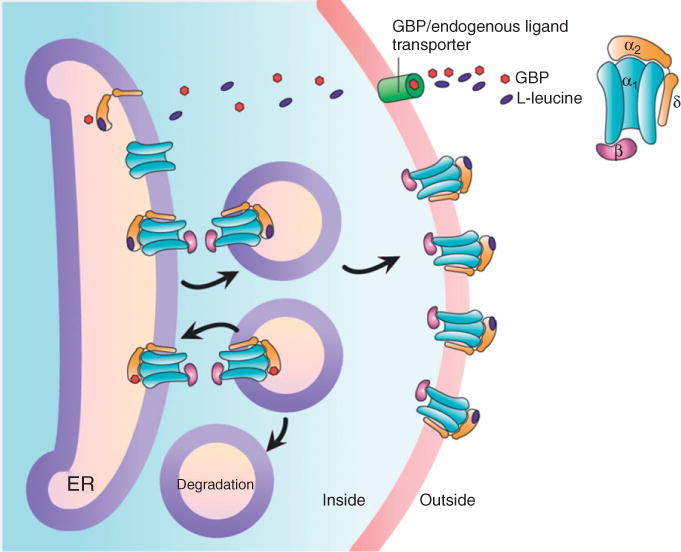
Hypothetical mechanisms of action of gabapentin (GBP). The effect of GBP may be to displace an endogenous ligand (L-leucine) and impair the ability of the auxiliary CaVα2δ subunit to increase the number of functional channels at the plasma membrane. GBP may be entering the cells using the system-L transporter protein LAT4 and might be exerting its effect on intracellular CaVα2δ subunits during assembly and trafficking of the CaV channel complex to the cell surface.
In addition, gabapentin has been shown to inhibit Rab11-dependent recycling of N-type CaV channels from the plasma membrane.106 However, not only the trafficking and recycling of recombinant CaV channels appears to be impeded by gabapentin but also the enhancement of native N-type channel at the cell surface associated with chronic pain.98 It is worth mentioning that endosomal trafficking may represent an important mechanism for maintaining proper plasma membrane expression of distinct native CaV channels,107,108 though the role of CaVα2δ in this process, if any, is yet to be determined.
Last, although investigations regarding the role of the auxiliary subunits on subcellular distributions of CaV channels have primarily focused on the CaVβ subunits in neurons,40,41 initial studies using L-type channels have revealed that the CaVα2δ subunits may also play a role in this process. Hence, it has been reported that the RalA GTPase regulates CaV channel activity, and acts as a chaperone for hormone secretory granules to L- and R-type channels on the plasma membrane, where these granules become tethered, in preparation for the formation of excitosome complexes with vesicle and plasma membrane SNARE proteins. Notably, this interaction occurs through a physical association between RalA and CaVα2δ-1,109 suggesting that the RalA/CaVα2δ-1 complex likely engage and traffic secretory granules to the plasma membrane-bound CaVα1 subunits to be able to then conduct Ca2+ through functional channels at precise sites where hormone release occurs.
ROLE OF THE AUXILIARY SUBUNITS ON HVA CaV CHANNEL REGULATED DEGRADATION
Recent studies have proposed that the CaVβ auxiliary subunits increase channel surface expression by preventing CaVα1 ubiquitination and proteasomal degradation.63,110 Indeed, CaV1.2 channels are tonically ubiquitinated and the degree of ubiquitination seems to be increased in the absence of the CaVβ subunits by an ER associated ubiquitin ligase called RFP2. Hence, in the absence of CaVβ, an interaction occurs between the channel and proteins related to the ER Associated Protein Degradation (ERAD) system. The end result of this is channel retrotranslocation and proteasomal degradation in the cytosol. On the other hand, in the absence of the CaVβ subunits, a proteasome inhibitor can rescue CaVα1 surface expression.110
These results suggest that the Cavβ subunits may be required to help CaVα1 (particularly of the CaV1.2 and CaV2.2 class) escape the ubiquitin proteasome system (UPS) degradation pathway.63,110 Consistent with this, reduced levels of bulk CaV2.2 and Ub-CaV2.2 proteins are observed in the absence of CaVβ subunits.111 In this regard, elements in the proximal C-terminus of a CaV2.2α1 alternative splice variant (e37b) predispose cloned and native channels to downregulation by the UPS.111 Therefore, the question arises as to whether sequences unique to e37b may destabilize the CaV2.2/CaVβ subunit interaction leading to increased ubiquitination and degradation by the UPS.
In a similar manner, it has been suggested that the CaVα2δ-1 subunit may play a role in stabilizing CaV channel functional expression once delivered to the plasma membrane. By examining the binding, internalization, and degradation kinetics of recombinant N-type (CaV2.2α1δ/CaVβ1b) channels in the presence or the absence of CaVα2/δ-1, Bernstein and Jones80 proposed that the CaVα2δ subunits may act, at least in part, reducing the entry of surface resident CaV channels into degradative pathways. Computer modeling of trafficking data showed that CaVα2δ stabilizes N-type channels after their delivery to the cell surface through a significant reduction in the rate constants for internalization and degradation.80 These kinetic changes help to explain the enhanced N-type channel expression levels and suggest that, in conjunction with forward trafficking effects (see the preceding section), the CaVα2δ subunits play a significant role in defining HVA CaV channel functional expression.
Last, it is well established that skeletal muscle L-type Ca2+ channels contain a CaVγ1 subunit which includes four transmembrane helices.1,3 It was subsequently shown that neurons express at least four homologues of CaVγ1,112 most notably CaVγ2 also known as stargazin,113,114 which in addition to regulate CaV channel function may also play a role in mediating AMPA receptor trafficking to the synapsis.115 In general, the γ subunits appear to be inhibitory and their physiological relevance is emphasized by the epileptic phenotype in the stargazer mouse, a spontaneous mutant that lacks the CaVγ2 subunit.52,112,113,115
Although it has been shown that CaVγ1, CaVγ2, and CaVγ7 drastically reduce Ca2+ current density in heterologous expression systems,116–119 it is worth noting that these effects may not be associated with altered channel trafficking but most likely occur as a result of changes in channel expression. Interestingly, the effect of the CaVγ subunits resemble the inhibition of the CaV2.2 currents induced by the coexpression of truncated CaV2.2 constructs,120 which occurs after the activation of PERK, a component of the unfolded protein response (UPR).118,121 When UPR is initiated, an immediate consequence is the inhibition of protein biosynthesis through phosphorylation of the translation initiation factor eIF2α.122
PERSPECTIVES
Molecular and cellular biological studies have begun to explore the intriguing question of the spatial and temporal control of CaV channel density in the plasma membrane. These studies will surely be followed with more mechanistic analyses of the trafficking, targeting, recycling, and degradation, as well as their interactions with the cytoskeleton and regulation by cellular activity. Future studies must consider also other important variables such as distinct cell phenotypes, developmental, and cell cycle stages, and the dynamic arrangement of microdomains within a given cell. Therefore, the molecular characterization of the possible roles of CaV channel auxiliary subunits is only one of the crucial early steps towards understanding how different cells regulate membrane expression and organization of CaV channels for functional purposes. Undoubtedly, the current shortage of knowledge will be the motivation for intensive work in this field in the near future.
Acknowledgments
This work was supported in part by a grant (128707-Q) from The National Council for Science and Technology (Conacyt, Mexico) to RF and from NIH (1K99MH099405-01) to AA. A doctoral fellowship from Conacyt to ACR is also gratefully acknowledged.
Footnotes
Conflict of interest: The authors have declared no conflicts of interest for this article.
References
- 1.Catterall WA. Voltage-gated calcium channels. Cold Spring Harb Perspect Biol. 2011;3:a003947. doi: 10.1101/cshperspect.a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Perez-Reyes E. Molecular physiology of low-voltage-activated t-type calcium channels. Physiol Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- 3.Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- 4.Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- 5.Richards KS, Swensen AM, Lipscombe D, Bommert K. Novel CaV2.1 clone replicates many properties of Purkinje cell CaV2.1 current. Eur J Neurosci. 2007;26:2950–2961. doi: 10.1111/j.1460-9568.2007.05912.x. [DOI] [PubMed] [Google Scholar]
- 6.Dolphin AC. Calcium channel auxiliary α2δ and β subunits: trafficking and one step beyond. Nat Rev Neurosci. 2012;13:542–555. doi: 10.1038/nrn3311. [DOI] [PubMed] [Google Scholar]
- 7.Simms BA, Zamponi GW. Trafficking and stability of voltage-gated calcium channels. Cell Mol Life Sci. 2012;69:843–856. doi: 10.1007/s00018-011-0843-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- 9.Buraei Z, Yang J. The β subunit of voltage-gated Ca2+ channels. Physiol Rev. 2010;90:1461–1506. doi: 10.1152/physrev.00057.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hanlon MR, Berrow NS, Dolphin AC, Wallace BA. Modelling of a voltage-dependent Ca2+ channel β subunit as a basis for understanding its functional properties. FEBS Lett. 1999;445:366–370. doi: 10.1016/s0014-5793(99)00156-8. [DOI] [PubMed] [Google Scholar]
- 11.Oliva C, Escobedo P, Astorga C, Molina C, Sierralta J. Role of the MAGUK protein family in synapse formation and function. Dev Neurobiol. 2012;72:57–72. doi: 10.1002/dneu.20949. [DOI] [PubMed] [Google Scholar]
- 12.Buraei Z, Yang J. Structure and function of the β subunit of voltage-gated Ca2+ channels. Biochim Biophys Acta. 2013;1828:1530–1540. doi: 10.1016/j.bbamem.2012.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the α1−β subunit interaction of voltage-gated Ca2+ channels. Nature. 2004;429:675–680. doi: 10.1038/nature02641. [DOI] [PubMed] [Google Scholar]
- 14.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel β subunit functional core and its complex with the α1 interaction domain. Neuron. 2004;42:387–399. doi: 10.1016/s0896-6273(04)00250-8. [DOI] [PubMed] [Google Scholar]
- 15.Van Petegem F, Clark KA, Chatelain FC, Minor DL., Jr Structure of a complex between a voltage-gated calcium channel β-subunit and an α-subunit domain. Nature. 2004;429:671–675. doi: 10.1038/nature02588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang T, Xu X, Kernan T, Wu V, Colecraft H. Rem, a member of the RGK GTPases, inhibits recombinant CaV1.2 channels using multiple mechanisms that require distinct conformations of the GTPase. J Physiol. 2010;588:1665–1681. doi: 10.1113/jphysiol.2010.187203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang T, Puckerin A, Colecraft HM, Distinct RGK. GTPases differentially use α1- and auxiliary β-binding-dependent mechanisms to inhibit CaV1.2/CaV2.2 channels. PLoS One. 2012;7:e37079. doi: 10.1371/journal.pone.0037079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang T, Colecraft HM. Regulation of voltage-dependent calcium channels by RGK proteins. Biochem Biophys Acta. 2013;1828:1644–1654. doi: 10.1016/j.bbamem.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zou S, Jha S, Kim EY, Dryer SE. The β1 subunit of L-type voltage-gated Ca2+ channels independently binds to and inhibits the gating of large-conductance Ca2+ –activated K+ channels. Mol Pharmacol. 2008;73:369–378. doi: 10.1124/mol.107.040733. [DOI] [PubMed] [Google Scholar]
- 20.Foell JD, Balijepalli RC, Delisle BP, Yunker AM, Robia SL, Walker JW, McEnery MW, January CT, Kamp TJ. Molecular heterogeneity of calcium channel β-subunits in canine and human heart: evidence for differential subcellular localization. Physiol Genomics. 2004;17:183–200. doi: 10.1152/physiolgenomics.00207.2003. [DOI] [PubMed] [Google Scholar]
- 21.Lacerda AE, Kim HS, Ruth P, Perez-Reyes E, Flockerzi V, Hofmann F, Birnbaumer L, Brown AM. Normalization of current kinetics by interaction between the α1 and β subunits of the skeletal muscle dihydropyridine-sensitive Ca2+ channel. Nature. 1991;352:527–530. doi: 10.1038/352527a0. [DOI] [PubMed] [Google Scholar]
- 22.Mori Y, Friedrich T, Kim MS, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, et al. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- 23.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. The roles of the subunits in the function of the calcium channel. Science. 1991;253:1553–1557. doi: 10.1126/science.1716787. [DOI] [PubMed] [Google Scholar]
- 24.Varadi G, Lory P, Schultz D, Varadi M, Schwartz A. Acceleration of activation and inactivation by the β subunit of the skeletal muscle calcium channel. Nature. 1991;352:159–162. doi: 10.1038/352159a0. [DOI] [PubMed] [Google Scholar]
- 25.Williams ME, Feldman DH, McCue AF, Brenner R, Velicelebi G, Ellis SB, Harpold MM. Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron. 1992;8:71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- 26.Neely A, Wei X, Olcese R, Birnbaumer L, Stefani E. Potentiation by the β subunit of the ratio of the ionic current to the charge movement in the cardiac calcium channel. Science. 1993;262:575–578. doi: 10.1126/science.8211185. [DOI] [PubMed] [Google Scholar]
- 27.Stea A, Dubel SJ, Pragnell M, Leonard JP, Campbell KP, Snutch TP. A β-subunit normalizes the electrophysiological properties of a cloned N-type Ca2+ channel α1-subunit. Neuropharmacology. 1993;32:1103–1116. doi: 10.1016/0028-3908(93)90005-n. [DOI] [PubMed] [Google Scholar]
- 28.Bünemann M, Gerhardstein BL, Gao T, Hosey MM. Functional regulation of L-type calcium channels via protein kinase A-mediated phosphorylation of the β2 subunit. J Biol Chem. 1999;274:33851–33854. doi: 10.1074/jbc.274.48.33851. [DOI] [PubMed] [Google Scholar]
- 29.Fitzgerald EM. The presence of Ca2+ channel β subunit is required for mitogen-activated protein kinase (MAPK)-dependent modulation of α1B Ca2+ channels in COS-7 cells. J Physiol. 2002;543:425–437. doi: 10.1113/jphysiol.2002.022822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Viard P, Butcher AJ, Halet G, Davies A, Nürnberg B, Heblich F, Dolphin AC. PI3K promotes voltage-dependent calcium channel trafficking to the plasma membrane. Nat Neurosci. 2004;7:939–946. doi: 10.1038/nn1300. [DOI] [PubMed] [Google Scholar]
- 31.Grueter CE, Abiria SA, Dzhura I, Wu Y, Ham AJ, Mohler PJ, Anderson ME, Colbran RJ. L-type Ca2+ channel facilitation mediated by phosphorylation of the β subunit by CaMKII. Mol Cell. 2006;23:641–650. doi: 10.1016/j.molcel.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 32.Su SC, Seo J, Pan JQ, Samuels BA, Rudenko A, Ericsson M, Neve RL, Yue DT, Tsai LH. Regulation of N-type voltage-gated calcium channels and presynaptic function by cyclin-dependent kinase 5. Neuron. 2012;75:675–687. doi: 10.1016/j.neuron.2012.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G protein-dependent opioid modulation of neuronal calcium channels. Proc Natl Acad Sci USA. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of gβγ with a C-terminal gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G proteincoupled receptors. Proc Natl Acad Sci USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang Y, Chen YH, Bangaru SD, He L, Abele K, Tanabe S, Kozasa T, Yang J. Origin of the voltage dependence of G-protein regulation of P/Q-type Ca2+ channels. J Neurosci. 2008;28:14176–14188. doi: 10.1523/JNEUROSCI.1350-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Béguin P, Nagashima K, Gonoi T, Shibasaki T, Takahashi K, Kashima Y, Ozaki N, Geering K, Iwanaga T, Seino S. Regulation of Ca2+ channel expression at the cell surface by the small G-protein kir/Gem. Nature. 2001;411:701–706. doi: 10.1038/35079621. [DOI] [PubMed] [Google Scholar]
- 37.Béguin P, Mahalakshmi RN, Nagashima K, Cher DH, Ikeda H, Yamada Y, Seino Y, Hunziker W. Nuclear sequestration of β-subunits by Rad and Rem is controlled by 14-3-3 and calmodulin and reveals a novel mechanism for Ca2+ channel regulation. J Mol Biol. 2006;355:34–46. doi: 10.1016/j.jmb.2005.10.013. [DOI] [PubMed] [Google Scholar]
- 38.Béguin P, Ng Y, Krause C, Mahalakshmi R, Ng M, Hunziker W. RGK small GTP-binding proteins interact with the nucleotide kinase domain of Ca2+-channel β-subunits via an uncommon effector binding domain. J Biol Chem. 2007;282:11509–11520. doi: 10.1074/jbc.M606423200. [DOI] [PubMed] [Google Scholar]
- 39.Finlin BS, Correll RN, Pang C, Crump SM, Satin J, Andres DA. Analysis of the complex between Ca2+ channel β-subunit and the Rem GTPase. J Biol Chem. 2006;281:23557–23566. doi: 10.1074/jbc.M604867200. [DOI] [PubMed] [Google Scholar]
- 40.Xiea S, Wakamori M, Miki T, Uriu Y, Nonaka M, Bito H, et al. RIM1 confers sustained activity and neurotransmitter vesicle anchoring to presynaptic Ca2+ channels. Nat Neurosci. 2007;10:691–701. doi: 10.1038/nn1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiyonaka S, Nakajima H, Takada Y, Hida Y, Yoshioka T, Hagiwara A, Kitajima I, Mori Y, Ohtsuka T. Physical and functional interaction of the active zone protein CAST/ERC2 and the β-subunit of the voltage-dependent Ca2+ channel. J Biochem. 2012;152:149–159. doi: 10.1093/jb/mvs054. [DOI] [PubMed] [Google Scholar]
- 42.Gandini MA, Sandoval A, González-Ramírez R, Mori Y, de Waard M, Felix R. Functional coupling of Rab3-interacting molecule 1 (RIM1) and L-type Ca2+ channels in insulin release. J Biol Chem. 2011;286:15757–15765. doi: 10.1074/jbc.M110.187757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Weiss N, Sandoval A, Kyonaka S, Felix R, Mori Y, De Waard M. Rim1 modulates direct G-protein regulation of CaV2.2 channels. Pflugers Arch. 2011;461:447–459. doi: 10.1007/s00424-011-0926-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gregg RG, Messing A, Strube C, Beurg M, Moss R, Behan M, Sukhareva M, Haynes S, Powell JA, Coronado R, et al. Absence of the β subunit (cchb1) of the skeletal muscle dihydropyridine receptor alters expression of the α1 subunit and eliminates excitation-contraction coupling. Proc Natl Acad Sci USA. 1996;93:13961–13966. doi: 10.1073/pnas.93.24.13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weissgerber P, Held B, Bloch W, Kaestner L, Chien KR, Fleischmann BK, Lipp P, Flockerzi V, Freichel M. Reduced cardiac L-type Ca2+ current in CaVβ2–/– embryos impairs cardiac development and contraction with secondary defects in vascular maturation. Circ Res. 2006;99:749–757. doi: 10.1161/01.RES.0000243978.15182.c1. [DOI] [PubMed] [Google Scholar]
- 46.Burgess DL, Jones JM, Meisler MH, Noebels JL. Mutation of the Ca2+ channel β subunit gene Cchb4 is associated with ataxia and seizures in the lethargic (lh) mouse. Cell. 1997;88:385–392. doi: 10.1016/s0092-8674(00)81877-2. [DOI] [PubMed] [Google Scholar]
- 47.Murakami M, Fleischmann B, De Felipe C, Freichel M, Trost C, Ludwig A, Wissenbach U, Schwegler H, Hofmann F, Hescheler J, et al. Pain perception in mice lacking the β3 subunit of voltage-activated calcium channels. J Biol Chem. 2002;277:40342–40351. doi: 10.1074/jbc.M203425200. [DOI] [PubMed] [Google Scholar]
- 48.Hibino H, Pironkova R, Onwumere O, Rousset M, Charnet P, Hudspeth AJ, Lesage F. Direct interaction with a nuclear protein and regulation of gene silencing by a variant of the Ca2+–channel β4 subunit. Proc Natl Acad Sci USA. 2003;100:307–312. doi: 10.1073/pnas.0136791100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhang Y, Yamada Y, Fan M, Bangaru SD, Lin B, Yang J. The β subunit of voltage-gated Ca2+ channels interacts with and regulates the activity of a novel isoform of Pax6. J Biol Chem. 2010;285:2527–2536. doi: 10.1074/jbc.M109.022236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tadmouri A, Kiyonaka S, Barbado M, Rousset M, Fablet K, Sawamura S, Bahembera E, Pernet-Gallay K, Arnoult C, Miki T, et al. Cacnb4 directly couples electrical activity to gene expression, a process defective in juvenile epilepsy. EMBO J. 2012;31:3730–3744. doi: 10.1038/emboj.2012.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ronjat M, Kiyonaka S, Barbado M, De Waard M, Mori Y. Nuclear life of the voltage-gated Cacnb4 subunit and its role in gene transcription regulation. Channels (Austin) 7:119–125. doi: 10.4161/chan.23895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Felix R. Insights from mouse models of absence epilepsy into Ca2+ channel physiology and disease etiology. Cell Mol Neurobiol. 2002;22:103–120. doi: 10.1023/a:1019807719343. [DOI] [PubMed] [Google Scholar]
- 53.Namkung Y, Smith SM, Lee SB, Skrypnyk NV, Kim HL, Chin H, Scheller RH, Tsien RW, Shin HS. Targeted disruption of the Ca2+ channel β3 subunit reduces N- and L-type Ca2+ channel activity and alters the voltage-dependent activation of P/Q-type Ca2+ channels in neurons. Proc Natl Acad Sci USA. 1998;95:12010–12015. doi: 10.1073/pnas.95.20.12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Caddick SJ, Wang C, Fletcher CF, Jenkins NA, Copeland NG, Hosford DA. Excitatory but not inhibitory synaptic transmission is reduced in lethargic (Cacnb4(lh)) and tottering (Cacna1atg) mouse thalami. J Neurophysiol. 1999;81:2066–2074. doi: 10.1152/jn.1999.81.5.2066. [DOI] [PubMed] [Google Scholar]
- 55.Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation. 2007;115:442–449. doi: 10.1161/CIRCULATIONAHA.106.668392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cordeiro JM, Marieb M, Pfeiffer R, Calloe K, Burashnikov E, Antzelevitch C. Accelerated inactivation of the L-type calcium current due to a mutation in CACNB2b underlies Brugada syndrome. J Mol Cell Cardiol. 2009;46:695–703. doi: 10.1016/j.yjmcc.2009.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Escayg A, De Waard M, Lee DD, Bichet D, Wolf P, Mayer T, Johnston J, Baloh R, Sander T, Meisler MH. Coding and noncoding variation of the human calcium-channel β4 subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000;66:1531–1539. doi: 10.1086/302909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The I-II loop of the Ca2+ channel α1 subunit contains an endoplasmic reticulum retention signal antagonized by the β subunit. Neuron. 2000;25:177–190. doi: 10.1016/s0896-6273(00)80881-8. [DOI] [PubMed] [Google Scholar]
- 59.Dalton S, Takahashi SX, Miriyala J, Colecraft HM. A single CaVβ can reconstitute both trafficking and macroscopic conductance of voltage-dependent calcium channels. J Physiol. 2005;567:757–769. doi: 10.1113/jphysiol.2005.093195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tareilus E, Roux M, Qin N, Olcese R, Zhou J, Stefani E, Birnbaumer L. A Xenopus oocyte β subunit: evidence for a role in the assembly/expression of voltage-gated calcium channels that is separate from its role as a regulatory subunit. Proc Natl Acad Sci USA. 1997;94:1703–1708. doi: 10.1073/pnas.94.5.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.He LL, Zhang Y, Chen YH, Yamada Y, Yang J. Functional modularity of the β-subunit of voltage-gated Ca2+ channels. Biophys J. 2007;93:834–845. doi: 10.1529/biophysj.106.101691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fang K, Colecraft HM. Mechanism of auxiliary β-subunit-mediated membrane targeting of L-type (CaV1.2) channels. J Physiol. 2011;589:4437–4455. doi: 10.1113/jphysiol.2011.214247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Altier C, Garcia-Caballero A, Simms B, You H, Chen L, Walcher J, Tedford HW, Hermosilla T, Zamponi GW. The CaVβ subunit prevents RFP2-mediated ubiquitination and proteasomal degradation of L-type channels. Nat Neurosci. 2011;14:173–180. doi: 10.1038/nn.2712. [DOI] [PubMed] [Google Scholar]
- 64.Sandoval A, Oviedo N, Tadmouri A, Avila T, De Waard M, Felix R. Two PEST-like motifs regulate Ca2+/calpain-mediated cleavage of the CaVβ3 subunit and provide important determinants for neuronal Ca2+ channel activity. Eur J Neurosci. 2006;23:2311–2320. doi: 10.1111/j.1460-9568.2006.04749.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen H, Puhl HL, Niu S, Mitchell D, Ikeda S. Expression of Rem2, an RGK family small GTPase, reduces N-type calcium current without affecting channel surface density. J Neurosci. 2005;25:9762–9772. doi: 10.1523/JNEUROSCI.3111-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Xu X, Marx S, Colecraft H. Molecular mechanisms, and selective pharmacological rescue, of Reminhibited CaV1.2 channels in heart. Circ Res. 2010;107:620–630. doi: 10.1161/CIRCRESAHA.110.224717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vendel AC, Terry MD, Striegel AR, Iverson NM, Leuranguer V, Rithner CD, Lyons BA, Pickard GE, Tobet SA, Horne WA. Alternative splicing of the voltage-gated Ca2+ channel β4 subunit creates a uniquely folded N-terminal protein binding domain with cell-specific expression in the cerebellar cortex. J Neurosci. 2006;26:2635–2644. doi: 10.1523/JNEUROSCI.0067-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Helton TD, Horne WA. Alterative splicing of the β4 subunit has α1 subtype-specific effects on Ca2+ channel gating. J Neurosci. 2002;22:1573–1582. doi: 10.1523/JNEUROSCI.22-05-01573.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gandini MA, Felix R. Functional interactions between voltage-gated Ca2+ channels and Rab3-interacting molecules (RIMs): new insights into stimulus-secretion coupling. Biochim Biophys Acta. 2012;1818:551–558. doi: 10.1016/j.bbamem.2011.12.011. [DOI] [PubMed] [Google Scholar]
- 70.Felix R. Voltage-dependent Ca2+ channel α2δ auxiliary subunit: structure, function and regulation. Receptors Channels. 1999;6:351–362. [PubMed] [Google Scholar]
- 71.Angelotti T, Hofmann F. Tissue-specific expression of splice variants of the mouse voltage-gated calcium channel α2/δ subunit. FEBS Lett. 1996;397:331–337. doi: 10.1016/s0014-5793(96)01205-7. [DOI] [PubMed] [Google Scholar]
- 72.Klugbauer N, Lacinová L, Marais E, Hobom M, Hofmann F. Molecular diversity of the calcium channel α2δ subunit. J Neurosci. 1999;19:684–691. doi: 10.1523/JNEUROSCI.19-02-00684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gurnett CA, Felix R, Campbell KP. Extracellular interaction of the voltage-dependent Ca2+ channel α2δ and α1 subunits. J Biol Chem. 1997;272:18508–18512. doi: 10.1074/jbc.272.29.18508. [DOI] [PubMed] [Google Scholar]
- 74.Shistik E, Ivanina T, Puri T, Hosey M, Dascal N. Ca2+ current enhancement by α2/δ and β subunits in Xenopus oocytes: contributions of changes in channel gating and α1 protein level. J Physiol. 1995;489:55–62. doi: 10.1113/jphysiol.1995.sp021029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gurnett CA, De Waard M, Campbell KP. Dual function of the voltage-dependent Ca2+ channel α2δ subunit in current stimulation and subunit interaction. Neuron. 1996;16:431–440. doi: 10.1016/s0896-6273(00)80061-6. [DOI] [PubMed] [Google Scholar]
- 76.Felix R, Gurnett CA, De Waard M, Campbell KP. Dissection of functional domains of the voltage dependent Ca2+ channel a2d subunit. J Neurosci. 1997;17:6884–6891. doi: 10.1523/JNEUROSCI.17-18-06884.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yasuda T, Chen L, Barr W, McRory JE, Lewis RJ, Adams DJ, Zamponi GW. Auxiliary subunit regulation of high-voltage activated calcium channels expressed in mammalian cells. Eur J Neurosci. 2004;20:1–13. doi: 10.1111/j.1460-9568.2004.03434.x. [DOI] [PubMed] [Google Scholar]
- 78.Cantí C, Nieto-Rostro M, Foucault I, Heblich F, Wratten J, Richards MW, Hendrich J, Douglas L, Page KM, Davies A, et al. The metal-ion-dependent adhesion site in the Von Willebrand factor-A domain of α2δ subunits is key to trafficking voltage-gated Ca2+ channels. Proc Natl Acad Sci USA. 2005;102:11230–11235. doi: 10.1073/pnas.0504183102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Andrade A, Sandoval A, González-Ramírez R, Lipscombe D, Campbell KP, Felix R. The α2δ subunit augments functional expression and modifies the pharmacology of CaV1.3 L-type channels. Cell Calcium. 2009;46:282–292. doi: 10.1016/j.ceca.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bernstein GM, Jones OT. Kinetics of internalization and degradation of N-type voltage-gated calcium channels: role of the α2δ subunit. Cell Calcium. 2007;41:27–40. doi: 10.1016/j.ceca.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 81.Davies A, Douglas L, Hendrich J, Wratten J, Tran Van Minh A, Foucault I, Koch D, Pratt WS, Saibil HR, Dolphin AC. The calcium channel α2δ-2 subunit partitions with CaV2.1 into lipid rafts in cerebellum: implications for localization and function. J Neurosci. 2006;26:8748–8757. doi: 10.1523/JNEUROSCI.2764-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Robinson P, Etheridge S, Song L, Armenise P, Jones OT, Fitzgerald EM. Formation of N-type (CaV2.2) voltage-gated calcium channel membrane microdomains: Lipid raft association and clustering. Cell Calcium. 2010;48:183–194. doi: 10.1016/j.ceca.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 83.Robinson P, Etheridge S, Song L, Shah R, Fitzgerald EM, Jones OT. Targeting of voltage-gated calcium channel α2δ-1 subunit to lipid rafts is independent from a GPI-anchoring motif. PLoS One. 2011;6:e19802. doi: 10.1371/journal.pone.0019802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Davies A, Kadurin I, Alvarez-Laviada A, Douglas L, Nieto-Rostro M, Bauer CS, Pratt WS, Dolphin AC. The α2δ subunits of voltage-gated calcium channels form GPI-anchored proteins, a posttranslational modification essential for function. Proc Natl Acad Sci U S A. 2010;107:1654–1659. doi: 10.1073/pnas.0908735107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kadurin I, Alvarez-Laviada A, Ng SF, Walker-Gray R, D’Arco M, Fadel MG, Pratt WS, Dolphin AC. Calcium currents are enhanced by α2δ-1 lacking its membrane anchor. J Biol Chem. 2012;287:33554–33566. doi: 10.1074/jbc.M112.378554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sandoval A, Oviedo N, Andrade A, Felix R. Glycosylation of asparagines 136 and 184 is necessary for the α2δ subunit-mediated regulation of voltage-gated Ca2+ channels. FEBS Lett. 2004;576:21–26. doi: 10.1016/j.febslet.2004.08.054. [DOI] [PubMed] [Google Scholar]
- 87.Andrade A, Sandoval A, Oviedo N, De Waard M, Elias D, Felix R. Proteolytic cleavage of the voltage-gated Ca2+ channel α2δ subunit: structural and functional features. Eur J Neurosci. 2007;25:1705–1710. doi: 10.1111/j.1460-9568.2007.05454.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Calderón-Rivera A, Andrade A, Hernández-Hernández O, González-Ramírez R, Sandoval A, Rivera M, Gomora JC, Felix R. Identification of a disulfide bridge essential for structure and function of the voltage-gated Ca2+ channel α2δ-1 auxiliary subunit. Cell Calcium. 2012;51:22–30. doi: 10.1016/j.ceca.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jay SD, Sharp AH, Kahl SD, Vedvick TS, Harpold MM, Campbell KP. Structural characterization of the dihydropyridine-sensitive calcium channel α2-subunit and the associated δ peptides. J Biol Chem. 1991;266:3287–3293. [PubMed] [Google Scholar]
- 90.Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, et al. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci. 2001;21:6095–6104. doi: 10.1523/JNEUROSCI.21-16-06095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Brill J, Klocke R, Paul D, Boison D, Gouder N, Klugbauer N, Hofmann F, Becker CM, Becker K. entla, a novel epileptic and ataxic Cacna2d2 mutant of the mouse. J Biol Chem. 2004;279:7322–7330. doi: 10.1074/jbc.M308778200. [DOI] [PubMed] [Google Scholar]
- 92.Wycisk KA, Zeitz C, Feil S, Wittmer M, Forster U, Neidhardt J, Wissinger B, Zrenner E, Wilke R, Kohl S, et al. Mutation in the auxiliary calcium-channel subunit CACNA2D4 causes autosomal recessive cone dystrophy. Am J Hum Genet. 2006;79:973–977. doi: 10.1086/508944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Edvardson S, Oz S, Abulhijaa FA, Taher FB, Shaag A, Zenvirt S, Dascal N, Elpeleg O. Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet. 2013;50:118–123. doi: 10.1136/jmedgenet-2012-101223. [DOI] [PubMed] [Google Scholar]
- 94.Burashnikov E, Pfeiffer R, Barajas-Martinez H, Delpón E, Hu D, Desai M, Borggrefe M, Häissaguerre M, Kanter R, Pollevick GD, et al. Mutations in the cardiac L-type calcium channel associated with inherited J-wave syndromes and sudden cardiac death. Hear Rhythm. 2010;7:1872–1882. doi: 10.1016/j.hrthm.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Templin C, Ghadri JR, Rougier JS, Baumer A, Kaplan V, Albesa M, Sticht H, Rauch A, Puleo C, Hu D, et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6) Eur Heart J. 2011;32:1077–1088. doi: 10.1093/eurheartj/ehr076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Luo ZD, Chaplan SR, Higuera ES, Sorkin LS, Stauderman KA, Williams ME, Yaksh TL. Upregulation of dorsal root ganglion α2δ calcium channel subunit and its correlation with allodynia in spinal nerve-injured rats. J Neurosci. 2001;21:1868–1875. doi: 10.1523/JNEUROSCI.21-06-01868.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Luo ZD, Calcutt NA, Higuera ES, Valder CR, Song YH, Svensson CI, Myers RR. Injury type-specific calcium channel α2δ-1 subunit up-regulation in rat neuropathic pain models correlates with antiallodynic effects of gabapentin. J Pharmacol Exp Ther. 2002;303:1199–1205. doi: 10.1124/jpet.102.041574. [DOI] [PubMed] [Google Scholar]
- 98.Bauer CS, Nieto-Rostro M, Rahman W, Tran-Van-Minh A, Ferron L, Douglas L, Kadurin I, Sri Ranjan Y, Fernandez-Alacid L, Millar NS, et al. The increased trafficking of the calcium channel subunit α2δ-1 to presynaptic terminals in neuropathic pain is inhibited by the α2δ ligand pregabalin. J Neurosci. 2009;29:4076–4088. doi: 10.1523/JNEUROSCI.0356-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Li CY, Zhang XL, Matthews EA, Li KW, Kurwa A, Boroujerdi A, Gross J, Gold MS, Dickenson AH, Feng G, et al. Calcium channel α2δ-1 subunit mediates spinal hyperexcitability in pain modulation. Pain. 2006;125:20–34. doi: 10.1016/j.pain.2006.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the α2δ subunit of a calcium channel. J Biol Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- 101.Kang MG, Felix R, Campbell KP. Long-term regulation of voltage-gated Ca2+ channels by gabapentin. FEBS Lett. 2002;528:177–182. doi: 10.1016/s0014-5793(02)03295-7. [DOI] [PubMed] [Google Scholar]
- 102.Vega-Hernández A, Felix R. Down-regulation of N-type voltage-activated Ca2+ channels by gabapentin. Cell Mol Neurobiol. 2002;22:185–190. doi: 10.1023/a:1019865822069. [DOI] [PubMed] [Google Scholar]
- 103.Zoidis G, Papanastasiou I, Dotsikas I, Sandoval A, Dos Santos RG, Papadopoulou-Daifoti Z, Vamvakides A, Kolocouris N, Felix R. The novel GABA adamantane derivative (AdGABA): design, synthesis, and activity relationship with gabapentin. Bioorg Med Chem. 2005;13:2791–2798. doi: 10.1016/j.bmc.2005.02.030. [DOI] [PubMed] [Google Scholar]
- 104.Hendrich J, Van Minh AT, Heblich F, Nieto-Rostro M, Watschinger K, Striessnig J, Wratten J, Davies A, Dolphin AC. Pharmacological disruption of calcium channel trafficking by the α2δ ligand gabapentin. Proc Natl Acad Sci USA. 2008;105:3628–3633. doi: 10.1073/pnas.0708930105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Martínez-Hernández E, Sandoval A, González-Ramírez R, Zoidis G, Felix R. Inhibition of recombinant N-type and native high voltage-gated neuronal Ca2+ channels by AdGABA: mechanism of action studies. Toxicol Appl Pharmacol. 2011;250:270–277. doi: 10.1016/j.taap.2010.10.030. [DOI] [PubMed] [Google Scholar]
- 106.Tran-Van-Minh A, Dolphin AC. The α2δ ligand gabapentin inhibits the Rab11-dependent recycling of the calcium channel subunit α2δ-2. J Neurosci. 2010;30:12856–12867. doi: 10.1523/JNEUROSCI.2700-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Green EM, Barrett CF, Bultynck G, Shamah SM, Dolmetsch RE. The tumor suppressor eIF3e mediates calcium-dependent internalization of the L-type calcium channel CaV1.2. Neuron. 2007;55:615–632. doi: 10.1016/j.neuron.2007.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Best JM, Foell JD, Buss CR, Delisle BP, Balijepalli RC, January CT, Kamp TJ. Small GTPase Rab11b regulates degradation of surface membrane L-type CaV1.2 channels. Am J Physiol Cell Physiol. 2011;300:C1023–C1033. doi: 10.1152/ajpcell.00288.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xie L, Kang Y, Liang T, Dolai S, Xie H, Parsaud L, Lopez JA, He Y, Chidambaram S, Lam PP, et al. RalA GTPase tethers insulin granules to L- and R-type calcium channels through binding α2δ-1 subunit. Traffic. 2013;14:428–439. doi: 10.1111/tra.12047. [DOI] [PubMed] [Google Scholar]
- 110.Waithe D, Ferron L, Page KM, Chaggar K, Dolphin AC. β-subunits promote the expression of CaV2.2 channels by reducing their proteasomal degradation. J Biol Chem. 2011;286:9598–9611. doi: 10.1074/jbc.M110.195909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Marangoudakis S, Andrade A, Helton TD, Denome S, Castiglioni AJ, Lipscombe D. Differential ubiquitination and proteasome regulation of CaV2.2 N-type channel splice isoforms. J Neurosci. 2012;32:10365–10369. doi: 10.1523/JNEUROSCI.0851-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kang MG, Campbell KP. γ subunit of voltage-activated calcium channels. J Biol Chem. 2003;278:21315–21318. doi: 10.1074/jbc.R300004200. [DOI] [PubMed] [Google Scholar]
- 113.Letts VA, Felix R, Biddlecome GH, Arikkath J, Mahaffey CL, Valenzuela A, Bartlett FS, 2nd, Mori Y, Campbell KP, Frankel WN. The mouse stargazer gene encodes a neuronal Ca2+-channel γ subunit. Nat Genet. 1998;19:340–347. doi: 10.1038/1228. [DOI] [PubMed] [Google Scholar]
- 114.Kang MG, Chen CC, Felix R, Letts VA, Frankel WN, Mori Y, Campbell KP. Biochemical and biophysical evidence for γ2 subunit association with neuronal voltage-activated Ca2+ channels. J Biol Chem. 2001;276:32917–32924. doi: 10.1074/jbc.M100787200. [DOI] [PubMed] [Google Scholar]
- 115.Osten P, Stern-Bach Y. Learning from stargazin: the mouse, the phenotype and the unexpected. Curr Opin Neurobiol. 2006;16:275–280. doi: 10.1016/j.conb.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 116.Moss FJ, Viard P, Davies A, Bertaso F, Page KM, Graham A, Cantí C, Plumpton M, Plumpton C, Clare JJ, et al. The novel product of a five-exon stargazin-related gene abolishes CaV2.2 calcium channel expression. EMBO J. 2002;21:1514–1523. doi: 10.1093/emboj/21.7.1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arikkath J, Chen CC, Ahern C, Allamand V, Flanagan JD, Coronado R, Gregg RG, Campbell KP. γ1 subunit interactions within the skeletal muscle L-type voltage-gated calcium channels. J Biol Chem. 2003;278:1212–1219. doi: 10.1074/jbc.M208689200. [DOI] [PubMed] [Google Scholar]
- 118.Sandoval A, Andrade A, Beedle AM, Campbell KP, Felix R. Inhibition of recombinant N-type CaV channels by the γ2 subunit involves unfolded protein response (UPR)-dependent and UPR-independent mechanisms. J Neurosci. 2007a;27:3317–3327. doi: 10.1523/JNEUROSCI.4566-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Sandoval A, Arikkath J, Monjaraz E, Campbell KP, Felix R. γ1-dependent down-regulation of recombinant voltage-gated Ca2+ channels. Cell Mol Neurobiol. 2007b;27:901–908. doi: 10.1007/s10571-007-9210-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Raghib A, Bertaso F, Davies A, Page KM, Meir A, Bogdanov Y, Dolphin AC. Dominant-negative synthesis suppression of voltage-gated calcium channel CaV2.2 induced by truncated constructs. J Neurosci. 2001;21:8495–8504. doi: 10.1523/JNEUROSCI.21-21-08495.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Page KM, Heblich F, Davies A, Butcher AJ, Leroy J, Bertaso F, Pratt WS, Dolphin AC. Dominantnegative calcium channel suppression by truncated constructs involves a kinase implicated in the unfolded protein response. J Neurosci. 2004;24:5400–5409. doi: 10.1523/JNEUROSCI.0553-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Harding HP, Calfon M, Urano F, Novoa I, Ron D. Transcriptional and translational control in the mammalian unfolded protein response. Annu Rev Cell Dev Biol. 2002;18:575–599. doi: 10.1146/annurev.cellbio.18.011402.160624. [DOI] [PubMed] [Google Scholar]