

Abstract
The etiology of Legg-Calvé-Perthes' disease (LCPD) is unknown. There are many insights however from epidemiologic/demographic information. A systematic medical literature review regarding LCPD was performed. The incidence ranges from 0.4/100,000 to 29.0/100,000 children <15 years of age. There is significant variability in incidence within racial groups and is frequently higher in lower socioeconomic classes. The typical age at presentation ranges from 4 to 8 years (average 6.5 years), except for children from the Indian subcontinent (average 9.5 years). There is a mild familial component. The children demonstrate impaired growth in height, skeletal age, and birth weight. This impaired growth coincides with an age appropriate reduced somatomedin A activity and decreased levels of IGF. LCPD can be associated with abnormalities in the coagulation cascade, including an increase in factor V Leiden mutation, low levels of protein C and/or S, and decreased antithrombin activity. There is decreased turnover in type I collagen and synthesis of type III collagen, as well as reduced levels of urinary glycosaminoglycans in the active phases of the disorder. Subtle abnormalities in the opposite hip and other minor/major congenital defects are reported. Children with LCPD are active and score abnormally in certain standardized psychological tests.
1. Introduction
Legg-Calvé-Perthes' disease (LCPD) is an idiopathic osteonecrosis of the proximal capital femoral epiphysis in children. The epiphysis undergoes collapse, resorption, reossification, and eventual healing. The healed hip may range from an essentially normal contoured femoral head (Stulberg I) to one with incongruous incongruity (Stulberg V). As with any pathologic process, LCPD goes through a course of disease denoted by the Waldenström stages, which are synovitic, avascular, fragmentation (collapse), reossification (healing), and healed (residual). The magnitude of epiphyseal involvement is determined by the Catterall class [1], Salter-Thompson group [2], and/or lateral pillar group [3]. The Catterall class is determined on both anteroposterior and frog-lateral radiographs during the stage of maximum fragmentation, the Salter-Thompson group is determined on the frog-lateral radiograph during the avascular/precollapse stage using the subchondral crescent fracture, and the lateral pillar classification is determined on the anteroposterior radiograph during early fragmentation.
2. Materials and Methods
There are many epidemiologic and demographic findings in LCPD. A systematic review of LCPD was performed. LCPD has been known by at least 22 different names since its first description in the late 19th and early 20th centuries [4]. Since 1963, the official medical subject heading (MESH) used by the National Library of Medicine is Legg-Perthes' disease, but many other names had been previously used. To ensure capture of all the published literature, older terms were also searched as keywords or keyword phrases. Therefore, the terms used to search for LCPD were arthritis deformans juvenilis, Calve-Perthes disease, coxa plana, femoral head necrosis, juvenile chondroepiphysitis, Legg-Calve-Perthes disease, Legg-Perthes disease, Legg's disease, osteochondritis deformans juvenilis, osteochondritis deformans juvenilis coxae, osteochondritis juvenilis, osteochondrosis of capital epiphysis of femur, Perthes disease, and pseudocoxalgia.
The databases searched were PubMed (http://www.ncbi.nlm.nih.gov/pubmed/), Ovid Medline, EMBASE, WorldCat (books and theses) (http://firstsearch.oclc.org/), and IndexCat (Index Catalogue of the Library of the Surgeon-General's Office) (http://www.indexcat.nlm.nih.gov/). Exclusion criteria were those manuscripts discussing surgery, therapy, rehabilitation, and any foreign language articles without an English abstract. Individual journals were also searched for articles published prior to 1996 that predate electronic Medline indexing, including Journal of Bone and Joint Surgery (American and British), Clinical Orthopaedics and Related Research, and Acta Orthopaedica Scandinavica. Age groups were limited to those <18 years old. Duplicate citations were removed. The dates for the search were 1880–1961 for IndexCat, 1900–2009 for WorldCat, 1948–1965 for OldMedline, and 1950–February 2010 for Ovid Medline.
This search resulted in 1124 unique citations. These 1124 manuscripts were reviewed to find those that discussed any of the topics regarding etiology, epidemiology, demographics, incidence, prevalence, race, gender, family history, genetics, inheritance, age, bone age, weight (either birth weight or normal weight), height, growth, maturation, other anthropometric characteristics, hormone/endocrine, smoking, coagulation, fibrinolysis, congenital anomalies, collagen, immunoglobulin, opposite hip, behavior/psychology, seasonal variation, and infection. Of these 1124 manuscripts, 144 provided ample information and are the contents of this paper.
3. Results
3.1. Incidence
The conventional quotation for the incidence of LCPD is the number per 100,000, usually for age < 15 years. The incidence of LCPD ranges widely, from 0.4 in Eastern India (Vellore-Taluk area) to 29.0 (Table 1) in the Faroe Islands (North Atlantic ocean). Significant variability exists within countries, cities, and ethnic groups. Race is classified using the definitions of Eveleth and Tanner: Caucasians, Africans in Africa and of African ancestry, Asiatics (Amerindians, Hispanics, Indonesian-Malays), Indo-Mediterraneans (inhabitants of the Near East, North Africa, and Indian subcontinent), and Australian Aborigines and Pacific Island peoples [23].
Table 1.
Incidence of Legg-Calvé-Perthes' disease*.
| Study | Year | City, country | Region | Ethnicity | No Pts | Incid |
|---|---|---|---|---|---|---|
| Joseph et al. [5] | 1988 | Vellore, India | Asia | Indo-Med (Indian) | 4 | 0.4 |
| Purry [6] | 1982 | Eastern Cape, South Africa | Africa | Black | 6 | 0.45 |
| Kim et al. [7] | 2006 | Japan | Asia | Indo-Malay (Japanese) | 711 | 0.9 |
| Purry [6] | 1982 | Eastern Cape, South Africa | Africa | Mixed | 11 | 1.73 |
| Ebong [8] | 1977 | Nigeria | Africa | Black | 10 | 1.8 |
| Rowe et al. [9] | 2005 | Chonnam, Korea | Asia | Indo-Malay (Korean) | 84 | 3.8 |
| Joseph et al. [5] | 1988 | Udupi, India | Asia | Indo-Med (Indian) | 138 | 4.4 |
| Wijesekera [10] | 1984 | Kurunegala, Sri Lanka (Ceylon) | Asia | Indo-Med (Indian) | 76 | 3.96 |
| Thompson and Leong [11] | 1978 | Hong Kong | Asia | Indo-Malay (Chinese) | 32 | 4.5 |
| Gray et al. [12] | 1972 | British Columbia, Canada | North America | White | 379 | 5.1 |
| Barker et al. [13] | 1978 | Wessex, England | British Isles | White | 34 | 5.5 |
| Molloy and MacMahon [14] | 1966 | Massachusetts | North America | White† | 86 | 5.7 |
| Hall and Barker [15] | 1989 | Yorkshire, England | British Isles | White | 101 | 6.1 |
| Barker et al. [13] | 1978 | Trent, England | British Isles | White | 78 | 7.6 |
| Moberg and Rehnberg [16] | 1964 | Zealand, Denmark | Scandinavia | White | NA | 8.0 |
| Moberg and Rehnberg [16] | 1992 | Uppsala, Sweden | Scandinavia | White | 51 | 8.5 |
| Moberg and Rehnberg [16] | 1964 | Jutland, Denmark | Scandinavia | White | NA | 9.0 |
| Wiig et al. [17] | 2006 | Norway | Scandinavia | White | 425 | 9.2 |
| Purry [6] | 1982 | Easter Cape, South Africa | Africa | White | 38 | 10.8 |
| Margetts et al. [18] | 2001 | Liverpool, England | British Isles | White | 122 | 11.1 |
| Barker et al. [13] | 1978 | Mersey, England | British Isles | White | 68 | 11.1 |
| Kealey et al. [19] | 2000 | Northern Ireland | British Isles | White | 313 | 11.6 |
| Pillai et al. [20] | 2005 | Dumfries, Scotland | British Isles | White | 40 | 15.4 |
| Hall et al. [21] | 1983 | Liverpool, England | British Isles | White | 157 | 15.6 |
| Niclasen [22] | 1974 | Faroe Islands, Denmark | Scandinavia | White | 43 | 29.0 |
*(per 100,000 children <15 yrs old).
†one of the 86 children was African.
NA: not available.
3.2. Whites
3.2.1. British Isles
The incidence ranges from 5.5 in Wessex, England [13], to 15.6 in Liverpool, England [21]. The incidence in 3 different regions of England [13] was 5.5 in the Wessex Health District, 7.6 in the Trent Health District, and 11.1 in the Mersey Health District (including Liverpool) (Figure 1(a)). In Liverpool, the incidence in the inner city was higher (21.1) compared to the surrounding areas (13.1—outer Liverpool, 14.6—Knowsley district, 11.9—Sefton district) [21] (Figure 1(b)) implying that the incidence is lower in less populated or more rural areas. However, in southwest Scotland [20], the incidence was higher in less populated areas (17 to 30) compared to more populated areas (4.5). In Yorkshire, England, which has a substantial rural population, the average incidence was 6.1, with large geographical variations unexplainable by differences between urban and rural populations [15]. The East Riding area of Yorkshire, located on the best agricultural land, had no cases [15].
Figure 1.
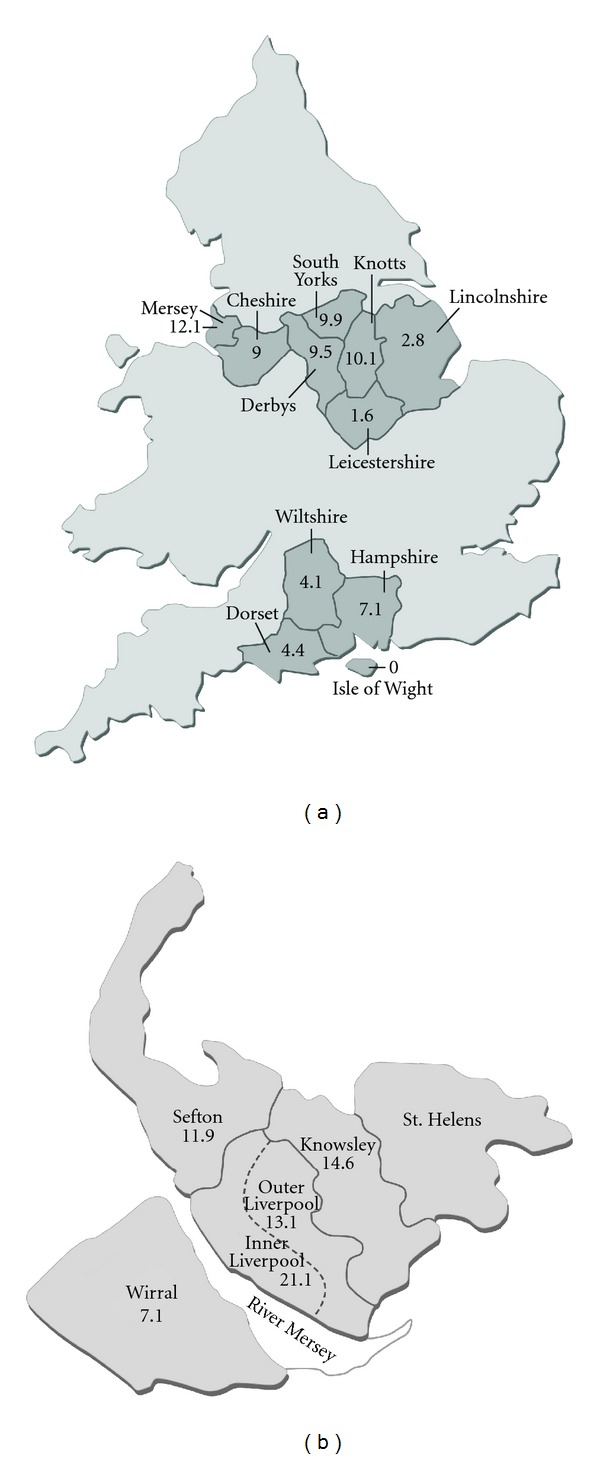
Maps demonstrating various incidences of LCPD in different regions of England. (a) Incidence of LCPD in 1976 per 100,000 children aged 14 years and under in three regions of England. Map of England taken and adapted from the National Policing Improvement Agency, located at http://maps.police.uk/, with permission (Data from [13]). (b) Average yearly incidence of LCPD per 100,000 children aged 14 and under in the Liverpool administrative area. Map of Merseyside area taken and adapted from the National Museums Liverpool located at http://www.liverpoolmuseums.org.uk/maritime/exhibitions/magical/placenames/index.asp, with permission (Data from Hall et al. [21] and Barker et al. [13] (Wirral)).
Many authors have noted differences in incidence by social class and/or inner city/urban/rural location. In the seminal epidemiologic study of 310 children in Edinburgh and Glasgow, Scotland [24], there was a higher than expected proportion of children with LCPD in lower socioeconomic classes; the same was noted in Liverpool [25] (Figure 2(a)). The incidence in the Liverpool inner city within the highest socially deprived area was 31.7 and 10.3 for the lowest; in the outer city the incidence was 21.8 within the highest socially deprived area and 7.4 for the lowest [21] (Figure 2(b)). In Northern Ireland [19], the highest incidence is in the most deprived rural location (16.1), over twice that in the least deprived rural location (7.1). In Southwest Scotland [20], the incidence was 33.6 in the most deprived areas and 7.8 in the least deprived areas; the 33.6 incidence is the highest found to date in any series/publications. However, in Glasgow, there was no association of LCPD incidence and social class [26]. In general, the incidence of LCPD in the British Isles is higher in lower socioeconomic classes and variable regarding rural/urban location (Figure 2(b)).
Figure 2.
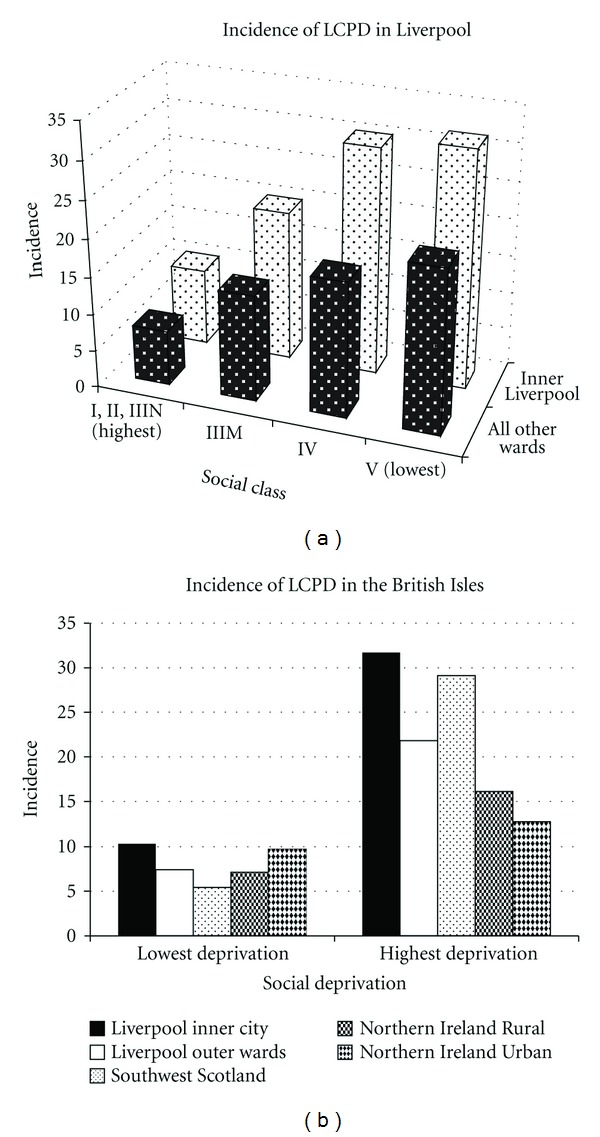
(a) Incidence of LCPD (100,000 children per year ≤14 years of age) by social class in location in Liverpool (Data from Hall et al. [21]). (b) Composite incidence of LCPD (100,000 children per year <14 years of age) by highest and lowest deprivation indices separated by rural and urban locations in the British Isles (Data from Hall et al. [21], Kealey et al. [19], and Pillai et al. [20]).
3.2.2. Scandinavia
The incidence is 8.5 in Uppsala, Sweden [16], and 9.2 in Norway [17]. Within Norway, similar to the British Isles, there is significant variability; the lowest incidence in the north (5.4) and the highest in the center and west (10.8 and 11.3).
3.2.3. North America
In British Columbia [12], the incidence was 5.10 and, in Massachusetts, [14] 5.7.
3.2.4. Africa
In Eastern Cape, South Africa [6], the incidence in Whites is 10.8; in the urban areas (Port Elizabeth and Uitenhage), it is ∼2 times greater than in rural areas. This urban-rural dichotomy was noted overall (3.85 versus 1.1) and when separated by race (12.6 versus 6.0 for Whites, 2.2 versus 1.4 for mixed African-White, and 0.7 versus 0.28 for Africans).
3.3. Indo-Malays
In Japan [7], the incidence was 0.90. In Bradford, England, the incidence was 4.6 in Caucasian children and 0.63 in Indo-Malay children [27]. In Korea [9], the incidence was 3.8 and lower in the greater Gwanju metropolitan areas compared to the rural Chonnam province (3.2 versus 4.3).
3.4. Indo-Mediterraneans
3.4.1. India/Sri Lanka
There is a 10-fold variability in incidence in India; 0.4 in the east (Vellore Taluk) [5] to 4.4 in the west (Udupi Taluk). In Sri Lanka (Kurunegala district) the incidence is 3.96 [10], and all 76 children with LCPD were from lower income groups [10].
3.5. Africans
True LCPD (excluding sickle cell hemoglobinopathy) is extremely rare in Africans. In Eastern Cape, South Africa [6], the incidence is 0.45 and rises to 1.73 in children of mixed African/Caucasian ancestry. The incidence is 1.8 in Nigeria [8]. In Togo, there were 22 cases of LCPD in 29620 children attending two Togolese hospitals over a 7-year period, indicating the rarity of the disorder. One of 86 children in Massachusetts [14] was African, and two of the 188 children in Connecticut was African [28].
3.6. Other Demographics (Age, Gender, Laterality, Family History)
3.6.1. Age, Gender, Laterality, LCPD Severity
The average age is 6.5 years, with a typical age range of 4 to 8 years (Table 2). The average age for Indian children is 9.5 years, for Nigerian children 10.3 years, and for all others 6.3 years. LCPD is more common in boys (81.4%) than girls (18.6%) and mostly unilateral (89.2%). Right and left hip involvement is similar (46.5% and 53.5%). In 1638 hips (Table 3), 112 (6.8%) were Catterall class I, 295 (18.0%) class II, 710 (43.3%) class III, and 521 (31.8%) class IV. In 1671 hips, 236 (14.1%) were lateral pillar group A, 971 (58.1%) B and B/C border, and 464 (27.8%) C.
Table 2.
Demographics of 4166 children with Legg-Calvé-Perthes' disease.
| Study | Year | City, country | Region | Race | No Pts | Age (yrs) | M (%) | F (%) | Unil (%) | Bil (%) | R (%) | L (%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rosenfeld et al. [29] | 2007 | Dallas, TX | North America | 172 | 4.6 | 156 (90.7) | 16 (9.3) | |||||
| Catterall [1] | 1971 | London, England | British Isles | White | 121 | 4.66 | 96 (79.3) | 25 (20.7) | 109 (90.1) | 12 (9.9) | ||
| Gray et al. [12] | 1972 | British Columbia, Canada | North America | White | 379 | 5 | 322 (85.0) | 57 (15.0) | 317 (84.1) | 60 (15.9) | 150 (47.3) | 167 (52.7) |
| Kealey et al. [19] | 2000 | Northern Ireland | British Isles | White | 313 | 5.7 | 256 (81.8) | 57 (18.2) | 264 (84.3) | 49 (15.7) | ||
| Moberg and Rehnberg [16] | 1992 | Uppsala, Sweden | Scandinavia | White | 51 | 5.75 | 38 (74.5) | 13 (25.5) | 45 (88.2) | 6 (11.8) | ||
| Hall et al. [21] | 1983 | Liverpool, England | British Isles | White | 157 | 5.8 | 133 (84.7) | 24 (15.3) | ||||
| Wiig et al. [30] | 2008 | Norway | Scandinavia | White | 425 | 5.8 | 324 (76.2) | 101 (23.8) | 370 (87.1) | 55 (12.9) | 167 (45.4) | 201 (54.6) |
| Fulford et al. [31] | 1993 | Edinburgh, Scotland | British Isles | White | 94 | 5.8 | 85 (90) | 9 (10) | 89 (95) | 5 (5) | ||
| Fisher [28] | 1972 | Connecticut | North America | White | 203 | 6 | 153 (81.4) | 35 (18.6) | 163 (86.7) | 25 (913.3) | 83 (50.9) | 80 (49.1) |
| Rowe et al. [9] | 2005 | Chonnam province, Korea | Asia | Indo-Malay (Korean) | 84 | 6 | 79 (94) | 5 (6) | 46 (52) | 43 (48) | ||
| Pillai et al. [20] | 2005 | Dumfries, Scotland | British Isles | White | 40 | 6.5 | 31 (78) | 9 (22) | ||||
| Guille et al. [32] | 1998 | Wilmington, DE | North America | 575 | 6.9 | 470 (81.7) | 105 (18.3) | 497 (86.4) | 78 (13.6) | |||
| Wang et al. [33] | 1990 | Taipei, Taiwan | Asia | Indo-Malay (Chinese) | 57 | 7 | 47 (83) | 10 (17) | 52 (91) | 5 (9) | 21 (40) | 31 (60) |
| Kim et al. [7] | 2006 | All Japan | Asia | Indo-Malay (Japanese) | 711 | 7.08 | 606 (86.4) | 95 (13.6) | 656 (92.3) | 55 (7.7) | 281 (43.0) | 327 (57.0) |
| Petrie and Bitenic [34] | 1971 | Montreal, Quebec | North America | White | 60 | 7.75 | 49 (82) | 11 (18) | 41 (84) | 8 (16) | ||
| Herring et al. [35] | 2004 | All USA | North America | 345 | 8 | 271 (80.4) | 66 (19.6) | 317 (91.9) | 28 (8.1) | 162 (47.0) | 183 (53.0) | |
| Joseph et al. [5] | 1988 | Karnataka and Vellore, India | Asia | Indo-Med (Indian) | 138 | 9.4 | 97 (70.3) | 41 (29.7) | ||||
| Chacko et al. [36] | 1986 | Karnataka, India | Asia | Indo-Med (Indian) | 165 | 9.6 | 119 (72.1) | 46 (27.9) | 155 (93.9) | 10 (6.1) | 92 (54.9) | 63 (40.6) |
| Wijesekera [10] | 1984 | Kurunegala, Sri Lanka (Ceylon) | Asia | Indo-Med (Sri Lankan) | 76 | 9.3 | 52 (68) | 24 (32) | 74 (97) | 2 (3) | 40 (53) | 34 (47) |
|
| ||||||||||||
| All | 6.5 | 4237 (81.4) | 969 (18.6) | 4039 (89.2) | 491 (10.8) | 1227 (46.5) | 1410 (53.5) | |||||
| Averages | Non-Indian | 6.3 | ||||||||||
| Indian | 9.5 | |||||||||||
Table 3.
Severity of epiphyseal involvement in Legg-Calvé-Perthes' disease.
| Study | Year | Location | Catterall class | Lateral pillar group | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| I | II | III | IV | A | B | BC | B + BC | C | |||
| Rosenfeld et al. [29] | 2007 | Dallas, TX | 7 | 108 | 30 | 138 | 43 | ||||
| Catterall [1] | 1971 | London, England | 31 | 31 | 22 | 13 | |||||
| Guille et al. [32] | 1998 | Wilmington, DE | 37 | 120 | 228 | 233 | 155 | 204 | 204 | 204 | |
| Wang et al. [33] | 1990 | Taipei, Taiwan | 0 | 7 | 19 | 21 | |||||
| Kim et al. [7] | 2006 | Japan | 30 | 103 | 352 | 210 | 68 | 350 | 350 | 157 | |
| Herring et al. [35] | 2004 | USA | 6 | 218 | 61 | 279 | 60 | ||||
| Wijesekera [10] | 1984 | Kurunegala, Sri Lanka | 13 | 21 | 33 | 11 | |||||
| Chacko et al. [36] | 1986 | Karnataka, India | 1 | 13 | 56 | 33 | |||||
|
| |||||||||||
| Total | 112 | 295 | 710 | 521 | 236 | 880 | 91 | 971 | 464 | ||
|
| |||||||||||
| Percentage | 6.8 | 18.0 | 43.3 | 31.8 | 14.1 | 52.7 | 5.4 | 58.1 | 27.8 | ||
3.6.2. Family History/Genetics
A positive family history has been noted by many [7, 24, 27, 28, 37–41]. Quoted percentages are 4.5% [7], 7% [28], and 8% [27]. There are also reports in siblings [42, 43]. The recurrence risk was 2.6% for siblings and offspring in a review of the family histories of 842 English children with LCPD [44], arguing for a multifactorial inheritance pattern. The proportion of the 842 children having a 1st degree relative with LCPD was 1.6%, a 2nd degree relative 0.27%, and a 3rd degree relative 0.27%; all higher than the average English incidence. In South Wales [45], the risk of LCPD in siblings was under 1% and of an affected parent 3%. There are several case reports of LCPD transmitted through several generations [40, 46]. In the Faroe Islands [47], an isolated genetic community, an accumulation of both LCPD and developmental dislocation of the hip was noted in certain families; it remains to be determined if this is genetic, environmental, or both. Others note no significant association with family history [24, 33].
There are case reports of LCPD in twins, both monozygotic [48–51] and dizygotic [52], as well as three female 1st degree relatives [53]. These dated studies could not assess for genetic markers, and thus it is unknown if this represents a true genetic pattern or simply the statistical chance of siblings developing the same disease. Wynne-Davies and Gormley [24] described 6 sets of twins with only one of the twins having LCPD.
Several studies show associations with certain HLA types. A positive association was noted with HLA-A1 [54, 55] and HLA-A9, HLA-A10, and HLA-B27 [56]. A protective effect was seen with HLA-A2 and HLA-Cw3; the incidence of LCPD was less in those types [57]. Two studies found no differences in HLA types [58, 59]. There is no apparent association between ABO and Rh blood groups [60].
3.7. Perinatal Factors (Parental Age, Birth Order/Presentation, Birth Weight)
Both parents of children with LCPD were older than the normal population (31.7 versus 28.8 years for the fathers, 28.9 versus 26.9 for the mothers) in one study [24], with no differences noted in parental age by others [28, 33, 61].
LCPD was more frequent in the 3rd born or older children [24] in one study, while others noted no differences [33]. Children with LCPD are more commonly born breech—10.7% compared to 2–4% in the normal population [24]. A lower birth weight was noted in children with LCPD [62, 63]; 7.1 lbs in 70 children with LCPD and 7.8 lbs in 70 control children without LCPD [62]. In 5 sets of twins, the smaller twin at birth developed the LCPD [63]; the average discordance in birth weight was 13.4% (range 7.1 to 23.5%). Others note no differences in birth weight [24, 33, 61].
3.8. Impaired Growth, Anthropometric Differences, and Skeletal Maturation
3.8.1. Impaired Growth and Anthropometric Differences
Height retardation was noted in 185 Ohio children with LCPD [64], even when accounting for parental height; body weight was average or above average. In Scottish children, a greater proportion of LCPD children have diminished height (<10th percentile) with no differences for weight [24]. In 76 Sri Lanka children with LCPD, 46% were below average height at presentation [10]. In 109 Japanese children with LCPD, 97 (89%) were below the mean in height [65]. Children with LCPD are shorter at birth and remained so during the phases of LCPD and adulthood [66, 67]; boys were 4.4 cm shorter and girls 2.5 cm shorter than their norms [66]. No height or weight differences were found in Irish [68] and Jewish children with LCPD [69].
Skeletal growth is progressively impaired in a caudal direction. Rostral sparing is documented by normal head growth [70] with increasing growth retardation in a caudal direction: biacromial width was less reduced than standing height; forearm and hand showed more impaired growth than the upper arm; the feet showed more impaired growth than the leg. This impaired growth most severely affects the feet [24, 71]. Growth retardation in LCPD children from rural India [72] is identical to English children.
3.8.2. Skeletal Maturation
Aside from one study in Jewish children with LCPD [69], all others note delayed bone age in LCPD. In 182 children with LCPD, many were <3rd percentile bone age, which was more common in boys than girls [76]. In 125 of 140 (89%) children with LCPD [28] bone age was delayed. Bone age was at least 3 months less in 83% of children [67]. This is seen [75] with both the Greulich-Pyle hand-wrist assessment [73] and the Oxford pelvis method [74] of determining bone age (Figure 3). The average chronologic age for both boys and girls was 8.2 years; for boys, the average hand-wrist bone age was 7.4 years and average pelvic bone age 5.9 years, and, for girls, the average hand-wrist bone age was 6.9 years and average pelvic bone 7.0 years [75]. Carpal maturation was delayed in 125 children with LCPD; the most severe delay was at 3 to 5 years of age [77]. In a study of 27 girls with LCPD at the time of diagnosis [78], bone age (Tanner Whitehouse 2 method) was delayed an average of 1.4 years for the radius/ulna and 1.9 years for the carpals. A greater delay in bone age is associated with more severe LCPD [79]. Children with transient synovitis show minimal delay in bone age compared to those with LCPD (7 months versus 23 months) [80].
Figure 3.
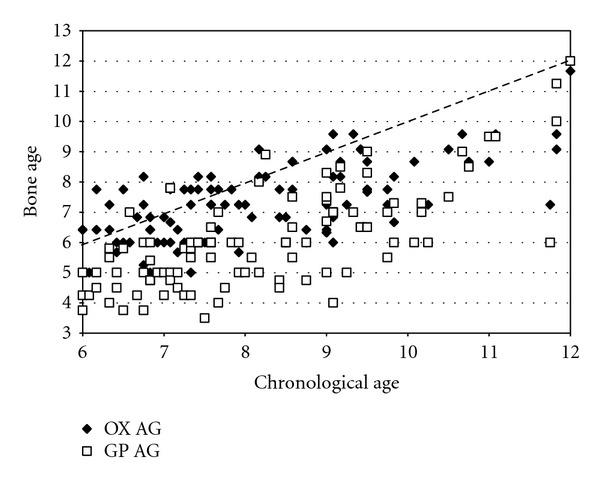
Chronologic age as a function of bone age in 100 children with LCPD. Both the hand-wrist bone of Greulich and Pyle (GP AG) [73] as well as the Oxford pelvic bone age (OX AG) [74] are shown (Data from the study of Loder et al. [75]).
In non-Caucasians, bone age was delayed 31.8 months in 17 of 25 Formosan (68%) children with LCPD [33]. In Hong Kong, all Chinese children with LCPD had a bone age lower than the mean [11]. In Korean children, bone age was delayed 10.4 months in boys and 4.6 months in girls [9]. In 21 Japanese LCPD children, delayed bone age was noted in all [65]. In 76 Sri Lankan children, 78% demonstrated skeletal retardation [10]. In Mexican children (Hispanic-Amerindian), bone age was delayed 28 months in children with LCPD between the ages of 6–10 years [81].
Skeletal standstill (no increase in bone age with increasing chronologic age) occurs in LCPD [75, 76] and resolves after the LCPD has healed [80].
3.9. Endocrine Dysfunction
Postnatal skeletal development is regulated by growth hormone, whose effects are partly mediated by somatomedins. Somatomedins stimulate cartilage activity resulting in cell proliferation and hypertrophy. In Japan, the incidence of LCPD was 70 in growth-hormone-deficient children [82] compared to 0.9 in the normal population [7]. Serum growth hormone response to insulin-induced hypoglycemia is reduced in boys with LCPD compared to those with constitutional short stature [83]. The primary somatomedin responsible for postnatal skeletal maturation is somatomedin C insulin-like growth factor (IGF-1). Somatomedin deficiency may result in impaired skeletal maturation, a well-known phenomenon in LCPD. Somatomedin A [84] and C [65] deficiency has been noted LCPD. Somatomedin activity normally increases with age in growing children, but this does not occur in children with LCPD [85, 86]. Plasma levels of IGF-1 were reduced the first 2 years after the diagnosis of LCPD [87], but with normal levels of IGF-binding protein [88]. Low levels of IGF-1 were confirmed by Crofton et al. [89], who also noted abnormal collagen turnover in the acute stages of LCPD. In plasma, nearly all the IGF-1 is bound to specific binding proteins, which for IGF-1 is the IGF-binding protein 3 (IGFBP3). Decreased levels of IGFBP in children with LCPD have been seen but with normal levels of IGF-1 [90]. No abnormalities in IGF-1 or IGFBP concentrations have been encountered by others [68, 83, 91, 92] in children with LCPD.
Early studies [93, 94] noted an association with hypothyroidism and LCPD but not seen in more recent studies [68, 83, 95–98]. No abnormalities in adrenal function (cortisol) [68, 83] or cholesterol [96] have been noted.
3.10. Smoking, Hypofibrinolysis, and LCPD
Passive smoke exposure during pregnancy has been correlated with LCPD. This was first noted in Massachusetts [62]; maternal smoking while pregnant was present in 63% of LCPD and 43% of control cases. This was confirmed in Sweden [99]; maternal smoking during pregnancy increased the odds of developing LCPD in the child by 1.44 if the mother smoked <10 cigarettes per day, and by 2.1 when ≥10 cigarettes per day. It was also noted that children with a birth weight <1500 gms had a 2.4 times increased risk of developing LCPD.
An increase in LCPD in children exposed to passive smoke after birth has also been noted. In children with LCPD [100], 63.9% had at least one smoker living in the child's household with a mean of 1.03 smoker years per year of life exposure to smoke; in control children, 39.6% had at least one smoker living in the child's house with a mean of 0.48 smoker years per year of life exposure to smoke. No association was noted between lower income and LCPD. This association with passive smoke exposure was corroborated in Spain [101], where 79% of LCPD children were passive smokers compared to 43% of controls. The odds ratio for a child, after controlling for age and gender, of developing LCPD when exposed to passive smoke was 5.3 (95% CI 2.9–9.7). There were no associations between passive smoking and age of child, Catterall class, or final Stulberg result. In another study of 39 children with LCPD [102], 24 had exposure to second hand smoke, some even in utero (17 of the 24). Of the children with LCPD and smoke exposure, 48% had low stimulated tissue plasminogen activator activity, compared to only 7% of the children without smoke exposure. In Georgia [103], children exposed to passive smoke were 5.6 (95% CI 2.0–12.0) times more likely to develop LCPD than those not exposed. This was strongly associated with a polymorphism in the β-fibrinogen gene G-455-A, which results in increased fibrinogen levels, thus leads to thrombotic/coagulation abnormalities in children with LCPD (Figure 4).
Figure 4.
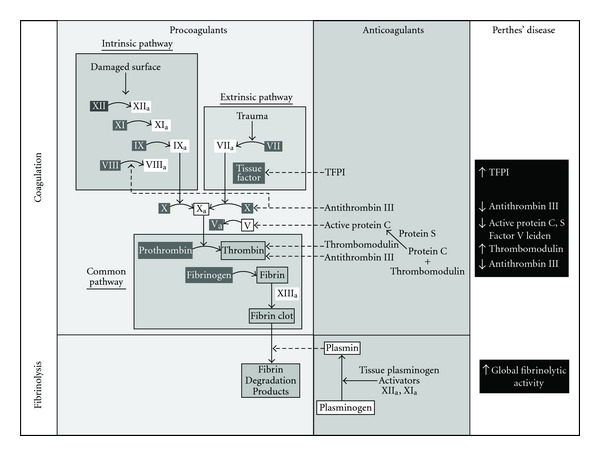
The coagulation and fibrinolytic cascade as it relates to children with LCPD. The abnormalities in this cascade in children with LCPD are shown in the far right column. TFPI: tissue factor pathway inhibitor.
Factor V Leiden mutation discovered in Leiden, Netherlands [104], results in production of factor V that cannot be inactivated by activated protein C. This leads to a persistence of circulating activated factor V with continued activation of the coagulation cascade and a hypercoagulable state. Families with LCPD and factor V Leiden mutations have been described [105, 106]. In nonfamilial LCPD, a factor V Leiden mutation has been noted by many; 12.5% [107] and 10.6% in children with LCPD [108] in studies without controls. In studies with controls, these values are 30% in LCPD and 1.87% in controls [109], 11% in LCPD and 4% in controls [110], 9% in LCPD and 5% in controls [111], and 4.9% in LCPD and 0.7% in controls [112]. Children with the most severe LCPD (Catterall IV) were homozygous for factor V Leiden mutation [108]. High levels of anticardiolipin antibodies (26% versus 11%) have also been noted [110]. The OR of developing LCPD with factor V Leiden mutation in two studies are 22.5 [109] and 3.3 [113]; the OR of developing LCPD with ≥ abnormalities in factor V or anticardiolipin antibody is 3.29 [110].
Other coagulation abnormalities exist in LCPD. Thrombophilia and hypofibrinolysis were noted in 8 children [114] in 1994. A subsequent investigation noted that 75% of 44 children with LCPD had coagulation abnormalities [115]; thrombophilia (a deficiency in antithrombotic factor C or S, with an increased tendency towards thrombosis) in 23 children; increased lipoprotien(a) (a thrombogenic lipoprotein associated with osteonecrosis in adults) in 7 children; hypofibrinolysis (reduced ability to lyse clots) in 3 children. In another study, only 14 of 64 children (22.5%) with LCPD had entirely normal coagulation measures [107] with resistance to activated protein C the most common abnormality (23 of 64). A 3.8 times increased risk for LCPD with low levels of protein C has been found [111]. Protein C activity is also lower in LCPD [116, 117]. Both protein C and antithrombin activities were lower in LCPD than controls [117]; a family history of hereditary thrombophilia was higher in LCPD than controls. LCPD was increased 2.8 times with protein S deficiency and 7.5 times with elevated factor VIII levels [113]. Others note no coagulation abnormalities in LCPD [116, 118–127].
Another fact supporting a hypercoagulable state in LCPD is tissue factor pathway inhibitor (TFPI). TFPI is an important natural anticoagulant molecule that downregulates the tissue factor dependent coagulation pathway. A deficiency leads to a prothrombotic state, and over expression may be a protective mechanism against ongoing local microvascular events. TFPI concentrations in children with LCPD were significantly higher (56.8 ng/mL) compared to controls (37.3 ng/mL) [128]. This is interpreted as a physiologic response to a hypercoagulable state; an increased TFPI is natural anticoagulation. Increased blood viscosity in LCPD is reported [129]; thus vascular occlusion may simply be due to fluid mechanic properties [129].
After thrombosis, the body attempts to lyse the clot. Fibrinolysis is mediated in part by thrombomodulin, an endothelial cell membrane-associated glycoprotein which functions in activation of the anticoagulant systems. In LCPD, thrombomodulin and global fibrinolytic activity are elevated [130]. This is interpreted as a compensatory reaction to the thrombosis in LCPD.
3.11. Other Associations with LCPD
3.11.1. Collagen Metabolism/Genetics and Bone Turnover
Type I collagen is found almost exclusively in bone and calcifying tissues. Markers of type I collagen degradation are urinary deoxypyridinoline (DPD) and type I collagen telopeptide (ICTP). ICTP is higher in children with LCPD compared to controls [89], indicating an increase in type I collagen degradation. The median urinary DPD/creatinine ratio in children with LCPD is reduced during the fragmentation stage and returns to normal (if not slightly higher) in the healed stage [131] (Figure 5). The DPD/creatinine decrease is greater with more severe LCPD (egg lateral pillar C > lateral pillar B). These findings support a systemic etiology in LCPD.
Figure 5.
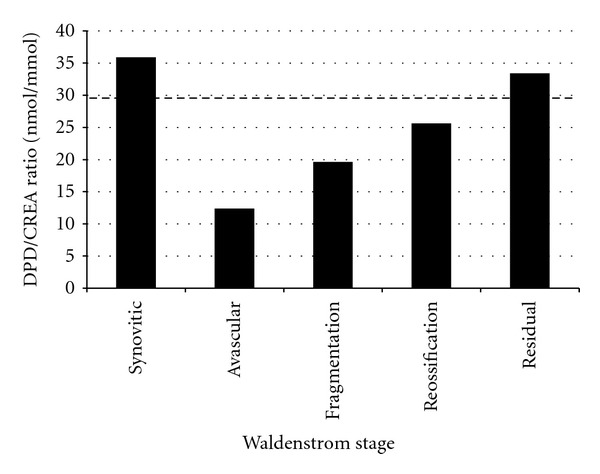
Urinary DPD/CREA (urinary deoxypyridinoline/creatinine ratio) in children with LCPD in different Waldenström stages. The control level is denoted by the hatched line. DPD is a degradation product of type I collagen; a decrease in its urinary excretion indicates a decrease in bone turnover. Thus, there is decreased bone turnover in children with LCPD during the avascular and fragmentation phases of the disease (Data from Westhoff et al. [131]).
A marker of type III collagen synthesis is the procollagen type III N-terminal propeptide (P3NP). Type III collagen synthesis is reduced at diagnosis in children with LCPD as demonstrated by very low levels of P3NP. However, there were no controls, and the differences in children with LCPD compared to otherwise normal children in the same geographic/ethnic/socioeconomic situation are not known.
A recurrent mutation in type II collagen (cartilage collagen) in a Japanese family with LCPD [38] has been noted. This mutation amino acid change (p.G1170S) perturbs the Gly-X-Y triple-helix of type II collagen. Similar findings were noted in a Chinese family where a p.Gly1170S mutation of COL2A1 resulted in premature hip osteoarthritis, avascular necrosis of the femoral head, or LCPD, depending upon the age at onset [39]. In a cohort of nonfamilial children with LCPD, no mutations in the COL2A1 gene were found [124].
3.11.2. Articular Cartilage Markers
Glycosaminoglycans (GAGs) are chains of repetitive disaccharide units linked with proteins in the cartilaginous extracellular matrix to form proteoglycans. Upon cartilage degradation, GAGs are eliminated by the kidneys. Elevated urinary GAG levels indicate increased articular cartilage degradation. Decreased levels of urinary GAGs in children with LCPD compared to normal children or those with transient synovitis have been noted [132]. This can be interpreted as either increased preservation of the GAGs within the hip or a decrease in the quantity of synovial fluid. Increased levels of proteoglycan fragments and stromelysin in the synovial fluid of children with LCPD have been noted, consistent with a synovitis [133].
3.11.3. The Opposite Hip in Unilateral LCPD
In a review of the radiographs of 153 children with unilateral LCPD [134], 48.4% demonstrated irregularity of the epiphyseal surface, flattening, or dimpling of the opposite “normal hip.” In most instances (37%), they were present in the initial radiograph. Similar changes were noted in only 10.4% of a control group of 153 age and gender-matched children using intravenous urograms. This was interpreted as the capital femoral epiphysis in the young child being very vulnerable to stress; the minimal contour irregularities in the “normal hip” represent one end of the spectrum and frank LCPD, the other as the stress response of the capital femoral epiphysis. Another study confirmed that the “unaffected” hip in LCPD demonstrates anterior and lateral flattening perhaps indicating a constitutional abnormality [135]. In a third study, 15% of the opposite “normal” hips demonstrated physeal changes, especially decalcification below the physis [136]. The initial radiographs of 125 Japanese children with unilateral LCPD demonstrate delayed ossification of the opposite epiphysis as seen by diminished epiphyseal height [137].
3.11.4. Behavioral/Psychological Issues
Children with LCPD are extremely busy and active. An early study (PhD thesis) discovered that children with LCPD demonstrated a motor-expressive personality, an active approach to life and had higher psychosomatic and visceral complaints [138]. A later study [139] reviewed the behavioral characteristics of 24 children with LCPD; 33% of children with LCPD had abnormally high scores in standard psychological child behavioral questionnaires for profiles associated with attention deficit hyperactivity disorder, greater than the 3–5% of age matched children. Certain epidemiologic characteristics of LCPD (gender, socioeconomic status, geographic location, and associated congenital anomalies) are also similar characteristics of attention deficit hyperactivity disorder. These findings were confirmed in a recent study of 19 children with LCPD [92]; 8 of 12 school-aged children had negative scores in neuropsychological tests and 5 of the 8 had learning difficulties at school.
3.11.5. Miscellaneous Findings
An increase in both major and minor congenital defects in children with LCPD is known [61]. These include anomalies of the genitourinary tract and inguinal region [140] and spina bifida occulta [10, 141, 142]. Sacral inclination, decreased lumbar lordosis, and an overall more negative spinal balance with vertebral end plate anomalies have been recently described in the spine of LCPD patients [143].
Low blood manganese levels were noted in children with LCPD in Liverpool [144], but refuted by others [145]. An increase in IgG and IgM, but not IgA serum immunoglobulin levels in LCPD, are described [146], suggesting that immunological mechanisms may mediate certain changes in LCPD. Rubella antibody titers are higher in both mothers and affected children with LCPD [147].
4. Conclusion and Unifying Possibilities
Can these epidemiologic and demographic findings be unified? There clearly is disharmony between cartilage and bone and growth in LCPD as evidenced by progressive caudal growth impairment and delays in skeletal maturation, both involving the wrist and the pelvis. The insult on skeletal maturation appears to occur early in life, perhaps even prenatally, since there is an increased frequency of minor congenital malformations in children with LCPD. These delays in maturation (both anthropometric and skeletal age) can be due to a combination of familial and environmental circumstances (lower socioeconomic class with malnutrition [148], underlying genetic/collagen defects, or some other unknown entity). The delay in skeletal ossification results in a weaker skeleton that is more susceptible to trauma. A highly active child incurs more skeletal injuries; this microtrauma in a biologically susceptible weaker skeleton creates microfractures in the proximal femoral epiphysis and metaphysis. A hypercoagulable state, due to underlying abnormalities in the clotting mechanisms and/or exposure to passive smoke, results in increased thrombosis in the proximal femur after microfractures with subsequent necrosis of the capital femoral epiphysis and the development of LCPD.
Conflict of Interests
The author's otherwise have no financial interests with any other organizations or bodies.
Disclosure
As a systematic literature review, Institutional Review Board approval is not applicable. This was the 2nd of three presentations on the epidemiology and demographics of Pediatric Hip Disorders given at the AO North American Symposium on Surgical Preservation of the Hip, Squaw Valley, California, January 2009.
Acknowledgment
This study was supported in part by the Garceau Professorship Endowment, Department of Orthopaedic Surgery, Indiana University School of Medicine, and the George Rapp Pediatric Orthopaedic Research Endowment, Riley Children's Foundation, Riley Children's Hospital, Indianapolis, Indiana.
References
- 1.Catterall A. The natural history of Perthes’ disease. Journal of Bone and Joint Surgery. British. 1971;53(1):37–53. [PubMed] [Google Scholar]
- 2.Salter RB, Thompson GH. Legg-Calve-Perthes disease. The prognostic significance of the subchondral fracture and a two-group classification of the femoral head involvement. Journal of Bone and Joint Surgery. American. 1984;66(4):479–489. [PubMed] [Google Scholar]
- 3.Herring JA, Neustadt JB, Williams JJ, Early JS, Browne RH. The lateral pillar classification of Legg-Calve-Perthes disease. Journal of Pediatric Orthopaedics. 1992;12(2):143–150. doi: 10.1097/01241398-199203000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Nevelos AB. Perthes’ disease: the family tree. Clinical Orthopaedics and Related Research. 1986;209:13–22. [PubMed] [Google Scholar]
- 5.Joseph B, Chacko V, Rao BS, Hall AJ. The epidemiology of Perthes’ disease in south India. International Journal of Epidemiology. 1988;17(3):603–607. doi: 10.1093/ije/17.3.603. [DOI] [PubMed] [Google Scholar]
- 6.Purry NA. The incidence of Perthes’ disease in three population groups in the Eastern Cape region of South Africa. Journal of Bone and Joint Surgery. British. 1982;64(3):286–288. doi: 10.1302/0301-620X.64B3.7096393. [DOI] [PubMed] [Google Scholar]
- 7.Kim WC, Hiroshima K, Imaeda T. Multicenter study for Legg-Calvé-Perthes disease in Japan. Journal of Orthopaedic Science. 2006;11(4):333–341. doi: 10.1007/s00776-006-1021-1. [DOI] [PubMed] [Google Scholar]
- 8.Ebong WW. Legg Calve Perthes disease in Nigerians. International Surgery. 1977;62(4):217–218. [PubMed] [Google Scholar]
- 9.Rowe SM, Jung ST, Lee KB, Bae BH, Cheon SY, Kang KD. The incidence of Perthes’ disease in Korea. A focus on differences among races. Journal of Bone and Joint Surgery. British. 2005;87(12):1666–1668. doi: 10.1302/0301-620X.87B12.16808. [DOI] [PubMed] [Google Scholar]
- 10.Wijesekera C. Perthes’ disease in Kurunegala. The Ceylon Medical Journal. 1984;29(2):65–83. [PubMed] [Google Scholar]
- 11.Thompson AG, Leong JCY. Legg-Calve-Perthes' disease in Southern Chinese children. Journal of the Western Pacific Orthopaedic Association. 1978;15(1):63–70. [Google Scholar]
- 12.Gray IM, Lowry RB, Renwick DH. Incidence and genetics of Legg-Perthes disease (osteochondritis deformans) in British Columbia: evidence of polygenic determination. Journal of Medical Genetics. 1972;9(2):197–202. doi: 10.1136/jmg.9.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barker DJP, Dixon E, Taylor JF. Perthes’ disease of the hip in three regions of England. Journal of Bone and Joint Surgery. British. 1978;60(4):478–480. doi: 10.1302/0301-620X.60B4.711792. [DOI] [PubMed] [Google Scholar]
- 14.Molloy MK, MacMahon B. Incidence of Legg-Perthes disease (osteochondritis deformans) New England Journal of Medicine. 1966;275(18):988–990. doi: 10.1056/NEJM196611032751804. [DOI] [PubMed] [Google Scholar]
- 15.Hall AJ, Barker DJP. Perthes’ disease in Yorkshire. Journal of Bone and Joint Surgery. British. 1989;71(2):229–233. doi: 10.1302/0301-620X.71B2.2925740. [DOI] [PubMed] [Google Scholar]
- 16.Moberg A, Rehnberg L. Incidence of Perthes’ disease in Uppsala, Sweden. Acta Orthopaedica Scandinavica. 1992;63(2):157–158. doi: 10.3109/17453679209154813. [DOI] [PubMed] [Google Scholar]
- 17.Wiig O, Terjesen T, Svenningsen S, Lie SA. The epidemiology and aetiology of Perthes' disease in Norway. Journal of Bone and Joint Surgery. British. 2006;88(9):1217–1223. doi: 10.1302/0301-620X.88B9.17400. [DOI] [PubMed] [Google Scholar]
- 18.Margetts BM, Perry CA, Taylor JF, Dangerfield PH. The incidence and distribution of Legg-Calvé-Perthes’ disease in Liverpool, 1982-95. Archives of Disease in Childhood. 2001;84(4):351–354. doi: 10.1136/adc.84.4.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kealey WDC, Moore AJ, Cook S, Cosgrove AP. Deprivation, urbanization and Perthes’ disease in Northern Ireland. Journal of Bone and Joint Surgery. British. 2000;82(2):167–171. [PubMed] [Google Scholar]
- 20.Pillai A, Atiya S, Costigan PS. The incidence of Perthes’ disease in Southwest Scotland. Journal of Bone and Joint Surgery. British. 2005;87(11):1531–1535. doi: 10.1302/0301-620X.87B11.16744. [DOI] [PubMed] [Google Scholar]
- 21.Hall AJ, Barker DJP, Dangerfield PH, Taylor JF. Perthes’ disease of the hip in Liverpool. British Medical Journal. 1983;287(6407):1757–1759. doi: 10.1136/bmj.287.6407.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niclasen SD. Incidence of Legg-Calve-Perthes’ disease in the Faroe Islands. Annales Societatis Scientiarum Faroensis. 1974;22:9–65. [Google Scholar]
- 23.Eveleth PB, Tanner JM. Worldwide Variation in Human Growth. 2nd edition. Cambridge, UK: Cambridge University Press; 1990. [Google Scholar]
- 24.Wynne-Davies R, Gormley J. The aetiology of Perthes’ disease: genetic, epidemiological and growth factors in 310 Edinburgh and Glasgow patients. Journal of Bone and Joint Surgery. British. 1978;60(1):6–14. doi: 10.1302/0301-620X.60B1.564352. [DOI] [PubMed] [Google Scholar]
- 25.Barker DJP, Hall AJ. The epidemiology of Perthes’ disease. Clinical Orthopaedics and Related Research. 1986;209:89–94. [PubMed] [Google Scholar]
- 26.Sharma S, Sibinski M, Sherlock DA. A profile of Perthes’ disease in Greater Glasgow. Is there an association with deprivation? Journal of Bone and Joint Surgery. British. 2005;87(11):1536–1540. doi: 10.1302/0301-620X.87B11.16608. [DOI] [PubMed] [Google Scholar]
- 27.Faraj AA, Nevelos AB. Ethnic factors in perthes disease: a retrospective study among white and Asian population living in the same environment. Acta Orthopaedica Belgica. 2000;66(3):255–258. [PubMed] [Google Scholar]
- 28.Fisher RL. An epidemiological study of Legg-Perthes disease. Journal of Bone and Joint Surgery. American. 1972;54(4):769–778. [PubMed] [Google Scholar]
- 29.Rosenfeld SB, Herring JA, Chao JC. Legg-Calvé-Perthes disease: a review of cases with onset before six years of age. Journal of Bone and Joint Surgery. American. 2007;89(12):2712–2722. doi: 10.2106/JBJS.G.00191. [DOI] [PubMed] [Google Scholar]
- 30.Wiig O, Terjesen T, Svenningsen S. Prognostic factors and outcome of treatment in Perthes’ disease: a prospective study of 368 patients with five-year follow-up. Journal of Bone and Joint Surgery. British. 2008;90(10):1364–1371. doi: 10.1302/0301-620X.90B10.20649. [DOI] [PubMed] [Google Scholar]
- 31.Fulford GE, Lunn PG, Macnicol MF. A prospective study of nonoperative and operative management for Perthes’ disease. Journal of Pediatric Orthopaedics. 1993;13(3):281–285. doi: 10.1097/01241398-199305000-00001. [DOI] [PubMed] [Google Scholar]
- 32.Guille JT, Lipton GE, Szöke G, Bowen JR, Harcke HT, Glutting JJ. Legg-Calve-Perthes disease in girls. A comparison of the results with those seen in boys. Journal of Bone and Joint Surgery. American. 1998;80(9):1256–1263. doi: 10.2106/00004623-199809000-00002. [DOI] [PubMed] [Google Scholar]
- 33.Wang NH, Lee FT, Chin LS, Lo WH. Legg-Calve-Perthes disease: clinical analysis of 57 cases. Journal of the Formosan Medical Association. 1990;89(9):764–771. [PubMed] [Google Scholar]
- 34.Petrie JG, Bitenc I. The abduction weight-bearing treatment in Legg-Perthes’ disease. Journal of Bone and Joint Surgery. British. 1971;53(1):54–62. [PubMed] [Google Scholar]
- 35.Herring JA, Hui TK, Browne R. Legg-Calvé-Perthes disease. Part II: prospective multicenter study of the effect of treatment on outcome. Journal of Bone and Joint Surgery. American. 2004;86(10):2121–2134. [PubMed] [Google Scholar]
- 36.Chacko V, Joseph B, Seetharam B. Perthes’ disease in South India. Clinical Orthopaedics and Related Research. 1986;209:95–99. [PubMed] [Google Scholar]
- 37.O’Sullivan M, O’Rourke SK, MacAuley P. Legg-Calve-Perthes disease in a family: genetic or environmental. Clinical Orthopaedics and Related Research. 1985;199:179–181. [PubMed] [Google Scholar]
- 38.Miyamoto Y, Matsuda T, Kitoh H, et al. A recurrent mutation in type II collagen gene causes Legg-Calvé-Perthes disease in a Japanese family. Human Genetics. 2007;121(5):625–629. doi: 10.1007/s00439-007-0354-y. [DOI] [PubMed] [Google Scholar]
- 39.Su P, Li R, Liu S, et al. Age at onset-dependent presentations of premature hip osteoarthritis, avascular necrosis of the femoral head, or legg-calvé-perthes disease in a single family, consequent upon a p.Gly1170Ser mutation of COL2A1. Arthritis and Rheumatism. 2008;58(6):1701–1706. doi: 10.1002/art.23491. [DOI] [PubMed] [Google Scholar]
- 40.Stephens FE, Kerby JP. Hereditary Legg-Calve-Perthes’ disease. Journal of Heredity. 1946;37(5):153–160. doi: 10.1093/oxfordjournals.jhered.a105605. [DOI] [PubMed] [Google Scholar]
- 41.McComas E. Perthes’ disease and its occurrence as a familial condition. The Medical Journal of Australia. 1946;2:584–585. doi: 10.5694/j.1326-5377.1946.tb75972.x. [DOI] [PubMed] [Google Scholar]
- 42.Burlington H. Legg-Perthes disease in siblings; a report of two cases with simultaneous onset. Pennsylvania Medical Journal. 1958;61(7):887–888. [PubMed] [Google Scholar]
- 43.Tracy HW. Coxa plana in siblings. North Carolina Medical Journal. 1963;24:76–79. [PubMed] [Google Scholar]
- 44.Hall DJ. Genetic aspects of Perthes’ disease: a critical review. Clinical Orthopaedics and Related Research. 1986;209:100–114. [PubMed] [Google Scholar]
- 45.Harper PS, Brotherton BJ, Cochlin D. Genetic risks in Perthes’ disease. Clinical Genetics. 1976;10(3):178–182. doi: 10.1111/j.1399-0004.1976.tb00030.x. [DOI] [PubMed] [Google Scholar]
- 46.Wamoscher Z, Farhi A. Hereditary Legg-Calve-Perthes disease. American Journal of Diseases of Children. 1963;106:97–100. doi: 10.1001/archpedi.1963.02080050099016. [DOI] [PubMed] [Google Scholar]
- 47.Niclasen SD. Family studies of relation between Perthes disease and congenital dislocation of the hip. Journal of Medical Genetics. 1978;15(4):296–299. doi: 10.1136/jmg.15.4.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Inglis A. Genetic implications in coxa plana. The Journal of Bone and Joint Surgery. American. 1960;42-A:711–715. [PubMed] [Google Scholar]
- 49.Giannestras N. Legg-Perthes disease in twins. The Journal of Bone and Joint Surgery. American. 1954;36-A:149–152. [PubMed] [Google Scholar]
- 50.Dunn AW. Coxa plana in monozygotic male twins. The Journal of Bone and Joint Surgery. American. 1960;42-A:178–183. [PubMed] [Google Scholar]
- 51.Söderberg L. Simultaneous occurrence of coxa plana in enzygotic twins. Acta Orthopaedica Scandinavica. 1957;27:135–140. [PubMed] [Google Scholar]
- 52.Derian PS. Coxa plana in dizygotic male twins. The Journal of Bone and Joint Surgery. American. 1956;38-A:901–907. [PubMed] [Google Scholar]
- 53.Livesey JP, Hay SM, Bell MJ. Perthes disease affecting three female first-degree relatives. Journal of Pediatric Orthopaedics Part B. 1998;7(3):230–231. doi: 10.1097/01202412-199807000-00010. [DOI] [PubMed] [Google Scholar]
- 54.Bertrams J, Schiersmann P, Ritgen G. HLA antigens in Perthes’ disease. Tissue Antigens. 1978;12(2):157–158. doi: 10.1111/j.1399-0039.1978.tb01316.x. [DOI] [PubMed] [Google Scholar]
- 55.Zervas J, Theordorou S, Constantopoulos C. HLA-A1 probable association with Perthes' disease. Tissue Antigens. 1982;20(2):159–160. doi: 10.1111/j.1399-0039.1982.tb00341.x. [DOI] [PubMed] [Google Scholar]
- 56.Halwag IAE, El-Abbasy EMK, Zaki SA, Abou-Khatwn MA. Association between HLA-A and -B and Perthes’ disease. Journal of the Medical Research Institute. 1991;12:89–98. [Google Scholar]
- 57.Dostál C, Raušer V, Popelka S, Ivašková E, Macurová M, Zvárová J. HLA antigens in Perthes disease. Acta Chirurgiae Orthopaedicae et Traumatologiae Cechoslovaca. 1987;54:324–329. [PubMed] [Google Scholar]
- 58.Thompson SK, Woodrow JC. HLA antigens in Perthes’ disease. Journal of Bone and Joint Surgery. British. 1981;63(2):278–279. doi: 10.1302/0301-620X.63B2.7217156. [DOI] [PubMed] [Google Scholar]
- 59.Mach J, Wegener S. HLA-examinations at Perthes disease. Beitrage zur Orthopadie und Traumatologie. 1985;32(12):590–592. [PubMed] [Google Scholar]
- 60.Cameron JM, Izatt MM. The ABO and rhesus blood groups in Perthes’ disease. Journal of Clinical Pathology. 1962;15:163–168. doi: 10.1136/jcp.15.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall DJ, Harrison MHM, Burwell RG. Congenital abnormalities and Perthes’ disease. Clinical evidence that children with Perthes’ disease may have a major congenital defect. Journal of Bone and Joint Surgery. British. 1979;61(1):18–25. doi: 10.1302/0301-620X.61B1.33996. [DOI] [PubMed] [Google Scholar]
- 62.Molloy MK, Macmahon B. Birth weight and Legg-Perthes disease. Journal of Bone and Joint Surgery. American. 1967;49(3):498–506. [PubMed] [Google Scholar]
- 63.Lappin K, Kealey D, Cosgrove A, Graham K. Does low birthweight predispose to Perthes’ disease? Perthes’ disease in twins. Journal of Pediatric Orthopaedics Part B. 2003;12(5):307–310. doi: 10.1097/01.bpb.0000079203.23239.2d. [DOI] [PubMed] [Google Scholar]
- 64.Weiner DS, O’Dell HW. Legg-Calvé-Perthes disease. Observations on skeletal maturation. Clinical Orthopaedics and Related Research. 1970;68:44–49. [PubMed] [Google Scholar]
- 65.Motokawa S. Effect of serum factors on skeletal growth in Perthes' disease. Journal of the Japanese Orthopaedic Association. 1990;64(9):790–797. [PubMed] [Google Scholar]
- 66.Eckerwall G, Wingstrand H, Hägglund G, Karlberg J. Growth in 110 children with Legg-Calve-Perthes’ disease: a longitudinal infancy childhood puberty growth model study. Journal of Pediatric Orthopaedics Part B. 1996;5(3):181–184. [PubMed] [Google Scholar]
- 67.Shapiro F. Legg-Calve-Perthes disease. A study of lower extremity length discrepancies and skeletal maturation. Acta Orthopaedica Scandinavica. 1982;53(3):437–444. doi: 10.3109/17453678208992238. [DOI] [PubMed] [Google Scholar]
- 68.Kealey WDC, Lappin KJ, Leslie H, Sheridan B, Cosgrove AP. Endocrine profile and physical stature of children with Perthes disease. Journal of Pediatric Orthopaedics. 2004;24(2):161–166. doi: 10.1097/00004694-200403000-00005. [DOI] [PubMed] [Google Scholar]
- 69.Laron Z, Axer A, Drezner Z. Growth and development of children with Legg Calve Perthes syndrome in Israel. Israel Journal of Medical Sciences. 1973;9(5):612–616. [PubMed] [Google Scholar]
- 70.Burwell RG, Dangerfield PH, Hall DJ. Perthes’ disease. An anthropometric study revealing impaired and disproportionate growth. Journal of Bone and Joint Surgery. British. 1978;60(4):461–477. doi: 10.1302/0301-620X.60B4.711791. [DOI] [PubMed] [Google Scholar]
- 71.Hall AJ, Barker DJP, Dangerfield PH, Osmond C, Taylor JF. Small feet and Perthes’ disease. A survey in Liverpool. Journal of Bone and Joint Surgery. British. 1988;70(4):611–613. doi: 10.1302/0301-620X.70B4.3403608. [DOI] [PubMed] [Google Scholar]
- 72.Rao BS, Joseph B, Chacko V, Hall AJ. Altered skeletal growth in Perthes’ disease: an anthropometric study of children from rural India. Journal of Pediatric Orthopaedics. Part B. 1995;4(1):91–94. [PubMed] [Google Scholar]
- 73.Greulich WW, Pyle SI. Radiographic Atlas of Skeletal Development of the Hand and Wrist. 2nd edition. Stanford, Calif, USA: Stanford University Press; 1959. [Google Scholar]
- 74.Acheson RM. The Oxford method of assessing skeletal maturity. Clinical orthopaedics. 1957;10:19–39. [PubMed] [Google Scholar]
- 75.Loder RT, Farley FA, Herring JA, Schork MA, Shyr Y. Bone age determination in children with Legg-Calve-Perthes disease: a comparison of two methods. Journal of Pediatric Orthopaedics. 1995;15(1):90–94. doi: 10.1097/01241398-199501000-00019. [DOI] [PubMed] [Google Scholar]
- 76.Harrison MHM, Turner MH, Jacobs P. Skeletal immaturity in Perthes’ disease. Journal of Bone and Joint Surgery. British. 1976;58(1):37–40. doi: 10.1302/0301-620X.58B1.178665. [DOI] [PubMed] [Google Scholar]
- 77.Kristmundsdottir F, Burwell RG, Hall DJ, Marshall WA. A longitudinal study of carpal bone development in Perthes’ disease: its significance for both radiologic standstill and bilateral disease. Clinical Orthopaedics and Related Research. 1986;209:115–123. [PubMed] [Google Scholar]
- 78.Kristmundsdottir F, Burwell RG, Harrison MHM. Delayed skeletal maturation in Perthes’ disease. Acta Orthopaedica Scandinavica. 1987;58(3):277–279. doi: 10.3109/17453678709146484. [DOI] [PubMed] [Google Scholar]
- 79.Lee ST, Vaidya SV, Song HR, Lee SH, Suh SW, Telang SS. Bone age delay patterns in legg-calvé-perthes disease: an analysis using the tanner and whitehouse 3 method. Journal of Pediatric Orthopaedics. 2007;27(2):198–203. doi: 10.1097/01.bpb.0000242381.89510.e5. [DOI] [PubMed] [Google Scholar]
- 80.Vila-Verde VMR, Da Silva KC. Bone age delay in Perthes disease and transient synovitis of the hip. Clinical Orthopaedics and Related Research. 2001;(385):118–123. doi: 10.1097/00003086-200104000-00019. [DOI] [PubMed] [Google Scholar]
- 81.Zarco NAN, Morán JM, Hernández MA. Delay in skeletal maturity in Mexican patients with Legg-Calvé-Perthes. Acta Ortopédica Mexicana. 2008;22(3):157–161. [PubMed] [Google Scholar]
- 82.Nishi Y, Tanaka T, Fujieda K, et al. Slipped capital femoral epiphysis, Perthes’ disease and scoliosis in children with growth hormone deficiency. Endocrine Journal. 1998;45:S167–S169. doi: 10.1507/endocrj.45.suppl_s167. [DOI] [PubMed] [Google Scholar]
- 83.Rayner PHW, Schwalbe SL, Hall DJ. An assessment of endocrine function in boys with Perthes’ disease. Clinical Orthopaedics and Related Research. 1986;209:124–128. [PubMed] [Google Scholar]
- 84.Tanaka H, Tamura K, Takano K. Serum somatomedin A in Perthes' disease. Acta Orthopaedica Scandinavica. 1984;55(2):135–140. doi: 10.3109/17453678408992324. [DOI] [PubMed] [Google Scholar]
- 85.Burwell RG, Vernon CL, Dangerfield PH. Raised somatomedin activity in the serum of young boys with Perthes’ disease revealed by bioassay: a disease of growth transition? Clinical Orthopaedics and Related Research. 1986;209:129–138. [PubMed] [Google Scholar]
- 86.Kitsugi T, Kasahara Y, Seto Y, Komai S. Normal somatomedin-C activity measured by radioimmunoassay in Perthes’ disease. Clinical Orthopaedics and Related Research. 1989;(244):217–221. [PubMed] [Google Scholar]
- 87.Neidel J, Zander D, Hackenbroch MH. Low plasma levels of insulin-like growth factor I in Perthes’ disease. A controlled study of 59 consecutive children. Acta Orthopaedica Scandinavica. 1992;63(4):393–398. doi: 10.3109/17453679209154752. [DOI] [PubMed] [Google Scholar]
- 88.Neidel J, Schonau E, Zander D, Rutt J, Hackenbroch MH. Normal plasma levels of IGF binding protein in Perthes’ disease. Follow-up of previous report. Acta Orthopaedica Scandinavica. 1993;64(5):540–542. doi: 10.3109/17453679308993688. [DOI] [PubMed] [Google Scholar]
- 89.Crofton PM, Macfarlane C, Wardhaugh B, et al. Children with acute Perthes’ disease have asymmetrical lower leg growth and abnormal collagen turnover. Acta Orthopaedica. 2005;76(6):841–847. doi: 10.1080/17453670510045462. [DOI] [PubMed] [Google Scholar]
- 90.Matsumoto T, Enomoto H, Takahashi K, Motokawa S. Decreased levels of IGF binding protein-3 in serum from children with Perthes’ disease. Acta Orthopaedica Scandinavica. 1998;69(2):125–128. doi: 10.3109/17453679809117611. [DOI] [PubMed] [Google Scholar]
- 91.Grasemann H, Nicolai RD, Hauffa BP, Reinhardt W, Nicolai H, Hövel M. Skeletal immaturity, IGF-I and IGFBP-3 serum concentrations in Legg-Calve-Perthes disease (skeletal immaturity, IGF-I and IGFBP-3 in LCPD) Klinische Padiatrie. 1996;208(6):339–343. doi: 10.1055/s-2008-1046494. [DOI] [PubMed] [Google Scholar]
- 92.Lahdes-Vasama TT, Sipilä ISJ, Lamminranta S, Pihko SH, Merikanto EOJ, Marttinen EJ. Psychosocial development and premorbid skeletal growth in legg-calve- perthes disease: a study of nineteen patients. Journal of Pediatric Orthopaedics Part B. 1997;6(2):133–137. doi: 10.1097/01202412-199704000-00009. [DOI] [PubMed] [Google Scholar]
- 93.Emerick RW, Corrigan KE, Joistad AH, Jr., Holly LE. Thyroid function in Legg-Calve-Perthes disease—a new approach to an old problem. Clinical Orthopaedics and Related Research. 1954;4:160–165. [PubMed] [Google Scholar]
- 94.Cavanaugh LA, Shelton EK, Barbara S, Sutherland R. Metabolic studies in osteochondritis of the capital femoral epiphysis. The Journal of Bone and Joint Surgery. American. 1936;18:957–968. [Google Scholar]
- 95.Gill AB. Relationship of Legg-Perthes disease to the function of the thyroid gland. The Journal of Bone and Joint Surgery. American. 1943;25-A:892–901. [Google Scholar]
- 96.Katz JF. Protein-bound iodine in Legg-Calvé-Perthes disease. The Journal of Bone and Joint Surgery. American. 1955;37(4):842–846. [PubMed] [Google Scholar]
- 97.Beiler DD, Love WH. Thyroid function in Legg-Perthes disease. The Journal of Bone and Joint Surgery. American. 1956;38(6):1320–1322. [PubMed] [Google Scholar]
- 98.Chapman EM. Thyroid function in Legg-Perthes disease. The New England journal of medicine. 1956;255(6):289–290. doi: 10.1056/NEJM195608092550609. [DOI] [PubMed] [Google Scholar]
- 99.Bahmanyar S, Montgomery SM, Weiss RJ, Ekbom A. Maternal smoking during pregnancy, other prenatal and perinatal factors, and the risk of legg-calvé-perthes disease. Pediatrics. 2008;122(2):e459–e464. doi: 10.1542/peds.2008-0307. [DOI] [PubMed] [Google Scholar]
- 100.Gordon JE, Schoenecker PL, Osland JD, Dobbs MB, Szymanski DA, Luhmann SJ. Smoking and socio-economic status in the etiology and severity of Legg-Calvé-Perthes’ disease. Journal of Pediatric Orthopaedics Part B. 2004;13(6):367–370. doi: 10.1097/01202412-200411000-00003. [DOI] [PubMed] [Google Scholar]
- 101.García Mata S, Ardanaz Aicua E, Hidalgo Ovejero A, Martinez Grande M. Legg-Calvé-Perthes disease and passive smoking. Journal of Pediatric Orthopaedics. 2000;20(3):326–330. [PubMed] [Google Scholar]
- 102.Glueck CJ, Freiberg RA, Crawford A, et al. Secondhand smoke, hypofibrinolysis, and Legg-Perthes disease. Clinical Orthopaedics and Related Research. 1998;(352):159–167. [PubMed] [Google Scholar]
- 103.Dilley A, Hooper WC, Austin H, et al. The beta fibrinogen gene G-455-A polymorphism is a risk factor for Legg-Perthes disease. Journal of Thrombosis and Haemostasis. 2003;1(11):2317–2321. doi: 10.1046/j.1538-7836.2003.00416.x. [DOI] [PubMed] [Google Scholar]
- 104.Bertina RM, Koeleman BPC, Koster T, et al. Mutation in blood coagulation factor V associated with resistance to activated protein C. Nature. 1994;369(6475):64–67. doi: 10.1038/369064a0. [DOI] [PubMed] [Google Scholar]
- 105.Glueck CJ, Tracy T, Wang P. Legg-calve-perthes disease, venous and arterial thrombi, and the factor V leiden mutation in a four-generation kindred. Journal of Pediatric Orthopaedics. 2007;27(7):834–837. doi: 10.1097/BPO.0b013e31815584bf. [DOI] [PubMed] [Google Scholar]
- 106.Gruppo R, Glueck CJ, Wall E, Roy D, Wang P. Legg-Perthes disease in three siblings, two heterozygous and one homozygous for the factor V Leiden mutation. Journal of Pediatrics. 1998;132(5):885–888. doi: 10.1016/s0022-3476(98)70326-9. [DOI] [PubMed] [Google Scholar]
- 107.Glueck CJ, Brandt G, Gruppo R, et al. Resistance to activated protein C and Legg-Perthes disease. Clinical Orthopaedics and Related Research. 1997;(338):139–152. doi: 10.1097/00003086-199705000-00021. [DOI] [PubMed] [Google Scholar]
- 108.Szepesi K, Pósán E, Hársfalvi J, et al. The most severe forms of Perthes’ disease associated with the homozygous Factor V Leiden mutation. Journal of Bone and Joint Surgery. British. 2004;86(3):426–429. doi: 10.1302/0301-620x.86b3.13442. [DOI] [PubMed] [Google Scholar]
- 109.Sanders LLO, Braga Júnior MB, Cima CWM, Mota RMS, Pardini MIMC, Rabenhorst SHB. Leiden's V-factor in Legg-Calvé-Perthes disease. Acta Ortopedica Brasileira. 2009;17(2):40–42. [Google Scholar]
- 110.Balasa VV, Gruppo RA, Glueck CJ, et al. Legg-Calvé-Perthes disease and thrombophilia. Journal of Bone and Joint Surgery. American. 2004;86(12):2642–2647. doi: 10.2106/00004623-200412000-00009. [DOI] [PubMed] [Google Scholar]
- 111.Eldridge J, Dilley A, Austin H, et al. The role of protein C, protein S, and resistance to activated protein C in Legg-Perthes disease. Pediatrics. 2001;107(6):1329–1334. doi: 10.1542/peds.107.6.1329. [DOI] [PubMed] [Google Scholar]
- 112.Arruda VR, Belangero WD, Ozelo MC, et al. Inherited risk factors for thrombophilia among children with Legg-Calve-Perthes disease. Journal of Pediatric Orthopaedics. 1999;19(1):84–87. [PubMed] [Google Scholar]
- 113.Vosmaer A, Pereira RR, Koenderman JS, Rosendaal FR, Cannegieter SC. Coagulation abnormalities in Legg-Calvé-Perthes disease. Journal of Bone and Joint Surgery. American. 2010;92(1):121–128. doi: 10.2106/JBJS.I.00157. [DOI] [PubMed] [Google Scholar]
- 114.Glueck CJ, Glueck HI, Greenfield D, et al. Protein C and S deficiency, thrombophilia, and hypofibrinolysis: pathophysiologic causes of Legg-Perthes disease. Pediatric Research. 1994;35(4):383–388. [PubMed] [Google Scholar]
- 115.Glueck CJ, Crawford A, Roy D, Freiberg R, Glueck H, Stroop D. Association of antithrombotic factor deficiencies and hypofibrinolysis with Legg-Perthes disease. Journal of Bone and Joint Surgery. American. 1996;78(1):3–13. doi: 10.2106/00004623-199601000-00002. [DOI] [PubMed] [Google Scholar]
- 116.Koo KH, Song HR, Ha YC, et al. Role of thrombotic and fibrinolytic disorders in the etiology of Perthes’ disease. Clinical Orthopaedics and Related Research. 2002;(399):162–167. doi: 10.1097/00003086-200206000-00019. [DOI] [PubMed] [Google Scholar]
- 117.Yilmaz D, Karapinar L, Karapinar B, Ozturk H, Kavakli K. Evaluation of anticoagulant system in Turkish children with Perthes disease. Pediatrics International. 2005;47(1):43–48. doi: 10.1111/j.1442-200x.2004.01995.x. [DOI] [PubMed] [Google Scholar]
- 118.Kealey WDC, Mayne EE, McDonald W, Murray P, Cosgrove AP. The role of coagulation abnormalities in the development of Perthe’s disease. Journal of Bone and Joint Surgery. British. 2000;82(5):744–746. doi: 10.1302/0301-620x.82b5.10183. [DOI] [PubMed] [Google Scholar]
- 119.Hresko MT, McDougall PA, Gorlin JB, Vamvakas EC, Kasser JR, Neufeld EJ. Prospective reevaluation of the association between thrombotic diathesis and legg-perthes disease. Journal of Bone and Joint Surgery. American. 2002;84(9):1613–1618. doi: 10.2106/00004623-200209000-00014. [DOI] [PubMed] [Google Scholar]
- 120.López-Franco M, González-Morán G, De Lucas JC, et al. Legg-Perthes disease and heritable thrombophilia. Journal of Pediatric Orthopaedics. 2005;25(4):456–459. doi: 10.1097/01.bpo.0000158781.29979.cf. [DOI] [PubMed] [Google Scholar]
- 121.Gallistl S, Reitinger T, Linhart W, Muntean W. The role of inherited thrombotic disorders in the etiology of Legg-Calve-Perthes disease. Journal of Pediatric Orthopaedics. 1999;19(1):82–83. [PubMed] [Google Scholar]
- 122.Mehta JS, Conybeare ME, Hinves BL, Winter JBM. Protein C levels in patients with Legg-Calve-Perthes disease: is it a true deficiency? Journal of Pediatric Orthopaedics. 2006;26(2):200–203. doi: 10.1097/01.bpo.0000194698.21645.37. [DOI] [PubMed] [Google Scholar]
- 123.Shiba R, Fujii S. Legg-Calve-Perthes in children. Letter to the editor. Clinical Orthopaedics and Related Research. 1999;363:268–269. [PubMed] [Google Scholar]
- 124.Kenet G, Ezra E, Wientroub S, et al. Perthes’ disease and the search for genetic associations: collagen mutations, Gaucher’s disease and thrombophilia. Journal of Bone and Joint Surgery. British. 2008;90(11):1507–1511. doi: 10.1302/0301-620X.90B11.20318. [DOI] [PubMed] [Google Scholar]
- 125.Hayek S, Kenet G, Lubetsky A, Rosenberg N, Gitel S, Wientroub S. Does thrombophilia play an aetiological role in Legg-Calve-Perthes disease? Journal of Bone and Joint Surgery. British. 1999;81(4):686–690. doi: 10.1302/0301-620x.81b4.9378. [DOI] [PubMed] [Google Scholar]
- 126.Sirvent N, Fisher F, El Hayek T, Appert A, Giudicelli H, Griffet J. Absence of congenital prethrombotic disorders in children with Legg-Perthes disease. Journal of Pediatric Orthopaedics Part B. 2000;9(1):24–27. doi: 10.1097/01202412-200001000-00005. [DOI] [PubMed] [Google Scholar]
- 127.Matos MA. The role of protein C deficiency in the etiology of Perthes disease. Ortopedia Traumatologia Rehabilitacja. 2008;10(3):274–278. [PubMed] [Google Scholar]
- 128.Aksoy MC, Aksoy DY, Haznedaroglu IC, Sayinalp N, Kirazli S, Alpaslan M. Enhanced tissue factor pathway inhibitor response as a defense mechanism against ongoing local microvascular events of Legg-Calve-Perthes disease. Pediatric Hematology and Oncology. 2005;22(5):391–399. doi: 10.1080/08880010590964273. [DOI] [PubMed] [Google Scholar]
- 129.Kleinman RG, Bleck EE. Increased blood viscosity in patients with Legg-Perthes disease: a preliminary report. Journal of Pediatric Orthopaedics. 1981;1(2):131–136. doi: 10.1097/01241398-198110000-00002. [DOI] [PubMed] [Google Scholar]
- 130.Aksoy MC, Aksoy DY, Haznedaroglu IC, Sayinalp N, Kirazli S, Alpaslan M. Thrombomodulin and GFC levels in Legg-Calve-Perthes disease. Hematology. 2008;13(6):324–328. doi: 10.1179/102453308X343509. [DOI] [PubMed] [Google Scholar]
- 131.Westhoff B, Krauspe R, Kalke AE, et al. Urinary excretion of deoxypyridinoline in Perthes’ disease. A prospective, controlled comparative study in 83 children. Journal of Bone and Joint Surgery. British. 2006;88(7):967–971. doi: 10.1302/0301-620X.88B7.16564. [DOI] [PubMed] [Google Scholar]
- 132.Orfanos I, Magkou C, Anastasopoulos I, Karanikas E, Sitaras NM. Urine glycosaminoglycans in children with transient hip synovitis and Perthes disease. Journal of Pediatric Orthopaedics Part B. 2005;14(2):92–96. doi: 10.1097/01202412-200503000-00006. [DOI] [PubMed] [Google Scholar]
- 133.Eckerwall G, Stefan Lohmander L, Wingstrand H. Increased levels of proteoglycan fragments and stromelysin in hip joint fluid in Legg-Calve-Perthes disease. Journal of Pediatric Orthopaedics. 1997;17(2):266–269. doi: 10.1097/00004694-199703000-00023. [DOI] [PubMed] [Google Scholar]
- 134.Harrison MHM, Blakemore ME. A study of the ’normal’ hip in children with unilateral Perthes’ disease. Journal of Bone and Joint Surgery. British. 1980;62(1):31–36. doi: 10.1302/0301-620X.62B1.7351433. [DOI] [PubMed] [Google Scholar]
- 135.Arie E, Johnson F, Harrison MHM. Femoral head shape in Perthes' disease: is the contralateral hip abnormal? Clinical Orthopaedics and Related Research. 1986;209:77–88. [PubMed] [Google Scholar]
- 136.Kandzierski G, Karski T, Kozlowski K. Capital femoral epiphysis and growth plate of the asymptomatic hip joint in unilateral Perthes disease. Journal of Pediatric Orthopaedics Part B. 2003;12(6):380–386. doi: 10.1097/01.bpb.0000079200.23239.f6. [DOI] [PubMed] [Google Scholar]
- 137.Kitoh H, Kitakoji T, Katoh M, Takamine Y. Delayed ossification of the proximal capital femoral epiphysis in Legg-Calvé-Perthes’ disease. Journal of Bone and Joint Surgery. British. 2003;85(1):121–124. doi: 10.1302/0301-620x.85b1.13426. [DOI] [PubMed] [Google Scholar]
- 138.Fitzgerald WE. A psychological factor in Legg-Perthes-Calvé disease. Boston, Mass, USA: Harvard University; 1961. Ph.D. thesis. [Google Scholar]
- 139.Loder RT, Schwartz EM, Hensinger RN. Behavioral characteristics of children with Legg-Calve-Perthes disease. Journal of Pediatric Orthopaedics. 1993;13(5):598–601. [PubMed] [Google Scholar]
- 140.Catterall A, Roberts GC, Wynne-Davies R. Association of Perthes' disease with congenital anomalies of genitourinary tract and inguinal region. Lancet. 1971;1(7707):996–997. doi: 10.1016/s0140-6736(71)91389-4. [DOI] [PubMed] [Google Scholar]
- 141.Makin M, Meyer S, Gilai AN. Perthes’ disease and posterior lumbosacral union. Journal of Pediatric Orthopaedics. 1992;12(5):607–609. [PubMed] [Google Scholar]
- 142.Katz JF. Spina bifida occulta in Legg-Calvé-Perthes disease. Clinical Orthopaedics and Related Research. 1959;14:110–118. doi: 10.1007/s11999-011-1803-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kitoh H, Kitakoji T, Katoh M, Ishiguro N. Sagittal spinal alignment in patients with Legg-Calve-Perthes disease. Pediatrics International. 2007;49(5):612–617. doi: 10.1111/j.1442-200X.2007.02428.x. [DOI] [PubMed] [Google Scholar]
- 144.Hall AJ, Margetts BM, Barker DJP, et al. Low blood manganese levels in Liverpool children with Perthes' disease. Paediatric and Perinatal Epidemiology. 1989;3(2):131–136. doi: 10.1111/j.1365-3016.1989.tb00505.x. [DOI] [PubMed] [Google Scholar]
- 145.Perry CA, Taylor JF, Nunn A, Dangerfield PH, Delves H. Perthes’ disease and blood manganese levels. Archives of Disease in Childhood. 2000;82(5):429–430. doi: 10.1136/adc.82.5.428f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Joseph B. Serum immunoglobulin in Perthes’ disease. Journal of Bone and Joint Surgery. British. 1991;73(3):509–510. doi: 10.1302/0301-620X.73B3.1670460. [DOI] [PubMed] [Google Scholar]
- 147.Matsoukas JA. Viral antibody titers to rubella in coxa plana or Perthes' disease. Perthes' disease: is it the late osseous residua of a minor prenatal rubella? Acta Orthopaedica Scandinavica. 1975;46(6):957–962. doi: 10.3109/17453677508989284. [DOI] [PubMed] [Google Scholar]
- 148.Jansen W, Hazebroek-Kampschreur AAJM. Differences in height and weight between children living in neighbourhoods of different socioeconomic status. Acta Paediatrica. 1997;86(2):224–225. doi: 10.1111/j.1651-2227.1997.tb08875.x. [DOI] [PubMed] [Google Scholar]