

Significance
X-ray crystallography is the primary technique used to obtain high-resolution structures of proteins. This method relies on diffracting large crystals that are identified by bright-field microscopy and usually optimized from an initial smaller and lower quality crystalline hit. Because of the limits of the optical methods used to visualize and identify these crystals, smaller nanometer crystals are excluded from the results of typical evaluations. However, the field of nanocrystallography, which utilizes a free electron laser to solve structures from nanocrystal (NC) slurries, makes these unidentified crystals highly useful. This paper presents a method, relying on transmission electron microscopy, to identify NCs, determine if they are protein, and evaluate their quality.
Keywords: nanocrystal detection, crystal optimization, crystal characterization, femtosecond diffraction, structural biology
Abstract
The current practice for identifying crystal hits for X-ray crystallography relies on optical microscopy techniques that are limited to detecting crystals no smaller than 5 μm. Because of these limitations, nanometer-sized protein crystals cannot be distinguished from common amorphous precipitates, and therefore go unnoticed during screening. These crystals would be ideal candidates for further optimization or for femtosecond X-ray protein nanocrystallography. The latter technique offers the possibility to solve high-resolution structures using submicron crystals. Transmission electron microscopy (TEM) was used to visualize nanocrystals (NCs) found in crystallization drops that would classically not be considered as “hits.” We found that protein NCs were readily detected in all samples tested, including multiprotein complexes and membrane proteins. NC quality was evaluated by TEM visualization of lattices, and diffraction quality was validated by experiments in an X-ray free electron laser.
The emergence of X-ray free electron laser (X-FEL)–based serial femtosecond crystallography holds the promise of solving the 3D structure of proteins that can only crystallize as “nanocrystals” (NCs) or are highly sensitive to radiation damage (1–5). NCs appropriate for X-FEL experiments are considered to be 200 nm to 2 μm in size (6). This size is constrained primarily by the requirements of the NC delivery system to the X-FEL beam. In addition to allowing for structure resolution of NCs by X-FEL experiments, they provide the advantage of requiring no crystal cryoprotection because these experiments are performed at room temperature (3, 7). Given the opportunities that X-FELs offer to the field of crystallography, efficient methodologies to detect NCs from single crystallography drops and to optimize these identified conditions yielding NCs will be essential for future developments in structural biology. Current methods to detect the presence of NCs include dynamic light scattering (DLS), bright-field microscopy, birefringence microscopy, and intrinsic tryptophan UV fluorescence imaging, as well as technologies that rely upon second harmonic generation, such as second order nonlinear imaging of chiral crystals (SONICC) (8, 9) and X-ray powder diffraction. However, limitations of these imaging techniques include (i) ineffective detection of crystals smaller than 5 μm (8, 10), (ii) false-positive conditions as a result of interference from precipitate backgrounds (8, 10), and (iii) false-negative conditions resulting from the lack of tryptophan residues in the case of UV fluorescence and from the lack of chiral centers in the case of SONICC (11). Although DLS can accurately measure the size distribution of nanometer-sized protein aggregates, it is unable to distinguish unambiguously between amorphous and crystalline (12). Finally, X-ray powder diffraction, a method that has been applied to evaluate samples for the presence and concentration of NCs, requires more material than is produced in a single crystallization screening drop, and synchrotron radiation is usually required to produce measurable diffraction (13). In this study, we use UV fluorescence microscopy and DLS to detect crystallization drops containing NCs, followed by transmission electron microscopy (TEM) to identify protein NCs accurately and determine NC quality by evaluating the reciprocal lattice reflections in diffraction patterns calculated from images.
Results
To develop a comprehensive method for identifying NCs, a four-step pathway was devised (Fig. 1A). A variety of targets from three different systems were chosen as test cases, including soluble proteins, membrane proteins, and multiprotein complexes (Fig. S1). Crystallization of protein samples using commercially available screens typically yields a spectrum of morphologies, including (i) clear drops (Fig. S2A), (ii) drops with granular aggregates (which can be amorphous or composed of well-differentiated individual particles) (Fig. S2B), (iii) drops with large solid aggregates (probably associated with sample denaturation) (Fig. S2C), and (iv) drops with phase separation. Such morphologies depend on the protein sample and the chemical nature of the precipitant (Table 1). For this study, conditions yielding granular aggregates (Fig. 1B and Fig. S2B) and clear drops (Fig. S2A) were selected for further analysis.
Fig. 1.
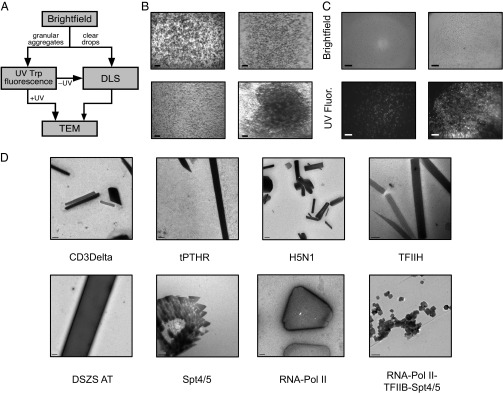
(A) Flow chart of the protocol used to identify protein NCs from crystallization drops. (B) Representative images of granular aggregates used for TEM. (C) Brightfield and UV fluorescence of granular aggregates, which can comprise UV-positive, well-differentiated nanoparticles (Left) or UV-positive, diffuse nanoaggregates (Right). (D) Examples of NCs identified by TEM. [Scale bars: (B) 300 μm, (C) 50 μm, (D) CD3Delta, 0.5 μm; tPTHR, 0.5 μm; RNA-Pol II-TFIIB-Spt4/5, 200 nm; TFIIH, 200 nm; DSZS AT, 0.5 μm; Spt4/5, 200 nm; RNA-Pol II, 0.2 μm; H5N1, 0.2 μm.]
Table 1.
Occurrence of clear drops, granular aggregates, phase separation, and denatured protein for the PTHR1 (3 mg/mL) and RPBlI–TFIIB complex (8 mg/mL)
| Screen | Protein | Clear drops, % | Granular aggregates, % | Phase separation, % | Denatured protein, % |
| HR Crystal Screen 2 | PTHR1 | 35 | 46 | 4 | 7 |
| HR SaltRX | Pol II complex | 52 | 41 | 0 | 7 |
| HR PEG suite II | Pol II complex | 42 | 46 | 8 | 4 |
The most sensitive parameter determining drop morphology is usually protein concentration. Protein concentration should be adjusted empirically to maximize the number of granular hits for each target. HR, Hampton Research.
To determine whether granular aggregates were proteinaceous in nature, UV tryptophan fluorescence microscopy was used (JANSi UVEX; JAN Scientific) (14). Drops with UV-positive granular aggregates were selected for direct TEM visualization (Fig. 1C). Because clear drops have been shown to yield NCs (15) and UV tryptophan fluorescence microscopy can yield false-negative results (16), UV-negative granular aggregates and clear drops were further processed using DLS to assess the presence of nanoparticles (Fig. S3). DLS measurements were performed using a Wyatt DynaPro Plate Reader Plus in batch mode (ideal for screening large numbers of crystallization conditions). To assess the size of the primary nanospecies of the sample accurately, calibration studies using nanospheres (Polysciences, Inc.) with particle radii of 50, 100, and 1,000 nm were compared with the sample’s autocorrelation function and decay time (Fig. S3A). DLS measurements of clear drops and UV-negative granular aggregates allowed detection of particles with diameters of 50–1,000 nm (Fig. S3B). To determine whether these particles were crystalline or merely protein aggregates, further assessment using TEM was performed.
UV- and DLS-positive samples were applied to a copper grid with continuous carbon film, stained with a 2% (wt/vol) uranyl acetate solution, and imaged using an FEI Tecnai T12 transmission electron microscope. Samples were taken directly from the crystal tray before transferring to a grid. In most circumstances, a single crystal drop containing thick aggregates was of sufficient concentration for imaging. For the majority of samples tested, TEM visualization (Fig. 2 and Fig. S4) provided an efficient method to reveal whether samples contained NCs or large protein aggregates. In addition, the presence of detergents in crystallization buffers did not appear to have a negative impact on visualization, and we were able to detect NCs for three membrane proteins: T-cell surface glycoprotein CD3 delta chain (CD3Delta), thermostabilized parathyroid hormone receptor (tPTHR), and influenza virus hemagglutinin protein (H5N1) (Fig. 1D). Because TEM allows visualization of crystal lattices, protein NCs could be discriminated from salt crystals, including those coated with protein aggregates that generated false-positive UV signals (Fig. S5A). Lattice visualization of thick NCs was performed by fragmentation using 0.5-mm glass beads. This method was a highly successful means of expanding the size range of NCs examined by TEM (Fig. S6).
Fig. 2.

(A and B) TEM images of NCs (Center) and accompanying lattice (Upper Left) and a fast Fourier transform (Lower Left) from the same crystallography conditions. (Right) Accompanying X-FEL diffraction pattern. (A) DSZS AT with diffraction up to a resolution of 1.8 Å. (B) RPBlI–GFP with diffraction up to a resolution of 4 Å acquired at the LCLS. (Scale bars: A, Upper Left, 20 nm; A, Center, 1 μm; B, Upper Left, 50 nm; B, Center, 100 nm.)
X-FEL experiments were used to test diffraction of NCs with high-quality lattices. Both the trans-acting acyl transferase from the disorazole synthase (DSZS AT) (Fig. 2A) and RNA polymerase (RPBII)–GFP (Fig. 2B) yielded significant diffraction. On the other hand, NCs of parathyroid hormone receptor (PTHR1) with poorly ordered lattices showed no diffraction (Fig. S4).
Although most NCs identified by TEM came from crystallization drops with UV-positive granular aggregates, a few were identified from UV-negative granular drops using DLS. We were unable to find protein NCs in UV-negative granular drops or clear drops that were preselected via DLS; most particles observed by TEM corresponded to large protein aggregates and salt crystals. However, because our observations were performed in a relatively small number of samples (around 100 crystal drops), thorough DLS evaluation of all conditions should be pursued when UV-positive conditions are scarce.
Discussion
Crystallization screening of protein samples involves setting up hundreds and sometimes thousands of crystallization drops, often without a single macrocrystal “hit.” However, the emergence of X-FEL–based serial femtosecond crystallography demonstrates that 3D structures of protein samples can be solved from NCs (1–5, 13, 17). To this end, we have developed an efficient method to detect the presence of protein NCs in UV-positive granular aggregates from crystallization drops using classic negative-stain TEM.
Our investigations show that crystallization trials of most protein samples can potentially yield NCs even for challenging systems, such as multiprotein complexes and membrane proteins. In addition to NC identification, the use of TEM may provide other insights, including (i) the possibility of finding NCs with different crystals forms and (ii) the evaluation of NC diffraction quality. Because crystalline lattices can be directly visualized with TEM, calculating Fourier transforms from the images allows qualitative evaluation of electron diffraction patterns (Bragg spots). Three examples of clear lattices with two or higher order spots are evident in the reciprocal lattice reflections obtained by the Fourier transform (Fig. 3). However, the limitations of negative stain techniques using uranyl acetate, including room temperature diffraction, sample dehydration, and possibly “crystal cracking” as a result of dye interactions with the crystal lattice, restricted electron diffraction to low resolution (∼20 Å). Despite these limitations, our experiments suggest that selection of crystals with higher order diffraction spots could potentially yield higher resolution X-ray diffraction data.
Fig. 3.

High-quality lattices of NCs visualized by TEM with accompanying FFT on right: lysozyme (A), RPBlI–GFP (B), and Pol-TFIIF (C). (Scale bars: A, 50 nm; B, 100 nm; C, 20 nm.)
Our work is not the first to characterize NCs by TEM but rather shows that NCs are commonly observed in crystallization drops of proteins of high biological interest and that evaluation of the crystal lattices of negatively stained NCs can provide preliminary qualitative information on their diffraction potential. Recent efforts have provided detailed characterization of the electron diffraction of lysozyme NCs by calculating fast Fourier transforms from their highly ordered lattices (18). Moreover, electron diffraction experiments in which three lysozyme NCs were used to collect a full tilt series allowed lysozyme structure determination by molecular replacement and refinement to a resolution of 2.9 Å (19). Given these advances, our work offers the possibility to expand electron crystallography studies to proteins of high biological relevance. Moreover, electron crystallography techniques could complement X-FEL experiments for those proteins that cannot yield the high volume of NCs required for gas dynamic virtual nozzle injectors (20).
This report establishes a method for prescreening NCs with no previous large crystal hits for use at an X-FEL. In addition to the advantages that TEM brings to the field of nanocrystallography, NC imaging may be beneficial during conventional crystallization trials to improve crystallization conditions and provide useful crystal “seeds” for the production of macrocrystals. This work demonstrates the potential of TEM to serve as a fundamental tool for evaluating NCs, as essential as bright-field microscopy is for evaluating and optimizing traditional large crystals.
Methods
Protein Production and Crystallography Condition Screening.
Eight proteins were used for nanocrystallography screening, as shown in Fig. S1, along with the expression system, protein yield, and maximal concentration (while maintaining monodispersity) that were used to set up trays. The expression and purification of PTHR1, CD3Delta, and RPBII, as well as complex formation with RPBII, were performed as described (21), whereas DSZS AT expression and purification are described separately (22). The tPTHR was purified following the same protocol as described for full-length PTHR1. Transcription factor II F (TFIIF) was purified as previously described (23). GFP was expressed and purified by standard methods (24). RPBII–TFIIF and RPBII–GFP complexes were assembled by adding 2.5 M excess of TFIIF or GFP to RPBII. The RPBII–TFIIF complex was purified using calmodulin affinity chromatography. The RPBII–GFP complex was isolated using a Superdex 200 10/300 GL column (GE Healthcare) [50 mM Hepes (pH 7.5), 100 mM KCl, 4 mM DTT, 2 mM CaCl2, and 10 μM ZnCl2].
Spt4 and Spt5 were overexpressed in Escherichia coli Rosetta (DE3) competent cells. Cells were grown at 37 °C to an OD at 600 nm of 0.6 and induced with 0.5 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 4 h at 30 °C and 2 mM IPTG for 2 h at 37 °C, respectively. Approximately 30 g of cells (mixing 15 g of spt4 cell with 15 g of spt5 cell) was resuspended in 150 mL of buffer A containing 250 mM NaCl, 25 mM Tris (pH 7.5), 10 μm ZnCl2, 5 mm imidazole, 0.1 mm PMSF, 0.5 mm C13E8, 10% glycerol, 2 mM β-mercaptoethanol (β-met), and 1× protease inhibitors (10 μg/mL pepstatin, leupeptin, chymostatin) (PIs). After sonication for 2 min and centrifugation for 45 min at 90,000 × g to separate pellet and supernatant fractions, the supernatant was loaded onto a Ni2+NTA (Sigma) column. The column was washed with buffer A with 25 mM imidazole and eluted with 150 mM imidazole in buffer A. The protein was desalted into buffer B containing 150 mM NaCl, 25 mM Tris (pH 7.5), 10 μM ZnCl2, 0.1 mM PMSF, 0.5 mM C13E8, 10% (vol/vol) glycerol, 2 mM β-met, and 1× PIs, and then applied to a HiTrap Heparin HP (GE Healthcare) column. The elution was performed with a linear gradient up to 1.0 M NaCl in buffer B. The elution fraction at the last peak was collected, concentrated, and further purified by gel filtration using a Superdex 200 (10/300 GL) column (GE Healthcare) in buffer B. The fractions containing the single peak were pooled, concentrated, and stored at −80 °C.
TFIIB was overexpressed in DE3 cells. Cells were grown at 37 °C to an OD at 600 nm of 0.6, and induced with 0.25 mM IPTG for 6 h at 30 °C. Approximately 20 g of cells was resuspended in 100 mL of buffer A containing 500 mm KCl, 25 mm Hepes (pH 8.0), 0.1% Triton X-100, and 1× PIs. After sonication for 2 min and centrifugation for 45 min at 35,000 × g to separate pellet and supernatant fractions, the supernatant was loaded onto chitin beads that were preequilibrated with 0.3 M NaOH, H2O, and buffer A. The column was washed with buffer A (more than 10 column-volumes), and 50 mM β-met in buffer A (3 columns) was then added, with continuous rocking overnight. The protein was desalted into buffer B containing 100 mM KCl, 25 mM Hepes (pH 7.0), 2 mM β-met, and 1× PIs, and was then applied to a HiTrap SP HP (GE Healthcare) column. The elution was performed with a linear gradient up to 1 M KCl in buffer B. The elution fraction at the first peak was collected, concentrated, and further purified by gel filtration using a Superdex 200 (10/300 GL) column in buffer B. The fractions containing the single peak were pooled, concentrated, and stored at −80 °C.
A high concentration of H5N1 protein was obtained by first growing a large SF9 culture: 3.2–4 L of 2 × 106 cells per milliliter infected with H5N1–10×-his baculovirus at a multiplicity of infection of 2.0 to get 30–40 g of cells. The insect cells were harvested at 48–50 h postinfection. The cell pellet (15 g) was washed with PBS and then resuspended with buffer C [150 mM NaCl and 35 mM Hepes (pH 7.5)] plus 1× PIs. Cell lysis was performed by sonication, followed by the addition of DNase, a short incubation, and ultracentrifugation (110,000 × g for 1 h). The pellet obtained was used for the detergent extraction step. To solubilize the H5N1 protein from the membrane pellet, 30 mM Sarcosine (Anatrace) plus 10 mm FOS-Choline 12 (Anatrace) in buffer D [75 mm NaCl and 35 mm Hepes (pH 7.5)] was used to homogenize the pellet. The homogenized pellet was incubated at 4 °C, with continuous rocking overnight. After incubation, the sample was ultracentrifuged (110,000 × g for 1 h) to obtain the supernatant containing the solubilized protein, which was then diluted 1:1 with buffer D to reduce the detergent concentration by one-half. The diluted sample was mixed with 15 mL of Ni2+ beads (preequilibrated with 15 mM Sarcosine/5 mM Fos-Choline in buffer D) for batch binding overnight. A detergent exchange (from 15 mM Sarcosine/5 mM FOS-Choline to 7.5 mM Sarcosine/4 mM Fos-Choline) gradient was performed using an ÄKTAxpress (GE Healthcare Life Sciences). After the gradient was completed, the sample Ni2+ beads were washed with buffer D with the addition of 4 mM Fos-Choline. Finally, the beads were washed with an astringent buffer of 250 mM NaCl, 35 mM Hepes (pH 7.5), and 25 mM imidazole to remove all of the unspecific proteins and extra material. To elute the protein from the Ni2+ beads, 300 mM imidazole in buffer D was used.
After elution, the protein sample was concentrated by centrifugation with a 100-kDa molecular weight cutoff concentrator (Vivaspin 20; GE Healthcare) and subsequently loaded onto a desalting column (GE Healthcare) to remove imidazole. The desalting column flow-through containing the protein was loaded into an anion exchanger column (1-mL column, Resource Q Hi Trap; GE Healthcare) to remove the unspecific proteins eluted with the 300 mM imidazole elution together with the H5N1. Most of the unspecific proteins passed through the Q column, whereas the H5N1 protein remained attached to the column matrix. The elution was performed with high salt buffer (300 mM NaCl).
A variety of commercially available crystallography screens were used for nanocrystallography screening, including the Hampton Research screens Crystal Screens 1 and 2, Index, PEG/Ion, SaltRx, Silver Bullets, and MembFac, as well as the Qiagen JCSG. Both hanging (Hampton Research VDX Plates) and sitting (Hampton Research Cryschem Plates) drop methods were used for screening 4-μL drops; they were set up at a 1:1 ratio of protein to mother liquor so as to have sufficient volume for further DLS and TEM experiments. Commercial screening solution (350 μL) was used in the well for each condition.
Generating Crushed Crystals of Thick NCs.
Glass beads (0.5 mm; Research Products International) were used to crush NCs too thick for lattice visualization by TEM. Twenty milligrams of beads was placed in a 1.5-μL microcentrifuge with the addition of the NC sample directly from the crystal plate, along with 5 μL of reservoir solution. Samples were vortexed twice for 10 s each time before being used for TEM grid preparation.
Generating Crushed Crystals of the DSZS AT for X-FEL Analysis.
The DSZS AT was purified and crystallized as previously described (22), by mixing 10 μL of well solution with 10 μL of purified protein at 5 mg/mL and allowing crystals to grow at room temperature for up to 1 wk. The crystallization solution contained 18–24% (wt/vol) PEG 3350, 100 mM Na-Hepes (pH 7.5), and 40 mM ammonium acetate. Approximately 900 μL of drops containing crystals was collected. When the crystals had settled to the bottom of the tube, we estimated that the mixture contained ∼50% (wt/vol) crystals. The crystals were held at room temperature until crushing and exposure to the Linac Coherent Light Source (LCLS) X-FEL. The tube was inverted several times to resuspend the crystals, and the samples were crushed in 90-μL aliquots by vortexing at high speed for 3 min with a Teflon seed bead (HR2-320; Hampton Research). Examination of crystal samples under a standard light microscope revealed large crystal fragments (edges >10 μm). Therefore, samples were further processed through a 10-μm3 filter before exposure with the LCLS using the coherent X-ray imaging (CXI) instrument.
X-FEL Analysis of NCs.
Diffraction experiments were carried out at the CXI end station of the LCLS (25) using 10.5-keV X-ray pulses with a duration of 40 fs for measurements of RPBII–GFP and PTHR1 and 8.5-keV X-ray pulses with a duration of 50 fs for DSZS. Crystal delivery was performed using the gas dynamic virtual nozzle (20) with an injection rate of 20 μL/min and a pressure of 750 psi (nitrogen) and 300 psi (shield). Data were analyzed using the CCTBX.XFEL software package (26). The RPBII–GFP sample yielded a hit rate of ∼2.5% and an indexing rate of 0.6%, representing 25% of hits [using a threshold of 16 spots above 450 analog to digital units (ADUs) per image]. Diffraction on the best images was up to 4 Å. The DSZS sample yielded a hit rate of ∼2.7% and an indexing rate of 0.07%, representing 29% of hits (using a threshold of 16 spots above 450 ADUs per image). Diffraction on the best images was up to 1.8 Å.
NC Candidate Identification and UV Tryptophan Screening.
Visual selection using an Olympus SZX16 bright-field microscope and corresponding 2XPFC objective was performed to identify NC candidates. Drops that had either visible precipitation with individual aggregates or a lawn of precipitate were selected for UV fluorescence imaging, examples of which are shown in Fig. 1B. Candidates selected visually were then subjected to UV fluorescence imaging with UV exposure from 1 to 5 s, using a JAN Scientific JANSi UVEX microscope. Images were analyzed using JAN Scientific CrystalDetect software. Once UV-positive conditions were identified, drops were harvested and high-quality images were taken of each drop using an Infinity 2-3C camera and Infinity Capture software from Lumenera Scientific. Harvested drops were subsequently used for DLS and TEM experiments.
DLS.
To test the limits of the Wyatt DynaPro plate reader for detecting nanoparticles of various radii, experiments using silica sphere (PolySciences, Inc) were performed in a 384 Corning clear bottom plate using beads diluted 1:1,000 with Millipore water with 15 μL of diluted beads deposited in the well. DLS data were acquired by performing 20 acquisitions (6 s for each acquisition) at 18 °C.
To determine the uniformity, as well as the range of the precipitate size, of selected NC candidates, samples were taken directly from the drop of the crystallization plate and put into a Greiner Sensoplate (glass bottom) 1,536 well plate, and DLS data were collected for 20 acquisitions (6 s for each acquisition) at 18 °C. These plates allowed a low working volume for screening (3 μL), as well as enhanced data clarity due to their glass bottoms. Samples were diluted with mother liquor up to a volume of 3 μL when necessary. If low intensity was observed and sufficient protein was available, additional drops were set up using the same conditions and combined to increase intensity.
TEM.
Four hundred square mesh copper grids with continuous carbon film (Electron Microscopy Sciences) were freshly glow-discharged for 1 min at 25 mV (EmiTech KX100) before incubation with samples. Selected samples were applied to grids by two methods depending on the concentration of nanoparticles. When a sufficiently high concentration of particles was present (i.e., when particles were visible and dense, and gave a DLS intensity signal above 1.25), 5–8 μL of sample was applied and incubated for 30 s on a grid before blotting and staining with 2% (wt/vol) uranyl acetate. Lower density samples were applied to the grid by placing a drop of the sample in a parafilm-lined Petri dish along with at least 300 μL of mother liquor from the sample’s origin in close proximity to the sample drop. A grid was put on top of the drop of sample (carbon side down), the Petri dish was sealed, and the sample was allowed to incubate for 10–60 min before staining with uranyl acetate. TEM images were acquired using an FEI Tecnai T12 electron microscope operating at 120 kV using a single-tilt specimen holder. Images were collected with a 2-k × 2-k Gatan UltraScan 1000 CCD camera.
Supplementary Material
Acknowledgments
Portions of this research were carried out at the Linac Coherent Light Source (LCLS), a National User Facility operated by Stanford University on behalf of the US Department of Energy, Office of Basic Energy Sciences. We thank Sebastien Boutet, Marc Messerschmidt and Garth Williams of LCLS, and Robert L. Shoeman and Sabine Botha of the Max Plank Institute for Medical Research for support during data collection at coherent X-ray imaging (CXI). The CXI instrument was funded through the LCLS Ultrafast Science Instruments (LUSI) project funded by the US Department of Energy (DOE) Office of Basic Energy Sciences. Use of the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, is supported by the DOE Office of Science, Office of Basic Energy Sciences under Contract DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1400240111/-/DCSupplemental.
References
- 1.Aquila A, et al. Time-resolved protein nanocrystallography using an X-ray free-electron laser. Opt Express. 2012;20(3):2706–2716. doi: 10.1364/OE.20.002706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boutet S, et al. High-resolution protein structure determination by serial femtosecond crystallography. Science. 2012;337(6092):362–364. doi: 10.1126/science.1217737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kern J, et al. Simultaneous femtosecond X-ray spectroscopy and diffraction of photosystem II at room temperature. Science. 2013;340(6131):491–495. doi: 10.1126/science.1234273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koopmann R, et al. In vivo protein crystallization opens new routes in structural biology. Nat Methods. 2012;9(3):259–262. doi: 10.1038/nmeth.1859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Redecke L, et al. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science. 2013;339(6116):227–230. doi: 10.1126/science.1229663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chapman HN, et al. Femtosecond X-ray protein nanocrystallography. Nature. 2011;470(7332):73–77. doi: 10.1038/nature09750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kern J, et al. Room temperature femtosecond X-ray diffraction of photosystem II microcrystals. Proc Natl Acad Sci USA. 2012;109(25):9721–9726. doi: 10.1073/pnas.1204598109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kissick DJ, Wanapun D, Simpson GJ. Second-order nonlinear optical imaging of chiral crystals. Annu Rev Anal Chem (Palo Alto Calif) 2011;4:419–437. doi: 10.1146/annurev.anchem.111808.073722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haupert LM, Simpson GJ. Screening of protein crystallization trials by second order nonlinear optical imaging of chiral crystals (SONICC) Methods. 2011;55(4):379–386. doi: 10.1016/j.ymeth.2011.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Judge RA, Swift K, González C. An ultraviolet fluorescence-based method for identifying and distinguishing protein crystals. Acta Crystallogr D Biol Crystallogr. 2005;61(Pt 1):60–66. doi: 10.1107/S0907444904026538. [DOI] [PubMed] [Google Scholar]
- 11.Wampler RD, et al. Selective detection of protein crystals by second harmonic microscopy. J Am Chem Soc. 2008;130(43):14076–14077. doi: 10.1021/ja805983b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kadima W, McPherson A, Dunn MF, Jurnak FA. Characterization of precrystallization aggregation of canavalin by dynamic light scattering. Biophys J. 1990;57(1):125–132. doi: 10.1016/S0006-3495(90)82513-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hunter MS, et al. X-ray diffraction from membrane protein nanocrystals. Biophys J. 2011;100(1):198–206. doi: 10.1016/j.bpj.2010.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gill HS. Evaluating the efficacy of tryptophan fluorescence and absorbance as a selection tool for identifying protein crystals. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2010;66(Pt 3):364–372. doi: 10.1107/S1744309110002022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Du S, et al. Structure of the HIV-1 full-length capsid protein in a conformationally trapped unassembled state induced by small-molecule binding. J Mol Biol. 2011;406(3):371–386. doi: 10.1016/j.jmb.2010.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Desbois S, Seabrook SA, Newman J. Some practical guidelines for UV imaging in the protein crystallization laboratory. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2013;69(Pt 2):201–208. doi: 10.1107/S1744309112048634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunter MS, Fromme P. Toward structure determination using membrane-protein nanocrystals and microcrystals. Methods. 2011;55(4):387–404. doi: 10.1016/j.ymeth.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nederlof I, Li YW, van Heel M, Abrahams JP. Imaging protein three-dimensional nanocrystals with cryo-EM. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 5):852–859. doi: 10.1107/S0907444913002734. [DOI] [PubMed] [Google Scholar]
- 19.Shi D, Nannenga BL, Iadanza MG, Gonen T. Three-dimensional electron crystallography of protein microcrystals. Elife. 2013;2:e01345. doi: 10.7554/eLife.01345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePonte DP, et al. Gas dynamic virtual nozzle for generation of microscopic droplet streams. J Phys D Appl Phys. 2008;41(19):195505. [Google Scholar]
- 21.Pullara F, et al. A general path for large-scale solubilization of cellular proteins: From membrane receptors to multiprotein complexes. Protein Expr Purif. 2013;87(2):111–119. doi: 10.1016/j.pep.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong FT, Jin X, Mathews II, Cane DE, Khosla C. Structure and mechanism of the trans-acting acyltransferase from the disorazole synthase. Biochemistry. 2011;50(30):6539–6548. doi: 10.1021/bi200632j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takagi Y, et al. Head module control of mediator interactions. Mol Cell. 2006;23(3):355–364. doi: 10.1016/j.molcel.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 24.Dieryck W, Noubhani AM, Coulon D, Santarelli X. Cloning, expression and two-step purification of recombinant His-tag enhanced green fluorescent protein over-expressed in Escherichia coli. J Chromatogr B Analyt Technol Biomed Life Sci. 2003;786(1-2):153–159. doi: 10.1016/s1570-0232(02)00764-x. [DOI] [PubMed] [Google Scholar]
- 25.Boutet Sb & Williams GJ The coherent x-ray imaging (CXI) instrument at the Linac Coherent Light Source (LCLS) New J Phys. 2010;12(3):035024. [Google Scholar]
- 26.Sauter NK, Hattne J, Grosse-Kunstleve RW, Echols N. New Python-based methods for data processing. Acta Crystallogr D Biol Crystallogr. 2013;69(Pt 7):1274–1282. doi: 10.1107/S0907444913000863. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.