

Abstract
The glucocorticoid receptor (GR) regulates hypothalamic-pituitary-adrenal (HPA) axis activity during the stress response. The paraventricular nucleus (PVN) is a major site of negative feedback to coordinate the degree of the HPA axis activity with the magnitude of the exposed stressor. To define the function of endogenous PVN GR, we used Cre-loxP technology to disrupt different GR exons in Sim1-expressing neurons of the hypothalamus. GR exon 2-deleted mice (Sim1Cre-GRe2Δ) demonstrated 43% loss of PVN GR compared with an 87% GR loss in exon 3-deleted mice (Sim1Cre-GRe3Δ). Sim1Cre-GRe3Δ mice display stunted growth at birth but develop obesity in adulthood and display impaired stress-induced glucose release. We observed elevated basal and stress-induced corticosterone levels in Sim1Cre-GRe3Δ mice, compared with control and Sim1Cre-GRe2Δ mice, and impaired dexamethasone suppression, indicating an inability to negatively regulate corticosterone secretion. Sim1Cre-GRe3Δ mice also showed increased CRH mRNA in the PVN, increased basal plasma ACTH levels, and reduced locomotor behavior. We observed no differences in Sim1Cre-GRe2Δ mice compared with control mice in any measure. Our behavioral data suggest that GR deletion in Sim1-expressing neurons has no effect on anxiety or despair-like behavior under basal conditions. We conclude that loss of PVN GR results in severe HPA axis hyperactivity and Cushing's syndrome-like phenotype but does not affect anxiety and despair-like behaviors.
The paraventricular nucleus of the hypothalamus (PVN) is a key regulator of metabolism, stress responses, and physiologic homeostasis. The glucocorticoid receptor (GR) is of specific importance in these varied functions of the PVN. GR functions as the primary mediator of feedback regulation in hypothalamic-pituitary-adrenal (HPA) axis stress response systems. The parvocellular neurons of the PVN synthesize and secrete CRH and vasopressin in response to stress. CRH is secreted into the hypophyseal portal system and acts on the anterior pituitary where it stimulates the synthesis and secretion of ACTH. ACTH is released into the bloodstream and stimulates adrenal cortex secretion of cortisol in humans or corticosterone (CORT) in rodents. CORT acts on peripheral tissues and central nervous system GRs to mediate normal circadian function as well as the stress response. CORT then negatively feeds back at the level of the pituitary to inhibit ACTH secretion, as well as at the level of the PVN to inhibit CRH secretion and restore the system to homeostasis. CORT binding to GR in the PVN, pituitary, and other central nervous system sites in the forebrain, mediates the negative feedback inhibition of CORT. Dysregulation of the HPA axis, possibly due to impaired GR signaling, has been implicated in the pathogenesis of affective disorders, such as major depression, in humans (1–5).
Several genetically altered rodent models of GR disruption have demonstrated a role for GR in mediating neuroendocrine, physiologic, and behavioral responses (see Refs. 6–8 for review). Global GR deletion in mice targeting exon 2 of GR, GRhypo (9), or Exon 3, GRnull (10, 11) leads to perinatal and fetal mortality due to improper lung maturation. Interestingly, 20% of GRhypo mice survive into adulthood, whereas all the GRnull mice die within hours after birth, indicating differences in penetrance depending on the specific nature of targeting the GR locus (12). The fetal lethality limited further studies on stress-related behaviors in these mice, leading to the development of region-specific deletion models.
Conditional deletion of the floxed GR exon 3 allele in the whole brain using Nestin-Cre results in mice with HPA axis hyperactivity and consequent Cushing syndrome phenotypes (growth impairment, altered fat distribution, obesity), as well as reduced anxiety-like behaviors (13). When GR disruption is limited to adult forebrain neurons using the CaMKIIα-Cre transgene (FB-GRko), mild HPA axis dysregulation occurs, as well as increased anxiety and despair-like behaviors that can be reversed with antidepressant treatment (14, 15). In comparison, GR disruption in embryonic forebrain neurons, PVN and pituitary neurons (using BAC CaMKIIα-iCre transgenic and floxed exon 3 transgenic mice [GRCaMKCre]) leads to severe HPA axis hyperactivation, growth impairments, and death before postnatal day 10 (16). Disrupting GR in the pituitary, GRPOMCCre, results in elevated basal CORT levels, changes in PVN CRH and vasopressin mRNA, reduced body weight, all of which normalize in adulthood, and more active stress-coping behavior in the forced swim test (17). As one of the primary sites of GR feedback regulation, the PVN's specific role during HPA axis development and stress reactivity still needs to be analyzed.
We set out to isolate the specific contribution of PVN GR in mediating the aforementioned changes in regulation of glucocorticoid synthesis. We used Sim1-Cre BAC transgenic mice (18) to recombine floxed GR alleles, one removing exon 1C-2, and the other removing exon 3. These mice drive Cre expression during embryonic and postnatal development in the PVN, as well as the medial amygdala and nucleus of the lateral olfactory tract (18). This model has the unique potential to address several important questions in HPA-axis regulation and behavior. For instance, can other extrahypothalamic sites of GR-negative feedback modulate glucocorticoid production and compensate for GR disruption in the PVN? Does PVN GR loss lead to up-regulation of CRH expression, as it remains unclear whether a functional glucocorticoid response element exists in CRH gene locus. Additionally, does loss of GR in the PVN affect stress-related behaviors? The 2 major conventional GR knockouts, described earlier, differ in their phenotypes. Therefore, in addition to understanding these unresolved questions regarding PVN GR, we investigate whether there would be any differences in phenotype depending on what domain of GR was conditionally disrupted. To investigate this, we used mice that had GR floxed for the exon 1C-2 region (GRe1C-2), containing promoter elements, translation start site, and the transcriptional activation domain or the exon 3 region (GRe3), containing the zinc finger of the DNA-binding domain (19, 20), both in combination with the Sim1-Cre BAC transgenic mice.
Our physiologic data indicate that loss of the exon 3 region of GR in the PVN results in fat accumulation due to glucocorticoid excess and a more severe HPA axis dysregulation phenotype than loss of exon 2. Our behavioral data indicate that PVN GR alone does not mediate basal changes in anxiety and despair-related behaviors. We find that conditional deletion of GRe3 in the PVN leads to increased PVN CRH mRNA, increased plasma ACTH, and glucocorticoid excess. Unlike GR deletion in the pituitary (17), we do not find compensation, but persistent HPA hyperactivation in adulthood, where circadian rhythm of glucocorticoid secretion is retained. In contrast, deletion of GRe1C-2 region is associated with intact HPA axis regulation. These differences may be contributed to by altered deletion efficiency or residual production of truncated activity-retaining GR fragments.
Materials and Methods
Animals
All animal protocols were in accordance with National Institutes of Health guidelines and were approved by the Animal Care and Use Committees of Vanderbilt University (Nashville, Tennessee) and Cincinnati Children's Medical Center (Cincinnati, Ohio). Mice were housed on a 12-hour/12-hour (Vanderbilt) or 14-hour/10-hour (Cincinnati Children's Medical Center) light-dark cycle with ad libitum access to rodent chow and water. Mice used for studies were between 3 and 6 months old.
Sim1Cre-GRe3Δ mice.
Sim1Cre-GRe2Δ mice.
We bred Sim1Cre mice to floxed GR exon 1C-2 mice (21).
Controls.
Littermates that were Cre negative but had GR exon 1C-2 or exon 3 floxed alleles were used as controls. In figures displaying a comparison of Sim1Cre-GRe2Δ and Sim1Cre-GRe3Δ mice, controls represent littermates from both types of matings. Sim1Cre mice were the kind gift of Dr. Brad Lowell (Beth Israel Deaconess Medical Center, Boston, Massachusetts). Sim1Cre, floxed GR exon 3, and floxed GR exon 1C-2 lines have been maintained on an inbred C57BL/6 background.
Immunohistochemistry
We anesthetized mice with 2.5% Avertin, perfused them with diethylpyrocarbonate (DEPC) 1× PBS followed by 4% DEPC paraformaldehyde (PFA), and postfixed the brains and pituitaries in 4% DEPC PFA overnight. We then immersed the brains in 70% ethanol and processed them for paraffin embedding. Paraffin-embedded brains were sectioned, on a microtome, at 8 μm whereas the pituitaries were sectioned at 5 μm onto Superfrost+ slides (VWR International). The slides were placed at 60°C overnight and then stored at room temperature until later use. The slides were deparaffinized and rehydrated in decreasing concentrations of ethanol. Next, we performed antigen reconstitution of sections in 10 mM citrate buffer (pH 6.0), followed by PBS washes and 1-hour blocking in 3% normal goat serum/ 0.25% Triton-X-100 in PBS to prevent nonspecific binding.
GR.
Primary antibodies were 1:200 rabbit m20-anti-GR, directed against the N terminus of GR, (Santa Cruz Biotechnology; catalog no. sc-1004) and 1:200 mouse anti-NeuN; secondary antibodies were 1:250 biotinylated goat antirabbit IgG and 1:200 goat antimouse Alexa 488 IgG; Washed slides were incubated in avidin/biotin complex (Vectastain), cyanine-3 tyramide amplified (PerkinElmer) and coverslipped with 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). We quantified the number of GR-positive cells that colocalized with DAPI as a percentage of total DAPI-positive cells in the PVN.
ACTH.
Primary antibodies were 1:200 rabbit m20-anti-GR (Santa Cruz Biotechnology) and 1:200 guinea pig anti-ACTH (Bachem; catalog no. S-3029); secondary antibodies were 1:250 biotinylated goat antirabbit IgG and 1:100 goat antiguinea pig fluorescein isothiocyanate IgG; slides were incubated with ABC, cyanine-3 tyramide amplified, and coverslipped with DAPI as above. We quantified the number of GR-positive cells that colocalize with ACTH-positive nuclei as a percentage of total ACTH-positive cells in the anterior pituitary.
Body weight, gonadal fat, and adrenal gland measurements
We weighed mice weekly starting on weaning day, postnatal day 21 (P21) to 22 weeks of age. We dissected and weighed intragonadal fat pads and adrenal glands of mice 3–6 months old.
Corticosterone RIA
Mice were single housed for a week, and submandibular bleeds were performed in mice at nadir, peak, and 20 minutes after restraint stress, with 2 days between bleeds. The blood was centrifuged at 14 000 rpm for 6 minutes, and the plasma was removed and stored at −80°C until a RIA was performed. RIA was performed using the Corticosterone Double Antibody-125I RIA Kit (MP Biomedicals).
Dexamethasone suppression test
Mice were given saline or dexamethasone (0.1 mg/kg) injections in the morning and bleeds were performed 1 hour before lights out. The blood was centrifuged at 14 000 rpm for 6 minutes, and the plasma was removed and stored at −80°C until the CORT RIA was performed.
Plasma glucose concentration
We used the AVIVA plus glucose meter to measure plasma glucose levels
CRH in situ hybridization
We anesthetized mice with 2.5% Avertin, perfused them with DEPC 1× PBS followed by 4% DEPC PFA, and postfixed the brains in 4% DEPC PFA overnight. Brains were immersed in 20% DEPC sucrose solution for 48 hours, and then embedded in OCT. sections (16 μm)were obtained from brains at −15°C in a cryostat and mounted onto superfrost+ slides (VWR International), vacuum-dried overnight, and stored at −80°C until in situ hybridization was performed. P-33 UTP was used to label an RNA probe complementary to a 0.32-kb fragment on exon 2 of the CRH mRNA. Sections were hybridized to the labeled probe at 65°C for 16 hours and washed in 0.1× saline-sodium citrate buffer at 60°C for 30 minutes. Slides were exposed to Maximum resonance film (Kodax BioMAx) for 4 days. We scanned the autoradiographic images at 3200 dots/inch into Photoshop on an Epson Perfection V600 scanner and quantified using NIH Image J software.
Behavioral tests
All behavioral tests were performed between 8:00 am and 12:00 pm. Mice were allowed to acclimate in the testing room for 1 hour prior to behavioral testing. Male mice between 12 and 20 weeks of age were used in tests. All behavioral tests were performed in the Vanderbilt Murine Neurobehavioral laboratory at the Vanderbilt University Medical Center and the Cincinnati Children's Hospital behavior core.
We evaluated anxiety using the open field and the elevated zero maze. Both tests pit the innate aversion of rodents to open unsecure spaces against their natural curiosity to explore the environment.
Open field.
The open field activity was measured in chambers (Med Associates, 27 × 27× 20.5 cm) that were contained within a light- and air-controlled environment. Each trial lasted 30 minutes per mouse. During that time, the Activity Monitor v5.10 program (Med Associates, Inc) was used to measure behaviors such as time spent, distance traveled, and number of entries into the inner and outer zones of the chamber.
Elevated zero maze.
This maze (34-cm inner diameter; 46-cm outer diameter, placed 40 cm off the ground on 4 braced legs) consists of a round maze with opposing open and closed arms. Mice are placed in the center of the open arm, and activity is scored for 5 minutes using ANY-Maze software (San Diego Instruments). Time spent in the open and closed areas, as well as number of entries into the open and closed areas, were measured.
Forced swim and tail suspension tests are measures of learned helplessness and despair (due to their predictive validity).
Forced swim test.
This test utilizes a 2-L beaker, filled to 1.5 L with water at room temperature (25°C). The mouse was placed in the beaker for 5 minutes. Every 5 secomds, the mouse's activity was recorded by an observer blind to genotype. We quantified the total time inactive during the trial, in which more inactivity is indicative of increased despair.
Tail suspension tests.
The tail suspension test employed a platform from which mice are suspended by their tails; motion was scored with either an automated system with user-defined thresholds or manually with the observer blinded to genotype. Motion was scored for 5 minutes manually or for 7 minutes in the automated system. Time spent below a specified lower threshold was interpreted as inactivity.
Animals excluded from analysis after behavioral testing were those unable to be scored due to falling off the zero maze apparatus or sinking in the forced swim. These test failures did not differ in frequency between the different genotypes.
Statistical analysis
Results are presented as mean ± SEM. Data were subjected to Student's t test, one- or two-way ANOVA test (Prism 5.0 software; GraphPad Software, Inc.). Statistical significance was analyzed using the Tukey or Bonferroni post hoc test. Significant difference was defined at a P ≤ .05.
Results
Deletion of GRe1C-2 and GRe3 in the PVN neurons expressing Sim1
We bred Sim1-Cre BAC transgenic mice (18) to mice that have loxP sites flanking exons 1C and 2 (21) or exon 3 of the GR gene (13). We thus generated mice with GRe1c-2 deleted in the PVN (Sim1Cre-GRe2Δ) or mice with GRe3 deleted in the PVN (Sim1Cre-GRe3Δ). Control mice were negative for Cre-recombinase. We evaluated the specificity and efficiency of GR deletion by fluorescent immunohistochemistry and quantified the percent of GR-positive nuclei in the PVN. We demonstrated a 43% reduction in PVN GR expression in Sim1Cre-GRe2Δ mice (P = .03). In contrast, we found 87% decrease in PVN GR in Sim1Cre-GRe3Δ mice (P = .002) (Figure 1) when compared with control mice (one-way ANOVA F(2, 8) = 15.47; P = .0018).
Figure 1.
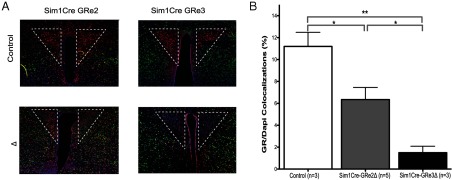
GR Immunoreactivity Is Reduced in the PVN (outlined area) of Mice with Disrupted GR. A, Immunofluorescent staining for GR protein in the PVN of controls, Sim1Cre-GRe2Δ, and Sim1Cre-GRe3Δ mice. GR (red), NeuN (green), DAPI (blue). B, Quantification of the amount of GR in the PVN that colocalized with DAPI as a percent of total DAPI. The bars represent mean + SEM. Sim1Cre-GRe2Δ mice have a 43% reduction in GR protein, whereas Sim1Cre-GRe3Δ mice have an 87% reduction compared with controls (ctrl). *, P < .05; and **, P < .01.
Sim1Cre-GRe3Δ, but not Sim1Cre-GRe2Δ, mice have increased body weight and fat mass in adulthood
In terms of body weight, Sim1Cre-GRe2Δ mice are phenotypically indistinguishable from control mice. On the other hand, Sim1Cre-GRe3Δ mice display characteristics of a glucocorticoid excess (cushingoid) phenotype. At birth, Sim1Cre-GRe3Δ mice are smaller than controls (P < .05), but in adulthood they display increased weight gain and obesity (Figure 2, A–C). We observed this in females (P < .05) (Figure 2A) and some male Sim1Cre-GRe3Δ mice, although there was enough variability between the males such that differences were not statistically different (Figure 2B).
Figure 2.

Sim1Cre-GRe3Δ Mice Have Altered Growth, Fat Accumulation, and Stress Glucose Response. A and B, The mean (±SEM) body weights of control and Sim1Cre-GRe3Δ mice over a 6-month period in females (A) and males (B). C, Images depicting phenotype differences between female control and Sim1Cre-GRe3Δ mice at 4 months of age. D and E, Ovarian (D) and testicular (E) fat pad weight in each group of mice. F and G, Plasma glucose levels measured at nadir, peak, and after 20 minutes of restraint stress. Control and Sim1Cre-GRe2Δ mice display increased plasma glucose after stress (***, P < .001), whereas Sim1Cre-GRe3Δ mice do not (P > .05 in females (F) and males (G)). *, P < .05; and **, P < .01; ***, P < .001; ****, P < .0001.
To determine contributors to increased body weight, we dissected and weighed perigonadal fat pads from female and male mice. Female Sim1Cre-GRe3Δ mice had a 14-fold increase in fat pad mass, whereas male Sim1Cre-GRe3Δ mice had a 3-fold increases in fat pad mass compared with control littermates (females, P = .0006; males, P = .001) (Figure 2, D and E). Due to this observed increase in adiposity and predicted high CORT levels, we tested serum glucose concentrations at baseline and following restraint stress. We observed that baseline blood glucose levels did not vary between genotypes in either gender (females, P = .12; males, P = .48) (Figure 2, F and G). However, Sim1Cre-GRe3Δ mice fail to exhibit the stress-induced elevations in blood glucose levels (females, P = .22; males, P = .3) observed in controls (females, P = .0007; males, P = .002) and Sim1Cre-GRe2Δ mice (females, P = .0005; males, P = .03) (Figure 2, F and G).
Sim1Cre-GRe3Δ mice, but not Sim1Cre-GRe2Δ mice, display increased HPA axis activity
To evaluate the cause of the Cushing-like phenotype, we measured serum CORT levels. We found significantly elevated basal plasma CORT levels in both female (P = .07 nadir; P = .0005 peak) and male (P < .0001; nadir and peak) Sim1Cre-GRe3Δ mice that maintained circadian rhythmicity (Figure 3, A and B). Twenty minutes of restraint stress further elevated CORT levels (Figure 3, A and B) in Sim1Cre-GRe3Δ mice (females, P = .04; males, P < .0001). Moreover, Sim1Cre-GRe3Δ mice fail to suppress CORT secretion in the dexamethasone suppression test (females, P = .45; males, P = .27) whereas their littermate controls suppressed CORT secretion by approximately 50% after dexamethasone administration (females, P = .02; males, P = .05) (Figure 3, C and D).
Figure 3.

Loss of PVN GR Results in Hypercorticosterolemia and Impaired Negative Feedback in Female (A and C) and Male (B and D) Mice. A and B, Plasma CORT levels measured at nadir, peak and after 20 minute of restraint stress. Sim1Cre-GRe3Δ mice display increased plasma CORT at nadir, peak, and after stress compared with controls and Sim1Cre-GRe2Δ mice. C and D, Dexamethasone suppression test was used to evaluate negative feedback. Control female mice show a 50% suppression of CORT after dexamethasone (P < .05, panel C), whereas males show a trend toward CORT suppression (#, P = .07 panel A, and P = .053, panel D). Sim1Cre-GRe3Δ mice do not show suppression of CORT after dexamethasone administration. *, P < .05; ***, P < .001.
To determine the mechanism by which loss of GR in the PVN leads to increased CORT, we evaluated CRH mRNA and protein expression in the PVN of control, Sim1Cre-GRe2Δ, and Sim1Cre-GRe3Δ mice. We observed a 3-fold increase in CRH mRNA expression in Sim1Cre-GRe3Δ (P = .01) but not Sim1Cre-GRe2Δ mice (P = .9) compared with controls (one-way ANOVA F(2, 9) = 30.22; P = .0001) (Figure 4, A and B). Interestingly, we found a significant effect of genotype on amygdala CRH mRNA expression (one-way ANOVA F(2, 9) = 6.202; P = .0203), where CRH mRNA in the central nucleus of the amygdala increased in Sim1Cre-GRe3Δ mice compared with Sim1Cre-GRe2Δ mice (t test, P = .016), with a similar trend in SimCre-GRe3Δ mice compared with control mice that did not achieve statistical significance (Supplemental Figure 1A published on The Endocrine Society's Journals online web site at http://endo.endojournals.org). Despite the significant increase in PVN CRH expression, pituitary ACTH immunoreactivity did not differ and GR protein in ACTH-producing cells of the pituitary remained intact (Figure 4C). Circulating nadir ACTH levels were significantly elevated whereas peak levels had a trend toward increase in male Sim1Cre-GRe3Δ mice compared with controls (two tailed t test: P = .03-nadir; P = .06 peak) but not in Sim1Cre-GRe2Δ mice compared with controls (Figure 4D). Stress levels of ACTH (Figure 4D) do not differ between genotypes. Female Sim1Cre-GRe3Δ mice show a trend toward increased adrenal weights (P = .059) (Supplemental Figure 1B), whereas male adrenal weights in Sim1Cre-GRe3Δ mice are significantly increased (P = .009) compared with controls and Sim1Cre-GRe2Δ mice (Supplemental Figure 1C).
Figure 4.
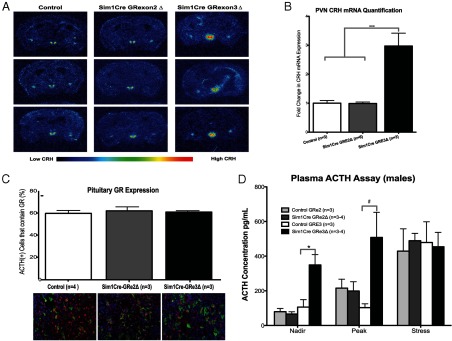
HPA Axis Hyperactivity Is Observed in Sim1Cre-GRe3Δ Mice. A and B, CRH mRNA expression is increased in the PVN of Sim1Cre-GRe3Δ mice. A, Representative images of pseudocolored CRH in situ hybridization in control mice (left), Sim1Cre-GRe2Δ mice (middle), and Sim1Cre-GRe3Δ mice (right). B, Quantification of pixel density showed a 3-fold increase in PVN CRH mRNA in Sim1Cre-GRe3Δ mice compared with controls and Sim1Cre-GRe2Δ mice; ***, P = .0001. C, Expression of GR in ACTH+ nuclei was quantified as a percentage of total ACTH+ nuclei (top panel) in the anterior pituitary. As visualized in the immunofluorescent images in the bottom panel, quantification revealed no differences between groups, implying intact GR in ACTH-producing cells GR (red), ACTH (green), DAPI (blue). D, Plasma ACTH levels measured at nadir, peak, and after 20 minutes of restraint stress. Sim1Cre-GRe3Δ mice display increased plasma ACTH at nadir; *, P = .03. They trend toward increased peak ACTH levels (#, P = .06), with no change in stress levels of ACTH compared with controls and Sim1Cre-GRe2Δ mice.
Sim1Cre-GRe3Δ mice exhibit reduced locomotor activity
Due to the role of brain GR in mediating anxiety and despair-related activity, we tested mice in different behavioral paradigms. Behaviorally, locomotor activity is reduced in male Sim1Cre-GRe3Δ mice (in terms of distance traveled in the open field; P = .02) (Figure 5, A and B). However, changes in anxiety (OF and elevated zero maze) (Supplemental Figure 2, A and B) and despair-like behaviors (tail suspension test and forced swim test) (Supplemental Figure 2, C and D) were not observed in Sim1Cre-GRe3Δ mice. Comparatively, Sim1Cre-2Δ mice have normal locomotor activity and display no anxiety or despair-like behavior (Supplemental Figure 3, A–D).
Figure 5.

Altered Locomotory Activity of Sim1Cre-GRe3Δ Mice in the OF Test. Sim1Cre-GRe3Δ mice show reduced distance traveled in outer zone and total distance (A) as well as distance traveled in 5-minute blocks (B); *, P < .05
Discussion
In this study, we investigate the neuroendocrine, metabolic, and behavioral consequences of disruption of GR in the PVN. Surprisingly, we find that the phenotype of Sim1-Cre-mediated deletion of floxed GR gene substrates depends upon the specific floxed GR allele targeted. Sim1Cre-GRe3Δ mice manifest elevated serum concentrations of CORT throughout the circadian cycle and in association with a stressor, compared with either Sim1Cre-GRe2Δ or nondeleted control mice. Because of the greater extent of deletion in the PVN of Sim1Cre-GRe3Δ compared with Sim1Cre-GRe2Δ mice, together with the potential for truncated carboxy-terminal products in Sim1Cre-GRe2Δ mice, the Sim1Cre-GRe3Δ line reflects a more complete phenotype for PVN GR loss. Despite elevated glucocorticoid concentrations and metabolic derangements, we did not find evidence for behavioral alterations in the Sim1Cre-GRe3Δ line. Although we did not observe differences between controls and Sim1Cre-GRe2Δ mice in HPA axis regulation under the conditions we analyzed, it is possible that a more extensive time series or different stress exposures could reveal a more subtle aspect of glucocorticoid feedback dysregulation.
Although the Sim1Cre line also expresses Cre-recombinase in the supraoptic nucleus, nucleus of the lateral olfactory tract, and other hypothalamic and medial amygdala nuclei (18), these sites are unlikely to influence the HPA axis regulation phenotype we report here. These Sim1Cre transgenics are the most selective mice that have been generated thus far for evaluating PVN-related functions of genes.
Differential HPA axis phenotype between Sim1Cre-GRe2Δ and Sim1Cre-GRe3Δ mice
In mice, the GR gene, Nr3c1, is greater than 110 kb in length and is the splice product of 9 exons, of which several alternative, noncoding first exons contribute (20). Exon 2 of GR encodes the usual translational start sites and the transactivation domain whereas exon 3 and 4 contain the zinc-finger of the DNA binding domain. The 5 C-terminal exons compose the ligand-binding domain of the receptor (20). Studies of GR disruption have mainly targeted the exon 2 and 3 regions.
The differences we observe in Sim1Cre-GRe2Δ compared with Sim1Cre-GRe3Δ mice may be the result of the level of GR deletion between the 2 lines. We measured a 43% reduction in GR-immunoreactive cells in Sim1Cre-GRe2Δ PVN, whereas Sim1Cre-GRe3Δ mice display an 87% reduction in PVN GR. This difference in deletion extent as being a contributor to the observed phenotypic differences is supported by previous findings in FBGRKO mice that at 2 months of age, when 50% of GR-immunoreactive neurons in the hippocampus are lost, demonstrate no significant differences in HPA axis activity or behavior, but at 4–6 months when there is 90%–100% deletion, have impaired HPA axis activity and increased anxiety and despair-like behaviors (14). These data suggest a critical threshold for GR that, when breached, disrupts normal functions.
An alternative, or additional contributor, to the differences between floxed lines may result from Sim1Cre-GRe2Δ mice having the capacity to produce truncated, function-retaining GR products. Should antibody-mediated detection of possible truncated carboxy-terminal products in Sim1Cre-GRe2Δ mice arise, the transcriptional activation abilities of these products may still be uncertain. Although conventional knockouts targeting exon 2, GRhypo (9) or exon 3, GRnull (11) result in fetal lethality, there is survival of 20% of the GRhypo mice into adulthood. Evidence suggests that insertion of a neomycin-resistance cassette into the second exon of GR results in incomplete gene inactivation due to the synthesis of a truncated C-terminal GR protein that maintains residual binding activity (9, 10, 19, 22). Truncated products were not revealed when GR exon 2 was disrupted by Cre-mediated excision of exon 1C to 2 of GR in thymocytes (21). Given the complexity of GR alternative exon 1 utilization and distal splicing, we cannot discount the existence of tissue-dependent alternative splicing that might yield activity-retaining GR products. In contrast to conventional GR exon 2 deletion, disruption of exon 3 results in complete inactivation of the GR gene due to out-of-frame splicing and premature translation termination (11, 13). We have been unsuccessful in identifying or generating antibodies against the C terminus of GR suitable for immunohistochemistry to directly demonstrate residual GR immunoreactivity in the exon 1C-2-deleted lines.
A recent study that used the same Sim1Cre transgenic mice crossed with floxed GR allele starting at an MfeI site 420 bp upstream of exon 2 region found that the mutant homozygotes die at birth (23). The difference between our PVN GR exon 2-deleted mice and theirs was the inclusion of additional promoter-regulatory regions in our line. Given the lack of mortality in either of our lines, the lethal phenotype observed by Jeannetteau et al. (23) does not arise from PVN deletion effects, but rather deletion in the lung, which was not observed in either of our 2 lines.
Consequences of glucocorticoid excess in Sim1Cre-GRe3Δ mice
Previous generation and characterization of GRNesCre mice, with GR deletion in all brain regions including the PVN, revealed a glucocorticoid excess, cushingoid phenotype (13). More selective GR inactivation in the PVN of our Sim1Cre-GRe3Δ mice also results in a cushingoid phenotype. Sim1Cre-GRe3Δ, compared with control and Sim1Cre-GRe2Δ mice, had reduced weight from weaning (P21) to the first 2 months of age. This pattern changed to increased weight gain throughout adulthood and an obesity phenotype, with thinning of the hair on the abdominal areas and areas of the face and back (Figure 2, A–C). We observed sexual dimorphism in dysregulation of adult body weight, with female Sim1Cre-GRe3Δ mice consistently weighing more than control mice. In comparison, 30% of male Sim1Cre-GRe3Δ mice did not weigh more than control mice, but maintained the truncal obesity phenotype. The obesity and increased body mass phenotype in Sim1Cre-GRe3Δ mice was largely contributed to by increased adiposity as reflected by perigonadal fat pad weights. Female and male Sim1Cre-GRe3Δ mice had approximately 14-fold and 3-fold increases in pelvic fat pad weight, respectively, compared with controls and Sim1Cre-GRe2Δ mice. This differential accumulation of adipose tissue may explain the differences observed in the growth curves of adult female and male mice. Given that males and females typically have altered patterns of fat distribution and accumulation, the influence of excess glucocorticoids interacting with sex hormones may account for the difference in adiposity. Similar to the growth phenotype in Sim1Cre-GRe3Δ mice, GRNesCre mice also demonstrate reduced body weight in the early period of life (24). However, whereas Sim1Cre-GRe3Δ mice display increased fat accumulation, adult GRNesCre mice exhibit significantly reduced body fat mass. Because CRH acts to inhibit food intake and drive energy expenditure, it was suggested that increased PVN CRH in the GRNesCre mice drives the reduced fat mass (24). Because we also find substantially increased PVN CRH in Sim1Cre-GRe3Δ mice, an alternative explanation may be that GR deletion in other brain regions regulating metabolism or sympathetic tone in GRNesCre mice account for the differences. Consistent with the phenotypic evidence of glucocorticoid excess, Sim1Cre-GRe3Δ mice displayed greater than 2.5-fold increases in nadir and peak levels of CORT with maintained circadian rhythmicity. The levels of CORT in Sim1Cre-GRe3Δ mice were further increased after restraint stress.
Interestingly, we found that Sim1Cre-GRe3Δ mice did not increase glucose secretion after restraint stress. This was quite surprising because high circulating glucocorticoids are known to induce hyperglycemia and insulin resistance in organisms (25, 26). It is possible that in Sim1Cre-GRe3Δ mice, the high basal levels of glucocorticoids have resulted in resistance to effects of further increases in glucocorticoids due to existing high occupation of receptors or receptor desensitization.
Consequences of GR disruption on hypothalamic and pituitary regulation
CRH has been implicated as the initiator of circadian and stress-induced increases in glucocorticoid production and is subject to glucocorticoid feedback modulation. Consistent with this role, CRH mRNA in the PVN of Sim1Cre-GRe3Δ mice, in which we find high glucocorticoid levels, is 3 times higher than levels in control and Sim1Cre-GRe2Δ mice.
We found no difference in GR expression in ACTH-immunoreactive cells in the anterior pituitary between control, Sim1Cre-GRe2Δ, or Sim1Cre-GRe3Δ mice. This intact pituitary GR, however, was not sufficient to drive feedback inhibition in the dexamethasone suppression test or prevent the elevated PVN CRH from increasing basal plasma ACTH concentrations in Sim1Cre-GRe3Δ mice. Although Sim1Cre-GRe3Δ mice exhibit higher basal ACTH levels than controls, they do not demonstrate the stress-induced elevations in ACTH observed in control and Sim1Cre-GRe2Δ mice. This is in divergence with CORT levels, where we observe both a circadian and a stress-induced rise in CORT. We tested whether adrenal hypertrophy had developed that would exaggerate smaller changes in ACTH in Sim1Cre-GRe3Δ mice. We found that female mice have trends toward increased adrenal weight compared with controls. Adrenal hypertrophy did occur in male Sim1Cre-GRe3Δ mice, demonstrating a 65% increase in adrenal weights compared with control and Sim1Cre-GRe2Δ mice.
PVN GR disruption and behavioral indices of anxiety and despair
In the current study, despite marked HPA axis dysregulation, we found no significant differences in anxiety-related behaviors measured in the OF or elevated zero maze tests. Neither did we observe significant differences in despair-like behavior in the tail suspension or forced swim tests. GR disruption in the entire brain, GRNesCre (13), and forebrain only, FBGRKO mice (14, 15), result in behavioral maladaptations in mice. The lack of behavioral changes in our PVN GR-disrupted mice supports the interpretation that altered GR signaling in limbic areas mediates the genesis of behavioral dysregulation. We did find that Sim1Cre-GRe3Δ mice have reduced locomotor behavior in the distance traveled in the OF test compared with control mice. This locomotor deficit may arise from the sustained effects of elevated glucocorticoids on muscle function.
In conclusion, we report here that PVN GR is needed for feedback inhibition of HPA axis function but is not required for circadian rhythmicity of adrenal activity. Surprisingly, GR loss in the PVN, with chronically elevated circulating glucocorticoid levels, does not lead to heightened anxiety or despair-like behavior. In many circumstances of glucocorticoid dysregulation with Cushing's syndrome, behavioral changes, particularly anxiety and depression-like symptoms, are common. One possibility suggested by our results is that loss of PVN GR in some manner prevents the induction of stress-related behaviors in the context of normal extrahypothalamic GR and elevated CORT. The whole brain knockout of GR using Nestin-Cre demonstrates that loss of GR, in the context of loss of GR in other brain regions, does lead to altered anxiety-related behaviors (13), as does intact PVN GR with disrupted forebrain GR (14, 15). Given our findings, the most parsimonious explanation synthesizing this information would be that PVN GR does not play a direct role in mediating anxiety and despair behavior. One intriguing possibility is that the extrahypothalamic brain regions in Sim1Cre-GRe3Δ mice are able to accommodate the elevated glucocorticoid levels because they still undergo a qualitatively similar pattern of circadian regulation.
Interesting new areas of investigation arise as a result of these studies. For example, given the normal anxiety and despair-related behaviors observed in Sim1Cre-GRe3Δ mice, can the brain compensate for chronically excess glucocorticoid exposure by altering CORT metabolism or functional GR signaling? Potential mechanisms include down-regulation of GR in extrahypothalamic regions, alteration of expression of coactivators or corepressors, or increases in 11β-hydroxysteroid dehydrogenase type 2 in other brain regions such that normalization of functional signaling occurs. Another area of interest is to investigate how PVN GR loss and excess glucocorticoid attenuate stress-induced increases in blood glucose levels. Future studies designed to answer these questions should prove informative for understanding normal glucocorticoid physiology, the pathogenesis of affective disorders, and the consequences of pharmacologic glucocorticoid therapy.
Acknowledgments
We thank Dr James Herman, Dr Lisa Muglia, and Dr. Melinda Arnett for manuscript review. The behavioral data presented were generated in the Vanderbilt Murine Neurobehavioral Core Laboratory and the Cincinnati Children's Hospital Behavior Core.
This work was supported by National Institutes of Health Grant MH079010.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- CORT
- corticosterone
- DAPI
- 4′,6-diamidino-2-phenylindole
- DEPC
- diethylpyrocarbonate
- GR
- glucocorticoid receptor
- HPA
- hypothalamic-pituitary-adrenal
- PFA
- paraformaldehyde
- PVN
- paraventricular nucleus.
References
- 1. Barden N, Reul JM, Holsboer F. Do antidepressants stabilize mood through actions on the hypothalamic-pituitary-adrenocortical system? Trends Neurosci. 1995;18:6–11 [DOI] [PubMed] [Google Scholar]
- 2. Holsboer F. The role of clinical research. Nat Med. 1995;1:492–493 [DOI] [PubMed] [Google Scholar]
- 3. Holsboer F. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology. 2000;23:477–501 [DOI] [PubMed] [Google Scholar]
- 4. Holsboer F, Barden N. Antidepressants and hypothalamic-pituitary-adrenocortical regulation. Endocr Rev. 1996;17:187–205 [DOI] [PubMed] [Google Scholar]
- 5. Zobel AW, Yassouridis A, Frieboes RM, Holsboer F. Prediction of medium-term outcome by cortisol response to the combined dexamethasone-CRH test in patients with remitted depression. Am J Psychiatry. 1999;156:949–951 [DOI] [PubMed] [Google Scholar]
- 6. Erdmann G, Berger S, Schütz G. Genetic dissection of glucocorticoid receptor function in the mouse brain. J Neuroendocrinol. 2008;20:655–659 [DOI] [PubMed] [Google Scholar]
- 7. Gass P, Reichardt HM, Strekalova T, Henn F, Tronche F. Mice with targeted mutations of glucocorticoid and mineralocorticoid receptors: models for depression and anxiety? Physiol Behav. 2001;73:811–825 [DOI] [PubMed] [Google Scholar]
- 8. Kolber BJ, Wieczorek L, Muglia LJ. Hypothalamic-pituitary-adrenal axis dysregulation and behavioral analysis of mouse mutants with altered glucocorticoid or mineralocorticoid receptor function. Stress. 2008;11:321–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cole TJ, Blendy JA, Monaghan AP, et al. Targeted disruption of the glucocorticoid receptor gene blocks adrenergic chromaffin cell development and severely retards lung maturation. Genes Dev. 1995;9:1608–1621 [DOI] [PubMed] [Google Scholar]
- 10. Finotto S, Krieglstein K, Schober A, et al. Analysis of mice carrying targeted mutations of the glucocorticoid receptor gene argues against an essential role of glucocorticoid signalling for generating adrenal chromaffin cells. Development. 1999;126:2935–2944 [DOI] [PubMed] [Google Scholar]
- 11. Tronche F, Kellendonk C, Reichardt HM, Schütz G. Genetic dissection of glucocorticoid receptor function in mice. Curr Opin Genet Dev. 1998;8:532–538 [DOI] [PubMed] [Google Scholar]
- 12. Kellendonk C, Gass P, Kretz O, Schütz G, Tronche F. Corticosteroid receptors in the brain: gene targeting studies. Brain Res Bull. 2002;57:73–83 [DOI] [PubMed] [Google Scholar]
- 13. Tronche F, Kellendonk C, Kretz O, et al. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103 [DOI] [PubMed] [Google Scholar]
- 14. Boyle MP, Brewer JA, Funatsu M, et al. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci USA. 2005;102:473–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Boyle MP, Kolber BJ, Vogt SK, Wozniak DF, Muglia LJ. Forebrain glucocorticoid receptors modulate anxiety-associated locomotor activation and adrenal responsiveness. J Neurosci. 2006;26:1971–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Erdmann G, Schütz G, Berger S. Loss of glucocorticoid receptor function in the pituitary results in early postnatal lethality. Endocrinology. 2008;149:3446–3451 [DOI] [PubMed] [Google Scholar]
- 17. Schmidt MV, Sterlemann V, Wagner K, et al. Postnatal glucocorticoid excess due to pituitary glucocorticoid receptor deficiency: differential short- and long-term consequences. Endocrinology. 2009;150:2709–2716 [DOI] [PubMed] [Google Scholar]
- 18. Balthasar N, Dalgaard LT, Lee CE, et al. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell. 2005;123:493–505 [DOI] [PubMed] [Google Scholar]
- 19. Mittelstadt PR, Ashwell JD. Disruption of glucocorticoid receptor exon 2 yields a ligand-responsive C-terminal fragment that regulates gene expression. Mol Endocrinol. 2003;17:1534–1542 [DOI] [PubMed] [Google Scholar]
- 20. Strähle U, Schmidt A, Kelsey G, et al. At least three promoters direct expression of the mouse glucocorticoid receptor gene. Proc Natl Acad Sci USA. 1992;89:6731–6735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Brewer JA, Khor B, Vogt SK, et al. T-cell glucocorticoid receptor is required to suppress COX-2-mediated lethal immune activation. Nat Med. 2003;9:1318–1322 [DOI] [PubMed] [Google Scholar]
- 22. Cole TJ, Myles K, Purton JF, et al. GRKO mice express an aberrant dexamethasone-binding glucocorticoid receptor, but are profoundly glucocorticoid resistant. Mol Cell Endocrinol. 2001;173:193–202 [DOI] [PubMed] [Google Scholar]
- 23. Jeanneteau FD, Lambert WM, Ismaili N, et al. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc Natl Acad Sci USA. 2012;109:1305–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kellendonk C, Eiden S, Kretz O, et al. Inactivation of the GR in the nervous system affects energy accumulation. Endocrinology. 2002;143:2333–2340 [DOI] [PubMed] [Google Scholar]
- 25. Phillips DI, Barker DJ, Fall CH, et al. Elevated plasma cortisol concentrations: a link between low birth weight and the insulin resistance syndrome? J Clin Endocrinol Metab. 1998;83:757–760 [DOI] [PubMed] [Google Scholar]
- 26. Reynolds RM, Walker BR. Human insulin resistance: the role of glucocorticoids. Diabetes Obes Metab. 2003;5:5–12 [DOI] [PubMed] [Google Scholar]